
An electrochemical sensor highly selective for lindane determination: a comparative study using three different α-MnO2 nanostructures†
M. U. Anu Prathap,
Shengnan Sun and
Zhichuan J. Xu*
School of Materials Science and Engineering, Nanyang Technological University, 50 Nanyang Avenue, Singapore 639798, Singapore. E-mail: xuzc@ntu.edu.sg
First published on 22nd February 2016
Abstract
Here we describe a simple, highly reproducible ultra-sensitive electrochemical sensor for lindane based on α-MnO2 nanostructures. The results showed that the α-MnO2 nanostructures effectively catalyzed the electrochemical reduction of lindane. A good linearity was obtained in the range of 1.1 to 510 μM with a detection limit of 114 nM. The proposed lindane sensor was successfully employed for the determination of lindane in tap water samples with good recoveries. Negligible amperometric currents are observed in the control experiments using triclosan (T), chlorobenzene (CB), benzene (B), 1,3,5-trichlorobenzene (1,3,5-TCB), and 4-chlorobenzaldehyde (4-CBA), suggesting a sensing specificity to lindane. The proposed sensor also exhibited good stability and reproducibility for lindane determination.
Introduction
Lindane (a gamma isomer of hexachlorocyclohexane) is an organochlorine insecticide named after the Dutch chemist Teunis van der Linden who was the first to isolate α-, β-, and γ-isomers of hexachlorocyclohexane in 1912.1,2 The insecticidal properties of lindane were first demonstrated by Ulmann, 1972.1,2 Lindane has been used to treat food crops and in forestry products, as a seed treatment, a soil treatment, and to treat livestock and pets.1–3 Lindane is reported to be a central nervous system stimulant. The symptoms of acute exposure include mental and motor retardation, central nervous system excitation, tonic–clonic convulsions, respiratory failure, pulmonary edema and dermatitis.2,3 Lindane enters animal tissues through the food chain, respiration or dermal contact.2,3 After being under review for nearly 30 years, the pesticide lindane was finally withdrawn by the Environmental Protection Agency (EPA) for use in agriculture.3,4 Lindane is recognized by the EPA as one of the most toxic, persistent, bio-accumulative pesticides ever registered.5 Despite a recent global ban on its agricultural use, the pesticide is still used and approved by the Food and Drug Administration (FDA) as a second-line therapy for the topical treatment of pediculosis capitis (head lice), pediculosis pubis (pubic lice), or scabies in patients greater than two years of age who cannot tolerate or have failed first-line treatment.5 Therefore, the quantitative analysis of lindane is a paramount area in the context of environmental protection. New methods for lindane detection include more sophisticated analytical approaches, such as gas chromatography, liquid chromatography or gas chromatographic-mass spectrometry.6,7 Although chromatographic methods are accurate, they are expensive and time consuming.6–8 Among the various biosensing approaches, available to date, electrochemical sensors are emerging as a technology of choice due to their rapid and sensitive response, low fabrication cost and operational convenience.7,8 An excellent study discussed the reductive dechlorination of lindane catalyzed by a bioreceptor probe.9 It was found that the specificity of dechlorination depended on the enzyme stability, pH and temperature, which is the major limitation.7 Lindane has a very low aqueous solubility and has a very high overpotential for reduction; as a result the direct reduction of lindane using an aqueous medium is a challenging task. Recent publications discussing the dechlorination of lindane in ethanol/water and lindane gave only one reduction wave and benzene as the reduction product with 100% yield.10–15 The results of other researchers showed that the chloride ion was liberated from the lindane structure more rapidly than hydrogen or carbon atom fragments.10–15 Few studies have been dedicated to the non-enzymatic sensing of lindane using metal oxide, and it appears to be more effective.12,15 Thus, it is important to pursue a suitable modified electrode for the efficient reduction of lindane.In recent years, the application of nanoscaled metal oxides has received great attention in biotechnology and bio-analytical chemistry.16–20 The transition metal element manganese can exist as a variety of stable oxides (MnO, Mn3O4, Mn2O3, MnO2) and crystallize in various types of crystal structures. Among them, α-MnO2 has been widely investigated and extensively used in adsorption, catalysis, sensors, rechargeable lithium batteries and ion exchanges owing to the existing unique layers or tunnels in the crystal lattices.21–23 It is generally accepted that the physicochemical properties of MnO2 are greatly affected by the crystallographic structure, morphology, and surface area.21
In this work, we report three synthetic pathways for the preparation of α-MnO2 nanostructures (e.g., NW (nanowire), NT (nanotube), and MS (microsphere), designated as: α-MnO2-NW, α-MnO2-NT, and α-MnO2-MS). We further demonstrated that α-MnO2 nanostructures exhibit an excellent electrocatalytic activity for lindane reduction. The prepared sensor was characterized by electrochemical impedance spectroscopy (EIS), differential pulse voltammetry (DPV), cyclic voltammetry (CV), pulsed amperometry (PA) and chronoamperometry. Under the optimized conditions, the prepared sensor exhibited a selective recognition and highly sensitive detection toward lindane.
Experimental
Materials
All chemicals were analytical grade and used without further purification. KMnO4, urea, γ-HCCH (lindane), cyclohexane, chlorobenzene, 4-chlorobenzaldehyde, triclosan, benzene and tetra-n-butyl ammonium bromide (TBAB) were obtained from Sigma-Aldrich. Deionized water from Millipore Milli-Q system (resistivity 18.2 MΩ cm) was used in the electrochemical studies. Solutions for all electrochemical experiments were deoxygenated with zero-grade argon.Preparation of solution
A stock solution of lindane was prepared in methanol. 0.05 M tetra-n-butyl ammonium bromide (TBAB) solution was prepared in 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 (v/v) methanol–water. It was used as the supporting electrolyte.
:40 (v/v) methanol–water. It was used as the supporting electrolyte.
Synthesis of α-MnO2 microspheres (α-MnO2-MS)
α-MnO2-MS was synthesized by a hydrothermal approach with a modified procedure first reported by our group.15 1.5 mmol (0.237 g) of KMnO4 was dissolved in 100 mL of deionized water (DW), followed by addition of 5 mL hydrochloric acid (11.65 M, 35% (w/w)), forming a solution. The resulting mixture was stirred for 0.5 h; the solution was transferred to a Teflon-lined autoclave (50 mL capacity). The hydrothermal process was conducted at 353 K for 12 h. The product was filtered, washed with water and ethanol, and dried in an oven at 353 K for 8 h.Synthesis of α-MnO2 nanowires (α-MnO2-NW)
α-MnO2-NW was prepared by a reported procedure with increased temperature for higher purity samples.24 In a typical procedure, 10 mmol (0.6 g) of urea was dissolved in 35 mL deionized water, followed by addition of 10 mmol (1.58 g) KMnO4, forming a solution. The resulting mixture was stirred for 30 min; the solution was transferred to a Teflon-lined autoclave (50 mL capacity). The hydrothermal process was conducted at 433 K for 20 hours. The product was filtered, washed with DW and dried at 353 K overnight.Synthesis of α-MnO2 hollow nanotubes (α-MnO2-NT)
α-MnO2-NT was synthesized by a hydrothermal approach by modifying the reported recipe.25 15 mmol (2.37 g) of KMnO4 was dissolved in 200 mL of deionized water (DW), followed by the addition of 5 mL hydrochloric acid (11.65 M, 35% (w/w)), forming a solution. The resulting mixture was stirred for 0.5 h; the solution was transferred to a Teflon-lined autoclave (50 mL capacity). The hydrothermal process was conducted at 383 K for 15 h. The product was filtered, washed with water and ethanol, and dried in an oven at 353 K for 8 h.Materials characterizations
The as-prepared materials were characterized with an X-ray powder diffractometer (XRD; Shimadzu XRD-6000, Cu Kα radiation) at a scan rate of 1° min−1. Scanning Electron Microscopy (FESEM, JSM-7600F) and transmission electron microscopy (TEM; JEOL, JEM-2100F) operating at 200 kV was used to observe the morphological features. A nitrogen adsorption measurement at 77 K was performed by a Tristar-3000 surface area analyzer. Samples were out-gassed at 423 K for 4 h in the degas port of the adsorption apparatus. The specific surface area was determined by the Brunauer–Emmett–Teller (BET) method using the data points of P/P0 in the range of about 0.05–0.3.Electrochemical measurements
Cyclic voltammetry (CV), differential pulse voltammetry (DPV), pulsed amperometry (PA) and continuous amperometric studies were performed using a computer-controlled Pine Instrument. A three electrode electrochemical cell was employed with Ag/AgCl as the reference electrode (3 M KCl), a metal oxide mounted glassy carbon electrode (GCE) (0.196 cm2) as the working electrode, and Pt foil as the counter electrode. Before modification of the GCE, the polished electrode was ultrasonicated in ethanol and deionized water for 5 minutes, respectively. The working electrodes were prepared as follows: 10 μL aliquot of the MnO2 suspension (a homogenous sonicated solution of 10 mg of MnO2 and a mixture of 0.1 mL of Nafion and 0.9 mL of water) was placed onto the electrode surface and the electrode was dried in air leaving the material mounted onto the GCE surface.Results and discussion
XRD analysis was primarily used to determine the structure of the α-MnO2-NW (Fig. 1a). All the diffraction peaks can be indexed to tetragonal α-MnO2 (JCPDS, file no. 44-0141). A similar XRD observation was also noticed for the α-MnO2-NT and α-MnO2-MS prepared in this study (figure not shown). Fig. 1b–h shows the SEM and HRTEM images with different magnifications of α-MnO2-MS, α-MnO2-NT, and α-MnO2-NW. The SEM morphology of the α-MnO2-MS is spherical with a flower-like morphology (Fig. 1b and c). The higher magnification image shows that the petal structure of microspheres is assembled by many nanosheets (Fig. 1c). Fig. 1d–f are the representative SEM and HRTEM images of the as-prepared α-MnO2-NT. Evidently, each MnO2 nanotube is of high quality; they are open-ended and have a length of hundreds of nanometers (Fig. 1e and f). Fig. 1g shows the low magnification SEM image of the α-MnO2-NW; it can be found that the product consists of nanowires. The MnO2 nanowires are further investigated by HRTEM (Fig. 1h). The diameters of the presented MnO2 nanowires are within 50 nm (Fig. 1h). The N2 sorption isotherm of the α-MnO2-NW shows a type IV curve with a sharp capillary condensation step in the relative pressure range of 0.6–1 and H1 hysteresis loop, indicative of uniform cylindrical mesopores (Fig. 2a).26 The α-MnO2-NT synthesized in this study exhibited a type IV isotherm with H3 hysteresis, which is characteristic of mesoporous materials. The high uptake of N2 adsorption at a low relative pressure (∼0.4) reveals abundant micropores located in the α-MnO2-NT (Fig. 2a). The textural properties for various materials obtained from the N2-adsorption study are provided in Table S1, ESI.† The specific BET surface areas for the α-MnO2-NW, α-MnO2-NT, and α-MnO2-MS were found to be 124.2 m2 g−1, 79.3 m2 g−1 and 12.2 m2 g−1, respectively. From the SEM images it can be seen that the α-MnO2-NW has a short fiber length and diameter of ∼50 nm, while α-MnO2-NT has a diameter of ∼110 nm and α-MnO2-MS has a larger diameter (sub-micrometer size); the decrease of the lateral dimensions (diameter) to the nanoscale offers a high specific surface area and a high surface site density to the α-MnO2-NW (Fig. 1).26 | ||
| Fig. 1 XRD pattern (a) of the α-MnO2-NW. FE-SEM images (b, c) of the α-MnO2-MS, FE-SEM images (d, e) and TEM image (f) of the α-MnO2-NT, and FE-SEM image (g) and TEM images (h) of the α-MnO2-NW. The inset of (h) is a close view of the α-MnO2-NW. | ||
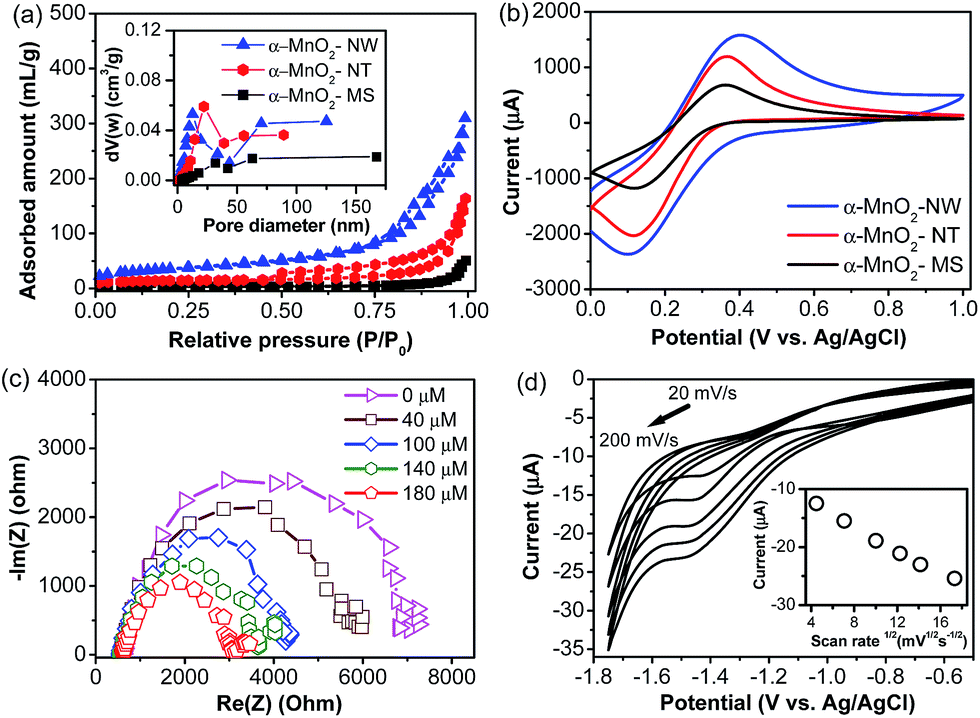 | ||
| Fig. 2 (a) N2 adsorption–desorption isotherms of materials synthesized in this study. The inset shows pore size distribution. (b) CV responses of various modified electrodes in 0.2 M KCl electrolyte containing 50 mM of [Fe(CN)6]3−/4− at a scan rate of 20 mV s−1. (c) Typical EIS response recorded for the α-MnO2-NW modified electrode in the presence of increasing concentrations of lindane, using 0.05 M TBAB solution in 60:40 methanol–water (20 mL) over the frequency range from 1 MHz to 10 mHz with a bias potential of −1.45 V. (d) CVs of lindane (200 μM) at various scan rates (20–200 mV s−1) for the α-MnO2-NW modified electrode. Inset shows the plot of peak currents vs. square root of scan rates. All scans are applied between −0.6 V and −1.8 V. | ||
The electrochemical behavior of different modified electrodes was investigated using potassium ferricyanide as the electrochemical probe by CV. The study was performed in 0.2 M KCl solution containing 50 mM K3[Fe(CN)6]/K4[Fe(CN)6] at a scan rate of 20 mV s−1 at different modified electrodes. The CV of various modified electrodes exhibited a pair of well-defined redox peaks corresponding to the Fe(CN)63−/4− redox couple (Fig. 2b). Among the various modified electrodes investigated in this study, the α-MnO2-NW exhibited the highest current response. The electroactive surface area of various electrodes was calculated according to the Randles–Sevčik equation (eqn (1)).23
| Ip = (2.69 × 105)n3/2AD1/2Cν1/2 | (1) |
The α-MnO2-NW modified electrode was constructed to obtain an optimized analysis parameter for lindane reduction. The electrochemical behaviour of lindane was first investigated using cyclic voltammetry in the absence and presence of 300 μM lindane. In the absence of lindane, no peak was observed. When lindane was added, a distinct peak at −1.45 V (versus Ag/AgCl) appeared, which is completely irreversible (Fig. S1, ESI†).10–15 The effect of the scan rate (range of 20–200 mV s−1) was investigated to predict the reaction kinetics process using cyclic voltammetry as shown in Fig. 2d. The results reveal that the reduction peak current of lindane was directly proportional to the square root of the scan rate up to 200 mV s−1; the negative intercept suggests that the electrode reaction is not a pure diffusion-controlled process (Fig. 2d, inset).10–15 The Randles–Sevčik equation for a reduction process with a zero intercept suggests a complete diffusion control process, thus, the negative intercept from the plot of the peak current versus the square root of the scan rate can be explained as the adsorption of the lindane molecule after diffusion to the electrode surface.15 The results of the voltammetric analyses are collected in Table S2.† The electron transfer coefficient α was calculated from the peak width (ΔEp/2 = Ep/2 − Ep). For all scan rates a very low value of α (less than 0.5) is obtained, showing that the rate-determining step of the electrode process is the dissociative electron transfer (ET), which is associated with the cleavage of the carbon–halogen bond.29,30 The dechlorination or displacement of chlorine of polychlorinated cyclohexanes is different from the dechlorination of polychlorinated benzenes.31,32 Earlier workers reported that the elimination of a chlorine atom by the reduction of polychlorinated benzenes always leads to a product that is more difficult to reduce.30,31 In contrast, the breaking of carbon–chlorine bonds in lindane occurs very fast and readily forms fully reduced benzene.31,32
Under the optimized conditions, pulsed amperometry and differential pulse voltammetry were used to determine lindane concentration. Fig. 3a shows the pulsed amperometric response of the α-MnO2-NW modified electrode to lindane. At a constant potential of −1.45 V (vs. Ag/AgCl), the measured current response is linear vs. the lindane concentration (Fig. 3a). The α-MnO2-NW modified electrode can detect lindane in the concentration range from 1.1 to 510 μM in the amperometric mode with a linear regression equation of I (μA) = −4.64–0.0346C (μmol L−1) (R2 = 0.9989) and a sensitivity of 0.18 μA μM−1 cm−2 and lower detection limit of 114 nM. The intercept of the calibration curve was not found to be zero, suggesting undesired non-faradaic processes (Fig. 3a, inset).33 DPV measurements were also carried out containing different lindane concentrations to obtain an analytical curve. Fig. 3b shows that the cathodic peak current was linearly dependent on the lindane concentration in the range from 2 to 520 μM with a linear regression equation of I (μA) = −6.208–0.022C (μmol L−1) (R2 = 0.9983) and a sensitivity of 0.14 μA μM−1 cm−2 and lower detection limit of 517 nM (Fig. 3b, inset). Similarly, linear relationships between oxidation peak currents and lindane concentrations were observed in the range of 4–500 μM and 15–350 μM for α-MnO2-NT and α-MnO2-MS modified electrodes (Fig. S2 and S3, ESI†). To obtain the limit of detection (LOD), we used an IUPAC (International Union of Pure and Applied Chemistry) recommended methodology, using the standard approach of alternative (SA).34
| LODSA = 3S/q | (2) |
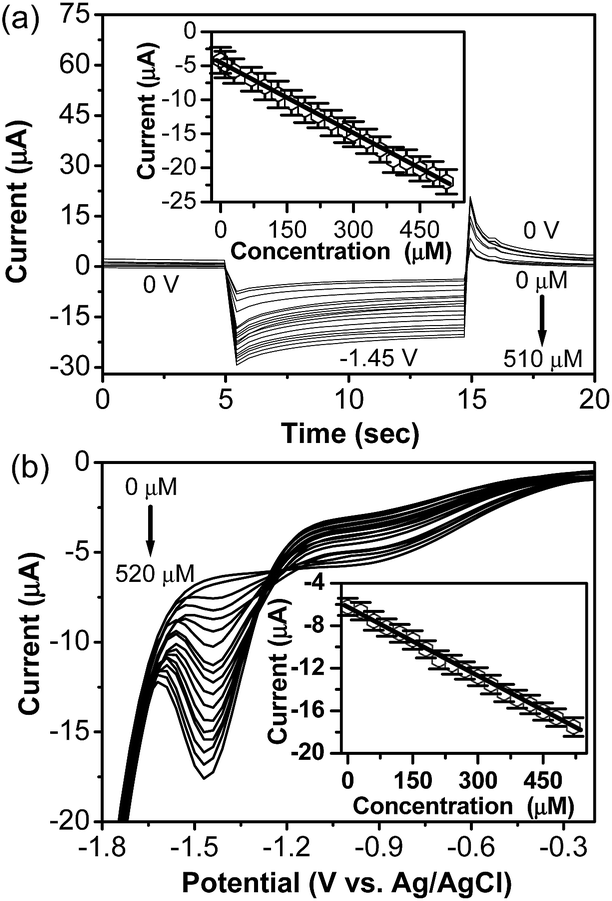 | ||
| Fig. 3 (a) Pulsed amperometric response of the α-MnO2-NW modified electrode to different concentrations of lindane at the specified voltage values. The inset shows the calibration plot. (b) DPVs of lindane at varying concentrations for the α-MnO2-NW modified electrode using 0.05 M TBAB solution in 60:40 methanol–water (20 mL). The inset shows the calibration plot. DPV parameters were selected as: peak height = 50 mV; peak width = 200 ms; peak period = 400 ms; increment = 20 mV; pre and post-pulse width = 3 ms. | ||
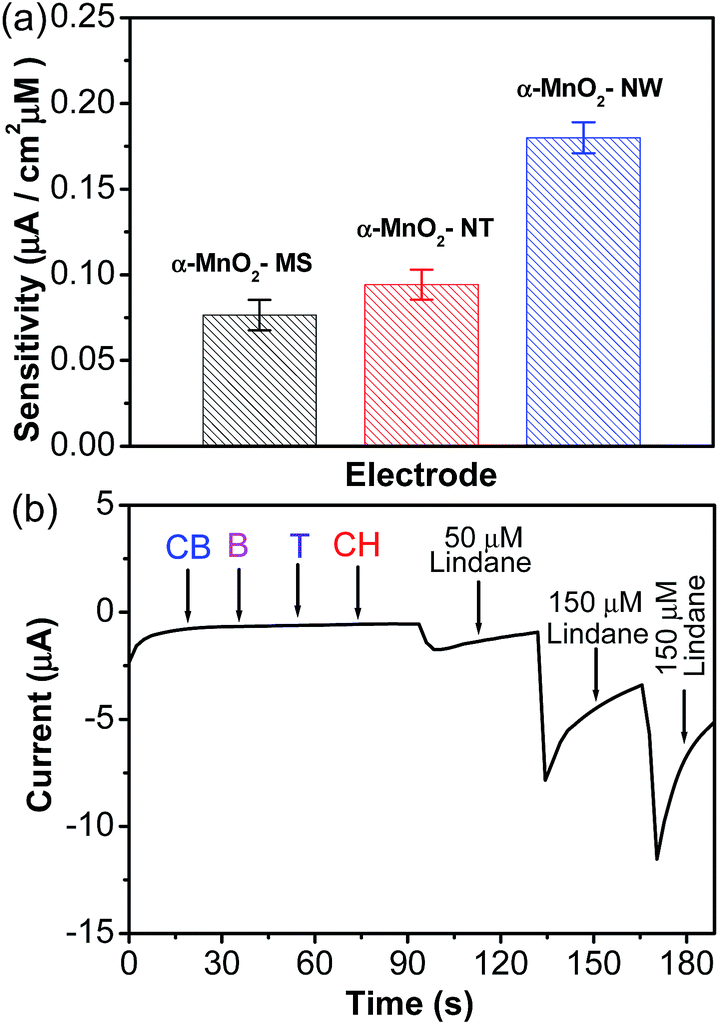 | ||
| Fig. 4 (a) Comparison of the sensitivity for lindane of the different modified electrodes investigated in this study. (b) Continuous amperometric response at the α-MnO2-NW modified electrode in the presence of a high concentration of interfering species, such as chlorobenzene (1 mM), benzene (1 mM), triclosan (1 mM), and cyclohexane (1 mM) as compared to the successive addition of lindane. | ||
The sensing ability of the prepared α-MnO2-NW modified electrode towards lindane was compared with the reported nanostructures (Table S3, ESI†).9,12,13,15,35–37 The performance of the α-MnO2-NW modified electrode is comparable if not superior to those reported in the literature (Table S3, ESI†). For the practical use of non-enzymatic lindane sensors, specificity is another important factor that needs attention. Under a rational design, the designed lindane sensor should have minimal interferences from water and soil samples from industrial areas and agricultural land. In the present work, interfering compounds were picked based on their structural similarity and solubility in water and soil.37–39 Therefore, inorganic ions, electroactive organic compounds and organic compounds containing chlorine groups were chosen which may potentially interfere with the determination of lindane.37–39 The amperometric response of the α-MnO2-NW modified electrode to the stepwise addition of lindane in the presence of 1 mM of each of the various interfering species cyclohexane (CH), triclosan (T), chlorobenzene (CB), and benzene (B) at an applied potential of −1.45 V is shown in Fig. 4b. The α-MnO2-NW modified electrode shows excellent anti-interference behaviour towards the interfering species (Fig. 4b). It may further be noted that 150 times higher concentrations of 4-CBA (0.5 M) and 1,3,5-TCB (0.5 M) interfere with the determination of lindane (Fig. 5a and b). The results also showed that 1 mM concentrations of inorganic ions (Ca2+, Co2+, Mg2+, Mn2+, Ni2+, and Zn2+) did not interfere with the detection of lindane (Fig. 5c).
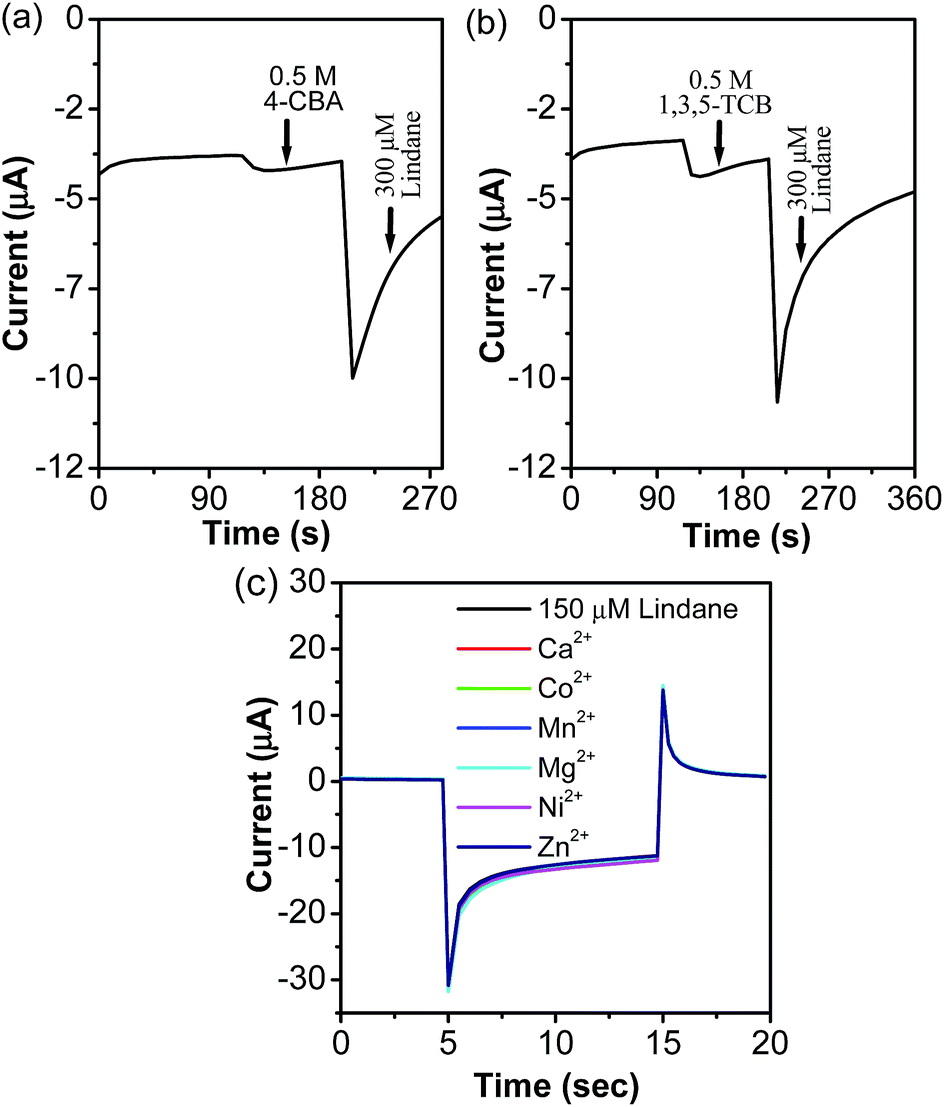 | ||
| Fig. 5 Continuous amperometric response at the α-MnO2-NW modified electrode in the presence of a high concentration of interfering species, such as (a) 4-chlorobenzaldehyde (0.5 M), and (b) 1,3,5-trichlorobenzene (0.5 M), compared to the addition of lindane (300 μM). (c) Pulsed amperometric response to 150 μM lindane spiked with 1 mM each of various metal ions at the α-MnO2-NW modified electrode (E1 = 0 V for 5 s; E2 = −1.45 V for 10 s and E3 = 0 V, for 5 s). | ||
Fig. 6a illustrates a pulsed amperometric response for a series of injections of standard lindane solution at a fixed potential (pulse sequence (n) = 5). Fig. 6a shows that when a fixed potential was applied (−1.45 V) the signal current increased relatively fast and no decrease of the signal was observed during the pulse length (relative standard deviation ∼1.02%), suggesting a very high reproducibility. After each measurement, the electrode was rinsed thoroughly with deionized water and was stored at room temperature when not in use and reactivated by dipping in the supporting electrolyte for 30 minutes before measurement. Furthermore, the α-MnO2-NW modified electrode could be used more than 13 times after subsequent cycles of washing and measuring operations. On the other hand, three fresh fabricated α-MnO2-NW modified electrodes were also used for determining 50 μM lindane. The RSD was calculated to be 2.71%. Therefore, our proposed lindane electrochemical sensor has a satisfactory repeatability. We examined the current stability for the α-MnO2-NW modified electrode by recording the amplitude of the pulse currents corresponding to the addition of lindane (200 μM) (Fig. 6b). The amplitude of the 40 pulse currents display insignificant changes (<0.5%), indicating a good stability of the α-MnO2-NW modified electrode. The storage stability was tested by storing the α-MnO2-NW modified electrode for two weeks. The result showed that the reduction peak potential of lindane had no shift and the current response only showed a 4.3% decrease compared with the original test.
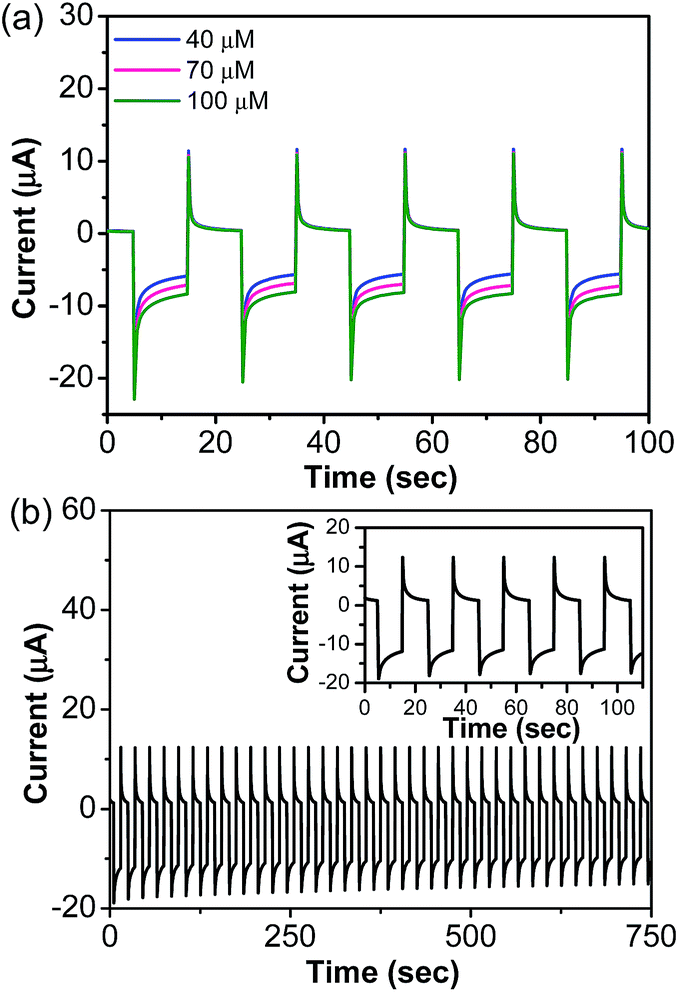 | ||
| Fig. 6 (a) Pulsed amperometric response (n = 5) of the α-MnO2-NW modified electrode to the increasing concentration of lindane (E1 = 0 V for 5 s, E2 = −1.45 V for 10 s and E3 = 0 V, for 5 s). (b) Pulsed amperometric response (n = 40) of the α-MnO2-NW modified electrode to 200 μM of lindane (E1 = 0 V for 5 s, E2 = −1.45 V for 10 s and E3 = 0 V, for 5 s). The inset shows an enlarged view (n = 5). | ||
To demonstrate the feasibility of the sensor in practicable applications, the proposed sensor was applied to the determination of lindane in tap water samples. No voltammetric response corresponding to lindane was observed in tap water, thus different quantities of lindane were added to tap water samples. The spiking method was adopted to evaluate the lindane content of three samples. The standard addition methodology was used to determine the average recovery of lindane as well as to validate the proposed method. The results are summarized in Table S4, ESI.† As can be seen in Table S4, ESI,† the recoveries were from 98.5% to 102.0%. The recovery calculations indicated the potential practical application of our proposed sensor.
Conclusions
In summary, we demonstrate a novel electrocatalyst for the non-enzymatic reduction of lindane based on α-MnO2 nanostructures. The sensors fabricated from the α-MnO2-NW exhibited the highest performance among the studied materials with a lower detection limit, high sensitivity, and wide linear range, making this novel sensing methodology an extremely promising one. We envision that this methodology reported here for lindane reduction encourages broad research in this direction.Acknowledgements
This work was supported by MOE Tier 1 Grants (RGT8/13 and RG131/14) of Singapore and the Singapore National Research Foundation under its Campus for Research Excellence And Technological Enterprise (CREATE) programme. Authors thank the Facility for Analysis, Characterisation, Testing and Simulation (FACTS) in Nanyang Technological University for materials characterizations.Notes and references
- C. L. Willett, E. M. Ulrich and R. A. Hites, Environ. Sci. Technol., 1998, 32, 2197 CrossRef.
- A. J. Durie, A. M. Z. Slawin, T. Lebl and D. O’ Hagan, Angew. Chem., Int. Ed., 2012, 51, 10086 CrossRef CAS PubMed.
- T. M. Phillips, A. G. Seech, H. Lee and J. Trevors, Biodegradation, 2005, 16, 363 CrossRef CAS PubMed.
- B. R. Garrido, T. A. L. Chau, G. Feijoo, F. Macias and M. C. Monterrroso, Environ. Sci. Technol., 2010, 44, 7063 CrossRef PubMed.
- S. Li, D. W. Elliott, S. T. Spear, L. Ma and W.-X. Zhang, Crit. Rev. Environ. Sci. Technol., 2011, 41, 1747 CrossRef CAS.
- S. L. Simonich and R. A. Hites, Science, 1995, 269, 1851 CrossRef CAS PubMed.
- M. Oturan, N. Oturan, C. Lahitte and S. Trevin, J. Electroanal. Chem., 2001, 507, 96 CrossRef CAS.
- G. Liu and Y. Lin, Anal. Chem., 2005, 77, 5894 CrossRef CAS PubMed.
- M. U. Anu Prathap, A. K. Chaurasia, S. N. Sawant and S. K. Apte, Anal. Chem., 2012, 84, 6672 CrossRef CAS PubMed.
- J. P. Merz, B. C. Gamoke, M. P. Foley, K. Raghavachari and D. G. Peters, J. Electroanal. Chem., 2011, 660, 121 CrossRef CAS.
- A. A. Peverly, J. A. Karty and D. G. Peters, J. Electroanal. Chem., 2013, 692, 66 CrossRef CAS.
- M. U. Anu Prathap and R. Srivastava, Electrochim. Acta, 2013, 108, 145 CrossRef CAS.
- A. Kumaravel, S. Vincent and M. Chandrasekaran, Anal. Methods, 2013, 5, 931 RSC.
- P. R. Birkin, A. Evans, C. Milhano, M. I. Montenegro and D. Pletcher, Electroanalysis, 2004, 16, 583 CrossRef CAS.
- M. U. Anu Prathap, S. Sun, C. Wei and Z. J. Xu, Chem. Commun., 2015, 51, 4376 RSC.
- J. F. Rusling, T. F. Connors and A. Owlia, Anal. Chem., 1987, 59, 2123 CrossRef CAS PubMed.
- H. B. Wu, J. S. Chen, H. H. Hng and X. W. D. Lou, Nanoscale, 2012, 4, 2526 RSC.
- M. U. Anu Prathap and R. Srivastava, Sens. Actuators, B, 2013, 177, 239 CrossRef CAS.
- M. U. Anu Prathap and R. Srivastava, Nano Energy, 2013, 2, 1046 CrossRef CAS.
- M. U. Anu Prathap, B. Kaur and R. Srivastava, J. Colloid Interface Sci., 2012, 381, 143 CrossRef CAS PubMed.
- C. Chen, Q. Xie, D. Yang, H. Xiao, Y. Fu, Y. Tan and S. Yao, RSC Adv., 2013, 3, 4473 RSC.
- C. Wei, L. Yu, C. Cui, J. Lin, C. Wei, N. Mathews, F. Huo, T. Sritharan and Z. Xu, Chem. Commun., 2014, 50, 7885 RSC.
- M. U. Anu Prathap, V. Anuraj, B. Satpati and R. Srivastava, J. Hazard. Mater., 2013, 262, 766 CrossRef CAS PubMed.
- X. C. Duan, J. Q. Yang, H. Y. Gao, J. M. Ma, L. F. Jiao and W. J. Zheng, CrystEngComm, 2012, 14, 4196 RSC.
- J. Luo, H. T. Zhu, H. M. Fan, J. K. Liang, H. L. Shi, G. H. Rao, J. B. Li, Z. M. Du and Z. X. Shen, J. Phys. Chem. C, 2008, 112, 12594 CrossRef CAS.
- L. Qian, L. Gu, L. Yang, H. Y. Yuan and D. Xiao, Nanoscale, 2013, 5, 7388 RSC.
- Y. Wang, Y. Zhu, J. Chen and Y. Zeng, Nanoscale, 2012, 4, 6025–6031 RSC.
- M. C. Rodriguez, A. N. Kawde and J. Wang, Chem. Commun., 2005, 4267 RSC.
- A. A. Isse, L. Falciola, P. R. Mussini and A. Gennaro, Chem. Commun., 2006, 344 RSC.
- A. A. Isse, S. Gottardello, C. Durante and A. Gennaro, Phys. Chem. Chem. Phys., 2008, 10, 2409 RSC.
- D. G. Peters, in Organic Electrochemistry, ed. H. Lund and O. Hammerich, Marcel Dekker, New York, 4th edn, 2000, ch. 8, pp. 341–377 Search PubMed.
- S. M. Kulikov, V. P. Plekhanov, A. I. Tsyganok, C. Schlimm and E. Heitz, Electrochim. Acta, 1996, 41, 527 CrossRef CAS.
- E. Grygolowicz-Pawlak and E. Bakker, Electrochem. Commun., 2010, 12, 1195 CrossRef CAS PubMed.
- J. Mocak, A. M. Bond, S. Mitchell and G. Scollary, Pure Appl. Chem., 1997, 69, 297 CrossRef CAS.
- P. Wang, L. Ge, M. Li, W. Li, L. Li, Y. Wang and J. Yu, J. Inorg. Organomet. Polym., 2013, 23, 703 CrossRef CAS.
- P. R. Birkin, A. Evans, C. Milhano, M. I. Montenegro and D. Pletcher, Electroanalysis, 2004, 16, 583 CrossRef CAS.
- T. S. Anirudhan and S. Alexander, Biosens. Bioelectron., 2015, 64, 586 CrossRef CAS PubMed.
- I. Ali and C. K. Jain, J. Environ. Hydrol., 2001, 9, 1 Search PubMed.
- C. Xu, K. Wu, S. Hu and D. Cui, Anal. Bioanal. Chem., 2002, 373, 284 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, figures. See DOI: 10.1039/c5ra26771d |
| This journal is © The Royal Society of Chemistry 2016 |