
Monolithic nitrogen-doped carbon as a water sorbent for high-performance adsorption cooling†
Abstract
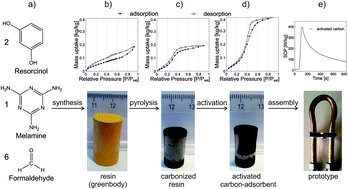
In the present study, we report on the development of carbon adsorbents for water adsorption heat pumps. Resorcinol-melamine-formaldehyde (RMF) resins were synthesized and molded into monolithic shapes before pyrolysis and chemical activation with KOH. The influence of the carbonization and activation treatments on the physicochemical properties and the water sorption behavior of the final adsorbent materials were investigated. Activated carbons with a one-to-one (C to KOH) impregnation mass ratio, an activation temperature of 800 °C and an activation time of one hour exhibited the highest water cycling ability. For isobaric adsorption at 23 mbar, the peak specific cooling power of the best monolithic activated carbon produced in this way was 192 W kg−1 for a temperature step from 90 °C to 50 °C compared to 255 W kg−1 for silica gel for a finned tube heat exchanger of comparable fin spacing. For a temperature step from 60 °C to 30 °C, the monolithic activated carbon exhibited a higher peak specific cooling power (389 W kg−1) compared to silica gel (240 W kg−1). In situ infrared thermography revealed superior thermal transport properties of the monolithic carbons compared to commercial silica gel.
 Please wait while we load your content...
Please wait while we load your content...

