
Oxidation of benzyl alcohol catalyzed by gold nanoparticles under alkaline conditions: weak vs. strong bases†
Camila P. Ferraza,
Marco Aurélio S. Garciaa,
Érico Teixeira-Netob and
Liane M. Rossi*a
aDepartamento de Química Fundamental, Instituto de Química, Universidade de São Paulo, Av. Prof. Lineu Prestes, 748, 05508-000, São Paulo, SP, Brazil. E-mail: lrossi@iq.usp.br
bLaboratório Nacional de Nanotecnologia, CNPEM/ABTLuS, Caixa Postal 6192, 13083-970, Campinas, SP, Brazil
First published on 1st March 2016
Abstract
Gold nanoparticles have shown excellent catalytic properties in selective oxidation of alcohols in the presence of base; however, the influence of the nature and concentration of the base on gold catalyst activity and selectivity is not completely understood. We here present a study of the effect of using weak and strong bases on the conversion and selectivity of PVA-stabilized gold nanoparticles supported on titania (AuPVA/TiO2) for benzyl alcohol oxidation. The increase in the concentration of base had little effect on conversion when a weak base was used (K2CO3, Na2B4O7 and Na(CH3COO)), due to the buffer effect, but strongly affected selectivity. The bases with higher pKa values provided higher conversions and increased production of benzoic acid. For the strong base NaOH, benzoic acid was always the major product, although conversion decreases in excess of base. The formation of benzoic acid is avoided by using K2CO3 in non-aqueous media; benzaldehyde is the main product in cyclohexane whereas benzyl benzoate is also formed in significant amounts in solvent-free conditions. The promotion effect observed in the presence of base was discussed in terms of reaction mechanism.
Introduction
Selective oxidation is one of the most important functional group transformations in organic chemistry, being crucial in industrial processes.1–5 Classes of organic compounds containing oxygen, such as epoxides, ketones, aldehydes, alcohols and acids serve as intermediates in the manufacture of plastics, detergents, paints, cosmetics, and food additives.1–4 Efficient industrial processes for commercial applications involving these chemicals have been pursued.6–8 Currently, special attention has been given to oxidation of substrates derived from biomass, owing to their potential to replace petroleum-derived chemicals.9–13 Due to their high extent of functionalities and hydrophilic properties, substrates derived from biomass should be processed in liquid-phase, typically in aqueous phase.12,13 Thus, the development of efficient liquid-phase technologies are welcome, as well as improved catalysts to work under mild reaction conditions.12,13Metal catalysts enable the use of green oxidants, such as molecular oxygen, which generates only water as a byproduct.4,5,14 Considered as catalytically inert, gold, when in nanometric dimensions, presents a number of new properties, including very high catalytic activity in reactions of industrial and environmental interest.3,5–8,15–20 Gold-based catalysts are more stable and selective for the aerobic oxidation of organic compounds in water than the conventional liquid-phase oxidation catalysts based on Pt and Pd, because the Au catalysts can offer better resistance to water and O2.9–11 Thus, gold offers unique opportunities for catalysis, besides an excellent alternative to the development of viable industrial process for the conversion of renewable biomass in aqueous phase.9–11 Previous studies with gold nanocatalysts in oxidation reactions of simple alcohols, such as benzyl alcohol, showed that an excellent selectivity can be achieved under mild conditions.4,5,9,21–40 Understanding the selective oxidation of benzyl alcohol can assist further application of catalytic oxidations for the valorization of renewable biomass substrates, such as glucose, HMF and glycerol.
Oxidation reactions in aqueous phase have been successful, but there is still room for further development, once water plays an important role in the efficiency of gold catalysts.4,9,21,27,28,33–35,38,39,41 The catalytic activity of gold catalysts for CO oxidation16–18 and for benzyl alcohol oxidation3,21 can be promoted significantly by water. Under identical reaction conditions, the activity of Au/TiO2 for benzyl alcohol oxidation in the presence of water is consistently higher than that in ethanol and p-xylene.21 Even in aqueous medium, the reaction rates are quite low.3,9,21,28,39 Acceptable conversions from a practical point of view for benzyl alcohol oxidation using gold catalysts are obtained in the presence of base promoters (aqueous3,4,9,27,33,35,38,39 and non-aqueous media9,22,30,32,37), by using a basic and/or nanosized support (non-aqueous media3,23–25,31,36,40,42) or by adding a second metal, in particular Pd (aqueous43,44 and non-aqueous media45–47). Aqueous oxidation of aromatic and aliphatic alcohols in general uses hydroxides and carbonates as base promoters and the amount of base has been arbitrarily chosen from sub-stoichiometric to equimolar or even large excess (from 0.5 to 3 equivalents). The catalytic activity in base-free conditions is generally an order of magnitude lower than the catalytic activity obtained in high-pH conditions,3,9,28,39 which emphasizes the critical role of the base as a promoter in gold catalysis. At the same time, in alkaline reaction conditions the selectivity is greatly affected towards benzoic acid.3–5,9,39,48,49 In this context, it would be interesting to ascertain how the amount of weak and strong bases affects the activity and selectivity of the catalytic oxidation of alcohols.
Besides the effect of base promoters in aqueous solution, marked examples underline the sensitivity of gold catalysts to the reaction media, that leads to different product distributions.3,5,9,39 As reported for benzyl alcohol oxidations, majority of gold catalysts in non-polar and solvent-free media favor selectivity towards benzaldehyde,3,23–25,36,40,42 while performing the oxidation in alcohols, such as MeOH, the corresponding ester is formed.26,31 In some cases, hydroxides and carbonates have been also applied as promoters;22,29,30,32,37 however, reports on the effect of the amount of base in these media are rather sparse.
Here we report on the effect of using weak and strong bases as promoters in the oxidation of benzyl alcohol catalyzed by AuPVA/TiO2. We evaluated how the nature of base and different [base]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[alcohol] molar ratios affect the reaction conversion and selectivity. The results were also compared to that obtained in non-aqueous media (cyclohexane and solvent-free).
:[alcohol] molar ratios affect the reaction conversion and selectivity. The results were also compared to that obtained in non-aqueous media (cyclohexane and solvent-free).
Results and discussion
Gold supported on titania (AuPVA/TiO2) catalyst was prepared using the well-known sol immobilization method with polyvinyl alcohol (PVA) stabilized Au NPs.40,50,51 PVA is a versatile stabilizing agent used to obtain colloidal metal dispersions. The presence of PVA has been demonstrated to affect the activity of AuPVA/TiO2 catalyst;51,52 but also to increase the catalyst stability. This method was chosen because of its high reproducibility. The catalyst was first tested in the aqueous oxidation of benzyl alcohol in the absence and presence of carbonate (K2CO3) as a base promoter. The conversion was monitored as a function of time, as shown in Fig. 1a. The catalyst exhibits some activity (33% conversion in 2.5 h) in the absence of a base promoter, because O2 adsorption on TiO2 could be facilitated in water.21 However, much higher conversions were achieved in the presence of K2CO3. The influence of the amount of base on the catalytic activity of AuPVA/TiO2 for benzyl alcohol oxidation was determined for a series of five different [K2CO3]:[alcohol] molar ratios (0.1, 0.5, 1, 2 and 4). In the aqueous oxidation of alcohols, the deprotonation of the alcohol to form an alkoxy intermediate is the first step and should be facilitated in alkaline media. Thus, the extent of the reaction should be directly affected by the amount of base, although the initial activation of the alcohol has also been suggested to occur on the catalyst surface.9,49 The results shown in Fig. 1a suggest that the conversion was little affected by varying the amount of K2CO3 from 0.5 to 4 equivalents, reaching 100% conversion within 2.5 h. The reaction performed with 0.1 equivalents of K2CO3 reached a maximum conversion of 70%. To analyze the effect of K2CO3 in the reaction, it must be considered the formation of a carbonate/bicarbonate buffer in the initial reaction solutions, producing HO−. Changes in pH values were monitored as a function of reaction time, as shown in Fig. 1b. Selected results are presented in Table 1 (entries 1, 8–12) and detailed data can be obtained in Table S1 (ESI†). The initial pH values of all reaction solutions were quite similar due to the buffer formation. The increase in the amount of base represents an increase in the buffer strength of the initial solutions. For [K2CO3]:[alcohol] > 0.5, the benzoic acid concentration increased along the reaction, but the final pH is little affected. However, for [K2CO3]:[alcohol] = 0.1, the formation of benzoic acid over the reaction time breaks the buffer solution. The decrease in the pH might be the reason for the lower conversion observed at long reaction times. In addition, the benzoic acid can adsorb on the catalyst surface, causing poisoning and reducing the reaction rates.22,25 The turnover frequencies at initial reaction times (TOF, h−1) were only slightly affected by the amount of K2CO3 (base to alcohol molar ratio) used with values ranging from 2400 to 3200 h−1 (Fig. 1c), which is an evidence of pH control under buffer conditions. The selectivity was strongly influenced by the amount of K2CO3 used (Fig. 1d–h). The conversion of benzyl alcohol into benzaldehyde is favored in the presence of sub-stoichiometric amounts of base (Fig. 1d), while benzoic acid is produced in higher yields in excess of base, reaching 99% selectivity (Fig. 1g and h). The formation of benzoic acid was observed since the beginning of reaction, in all amounts of base studied, and the selectivity to acid increased within the reaction time, while decreasing the selectivity to benzaldehyde. In excess of base ([K2CO3]:[alcohol] = 2 and 4), benzoic acid is always the major product. After 1 h of reaction, the selectivity to acid is already >90%. The formation of benzyl benzoate was minimal in all cases, not exceeding 8% selectivity. Moreover, the mass balance was very unfavorable for long reactions times (150 min) in all reaction conditions studied (Fig. 1d–h).
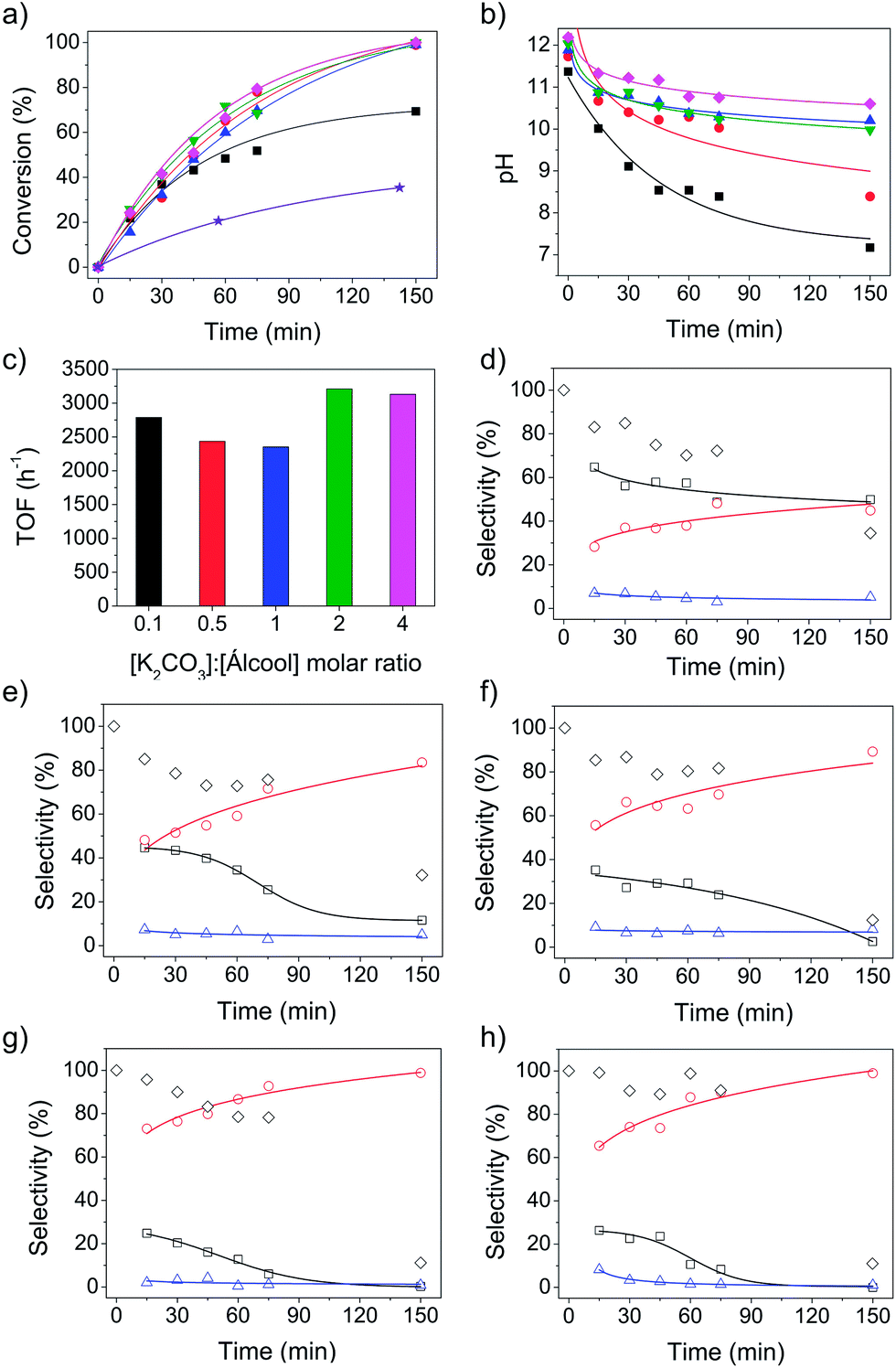 | ||
| Fig. 1 Aqueous oxidation of benzyl alcohol catalyzed by AuPVA/TiO2: (a) conversion as a function of time for ([K2CO3]:[alcohol] molar ratio = 0 (★), 0.1 (■), 0.5 (●), 1 (▲), 2 (▼) and 4 (♦)); (b) evolution of pH as a function of time for ([K2CO3]:[alcohol] molar ratio = 0.1, 0.5, 1, 2 and 4); (c) turnover frequencies (h−1); (d–h) selectivity (benzaldehyde (□), benzoic acid (○) and benzyl benzoate (△)) and mass balance (◇) as a function of time for [K2CO3]:[alcohol] molar ratio = (d) 0.1, (e) 0.5, (f) 1, (g) 2 and (h) 4. Reaction conditions: benzyl alcohol (2.9 mmol), catalyst (1.3 μmol Au), O2 (6 bar), 100 °C. | ||
| # | Base | pKa | [Base]:[alcohol]([HO−] mol L−1) |
Initial pH | Final pH | Conv. (%) | Selectivityb (%) | |
|---|---|---|---|---|---|---|---|---|
| BnCHO | BnCOOH | |||||||
| a Reaction conditions: benzyl alcohol (0.3 mL, 2.9 mmol), catalyst (15 mg, 1.3 μmol Au), O2 (6 bar), 100 °C, 1 h. BnCHO = benzaldehyde, BnCOOH = benzoic acid.b Mass balance > 85%. | ||||||||
| 1 | No base | 7.00 | 0 | 5.85 | 3.83 | 19 | 92 | 8 |
| 2 | Na(CH3COO) | 4.87 | 0.1 (1.28 × 10−5) | 9.11 | 5.74 | 12 | 82 | 18 |
| 3 | 1 (4.05 × 10−5) | 9.61 | 6.26 | 15 | 87 | 13 | ||
| 4 | 4 (8.10 × 10−5) | 9.91 | 6.57 | 28 | 92 | 8 | ||
| 5 | Na2B4O7 | 9.23 | 0.1 (2.15 × 10−3) | 11.33 | 8.67 | 31 | 68 | 28 |
| 6 | 1 (6.97 × 10−3) | 11.84 | 9.35 | 32 | 50 | 47 | ||
| 7 | 4 (14.02 × 10−3) | 12.15 | 9.84 | 43 | 41 | 59 | ||
| 8 | K2CO3 | 10.32 | 0.1 (2.35 × 10−3) | 11.37 | 8.54 | 48 | 57 | 38 |
| 9 | 0.5 (5.38 × 10−3) | 11.73 | 10.29 | 65 | 34 | 59 | ||
| 10 | 1 (7.65 × 10−3) | 11.88 | 10.37 | 60 | 29 | 63 | ||
| 11 | 2 (10.86 × 10−3) | 12.04 | 10.39 | 72 | 13 | 87 | ||
| 12 | 4 (15.40 × 10−3) | 12.19 | 10.77 | 66 | 11 | 88 | ||
| 13 | NaOH | — | 0.1 (2.89 × 10−2) | 12.46 | 5.56 | 36 | 53 | 47 |
| 14 | 1 (2.89 × 10−1) | 13.46 | 13.18 | 54 | 5 | 95 | ||
| 15 | 4 (1.16) | 14.06 | 13.60 | 34 | 0 | 100 | ||
The presence of hydroxide ions plays an important role during the oxidation reaction. Davis et al. have discussed how the product distribution depends on pH.9,49 In principle, the buffer effect can explain similar initial TOFs for the reactions; however, the increase in the buffer strength represents an increase in the concentration of HO− (6.5 times for [K2CO3]:[alcohol] from 0.1 to 4) and, consequently, can sustain the differences observed in the reaction selectivity. It is well known that aldehydes in basic aqueous solution can be oxidized to carboxylic acid more easily.9,49 The proposed mechanism involves the reversible hydration of the aldehyde to germinal diols, which is accelerated in high pH. The germinal diols will adsorb in the metal surface to form an alkoxide, that later undergoes a β-elimination to form carboxylic acid.9,49 Thus, the combination of high concentration of OH− and longer reaction times favors the oxidation to carboxylic acid.
Recently, Zheng and Stucky37 have shown that the addition of sub-stoichiometric amounts of either acetates, borates or carbonates, can enhance the activity of supported gold nanoparticles for the oxidation of alcohols in solvent-free medium. Considering the ability of these species with different pKa values to form buffered solution, we become interested in investigating their behavior in aqueous media and the consequences on the reaction selectivity.
The aqueous oxidation of benzyl alcohol was studied in the presence of different amounts of Na2B4O7 and Na(CH3COO) and the results are summarized in Table 1 (entries 2–7). The benzyl alcohol conversion after 60 min of reaction follows the same order as the pKa: K2CO3 > Na2B4O7 > Na(CH3COO), most probably associated to the concentration of benzyloxide species that are involved in the first step of the reaction. In fact, a linear correlation was found between the initial reaction pH or the amount of benzyloxide at initial pH and the reaction conversion (Fig. 2a). The conversion was only slightly improved with the increase in the [base]:[alcohol] molar ratio, due to the buffer formation, as observed previously in the experiments with potassium carbonate. That was not the case for the reaction with the strong base NaOH (Table 1, entries 13–15), wherein the conversion was greatly affected by the amount of base and does not follow the previous described tendency in Fig. 2a. For [NaOH]:[alcohol] = 0.1, the conversion was 35% and pH drops from 12.46 to 5.56. As the buffer is not formed, the formation of benzoic acid decreased the pH and then the reaction rate, which results in a lower conversion compared to the reaction with K2CO3. For [NaOH]:[alcohol] = 1 and 4, the final pH was not highly affected by the amount of acid formed in the reaction (13.18 and 13.60, respectively). An improvement in the conversion was observed for 1 equivalent of NaOH, reaching 54%, which was close and comparable to that obtained with K2CO3 (ca. 60%). However, for [NaOH]:[alcohol] = 4, the conversion dropped to 35%. In this condition, the concentration of HO− in the aqueous solution is about 75 times higher than it was produced by a similar amount of K2CO3, which suggests a lack of correlation between the reaction conversion and [HO−] (Fig. 2b). In order to rationalize these results, we should remember that the activation of O2 in the aqueous oxidation of alcohols proceeds through the formation and dissociation of peroxide (OOH*) and hydrogen peroxide (HOOH*) intermediates,9,49 which are necessary species to close the catalytic cycle (removal of excess electrons from the metal surface, oxidize metal-hydride bonds and regenerate hydroxide ions).9,25,27,42,49,53 It has been suggested that the oxidation of alcohols can be less effective in high concentration of NaOH (>0.6 M) due to the faster decomposition of peroxide.27 In order to confirm these assignments, the hydrogen peroxide formation was investigated for the aqueous oxidation reaction using K2CO3 and NaOH (Fig. 2b). When increasing the amount of NaOH from 0.1 to 1 equiv., the increase in the [HO−] favored the formation of H2O2,54 which caused an increase in the reaction conversion. However, for 4 equiv. of NaOH, the amount of H2O2 decreased as well as the reaction conversion. These results suggest that the excess of NaOH ([NaOH]:[alcohol] > 1) is detrimental to the oxidation reaction. Therefore, the fast decomposition of the peroxide in strong alkaline media can provide an explanation for the lack of correlation between the conversion and pH for NaOH.
 | ||
| Fig. 2 (a) Conversion as a function of initial pH (solid symbols) and initial concentration of benzyloxidea ([BnO−]) (empty symbols) for aqueous oxidation of benzyl alcohol catalyzed by AuPVA/TiO2 using K2CO3 (■), NaOH (●), Na2B4O7 (▼) and Na(CH3COO) (▲) as base for 60 min. (b) Conversion (solid symbols) and concentration of H2O2 (empty symbols) as a function of [base]:[alcohol] molar ratio (0.1, 1 and 4) for aqueous oxidation of benzyl alcohol catalyzed by AuPVA/TiO2 using K2CO3 (■) and NaOH (●) as base for 60 min. Reaction conditions: benzyl alcohol (2.9 mmol), catalyst (1.3 μmol Au), O2 (6 bar), 100 °C. aCalculated value using the Henderson Hasselbalch equation at each initial pH, according to ref. 52. | ||
A general tendency was observed in the selectivity for the reactions performed in the presence of weak bases, i.e., increasing the [HO−] decreases the selectivity to benzaldehyde, while favoring the formation of benzoic acid (Table 1, entries 2–12). Na(CH3COO) produces the lowest [HO−] and the best selectivity to benzaldehyde. In this case, the lower pH delays the further oxidation to acid, but also prevents the reaction to proceed to high conversions. However, K2CO3 was the most effective base to produce benzaldehyde or benzoic acid in aqueous media, producing benzaldehyde (57% selectivity at 48% conversion) in the [base]:[alcohol] = 0.1 and benzoic acid (87% selectivity at 72% conversion) in the [base]:[alcohol] = 2. The increase in the [base]:[alcohol] ratio represents little variation in the [HO−] (ca. 6.5 times), thus benzaldehyde was still observed in all reactions. For the strong base NaOH, the increase in the [base]:[alcohol] ratio represents an increase of up to 40 times in the [HO−], thus benzoic acid is the major product no matter the amount of NaOH used.
Further studies on the oxidation of benzyl alcohol using K2CO3 as a base promoter were conducted in cyclohexane and solvent-free conditions. Conversion and selectivity after 1 h reaction as a function of the amount of added base ([K2CO3]:[alcohol] = 0.1, 1 and 4) are presented in Table 2. Contrary to aqueous phase, only very low conversions were observed in absence of base for non-aqueous media (Table 2, entries 16 and 20). However, the use of K2CO3 as base promoter represented a marked improvement on reaction conversions. When comparing the use of K2CO3 in aqueous and cyclohexane media, the results suggest that the conversion was slightly affected by varying the reaction media, but selectivity has significantly changed towards benzaldehyde. In the solvent-free condition, the addition of base resulted also in high conversions, reaching 90% already with 1 equivalent of base, but selectivity was not as high as in cyclohexane. Differences in reactivity can be rationalized considering the proposed mechanism.9,25,42,49 The oxidation of alcohol proceeds through 3 steps: (1) metal-alkoxide formation; (2) β-elimination; (3) metal-hydride formation and catalyst regeneration. In the aqueous-phase, the basic solution favors the initial deprotonation of the alcohol to form an alkoxy intermediate in the first step and also decreases the energy barrier in step 2.9,49 In the non-aqueous phase, the support or a promoter must play an important role in the initial activation of the alcohol that might occur on the catalyst surface,9,49 just as the dehydration in the step 2. Reasonable conversions and the formation of benzaldehyde were only possible with the addition of K2CO3, indicating that both steps are favored in the presence of a base promoter. Both in aqueous or non-polar organic media, where K2CO3 is acting as a homogeneous base and as a solid base, respectively, similar conversions of benzyl alcohol were observed for the same amount of base (Table 1, entries 8–12 and Table 2, entries 16–19). In the solvent-free condition (Table 2, entries 20–22), when the neat alcohol was used, the conversion was boosted probably due to the high concentration of alcohol. Under this condition, a large majority of the metal sites will be coordinated forming the alkoxide.25
| # | Medium | [Base]:[alcohol] |
Conv. (%) | Selectivityc (%) | ||
|---|---|---|---|---|---|---|
| BnCHO | BnCOOH | BnBzO | ||||
| a Reaction conditions: benzyl alcohol (0.3 mL, 2.9 mmol), catalyst (15 mg, 1.3 μmol Au), O2 (6 bar), 100 °C, 1 h.b Benzyl alcohol (1 mL, 9.7 mmol), catalyst (52 mg, 4.4 μmol Au), O2 (6 bar), 100 °C, 1 h.c Mass balance > 93%.d [K2CO3]:[alcohol] = 4 was not performed because the mass of base would be higher than the mass of alcohol. This amount of base would not provide a liquid medium for the reaction. |
||||||
| 16 | Cyclohexanea | 0 | 2 | 100 | 0 | 0 |
| 17 | 0.1 | 44 | 89 | 0 | 11 | |
| 18 | 1 | 55 | 87 | 0 | 13 | |
| 19 | 4 | 72 | 85 | 0 | 15 | |
| 20 | Solvent-freeb | 0 | 3 | 100 | 0 | 0 |
| 21 | 0.1 | 61 | 70 | 0 | 30 | |
| 22 | 1 | 87 | 63 | 0 | 37 | |
| 23 | 4d | — | — | — | — | |
Differently from the aqueous media, in cyclohexane and solvent-free conditions, benzoic acid was not formed. We performed the oxidation of benzaldehyde under the same reaction conditions (see Table S2, ESI†). Benzaldehyde is converted in benzoic acid in the absence of catalyst in non-aqueous, but the formation of benzoic acid is inhibited by AuPVA/TiO2 catalyst. In aqueous media, benzaldehyde is converted in benzoic acid either with or without the catalyst. Thus, the results under non-aqueous media suggest that the formation of benzoic acid is not favorable on the catalyst surface, but its formation can still occur in alkaline aqueous medium once the HO− ions are available.
The only byproduct for the reaction in non-aqueous media is the benzyl benzoate, which is favored by the increase in the amount of K2CO3. As the conversions in water and in cyclohexane are similar, the difference relies on the selectivity: higher base concentration favors the formation of benzoic acid in aqueous media, while in cyclohexane, the ester is preferred with no formation of acid. The formation of benzyl benzoate is minimal in cyclohexane (up to 15% selectivity), but it is appreciable in solvent-free condition. In the presence of [K2CO3]:[alcohol] as low as 0.1, the selectivity for benzyl benzoate is already 30% and increases to 37% for [K2CO3]:[alcohol] = 1. This resulted in moderate selectivity to benzaldehyde (up to 70%). According to proposed mechanism for the oxidation of alcohols, the ester is formed by the attack of the alkoxy species (formed in step 1) into the aldehyde (formed in the step 2).48 Thus, the benzyl benzoate is more likely produced when the rate of formation of benzyloxide is high compared to the rate of formation of benzaldehyde. In the solvent-free medium, the benzyl ester is formed in the highest yields, indicating high rate of formation of benzyloxide, which is boosted by the base.
The stability of the AuPVA/TiO2 catalyst against aggregation after being exposed to different reaction conditions used in this study was investigated by Scanning Transmission Electron Microscopy (STEM). The STEM images (Fig. S1, ESI†) revealed that the gold nanoparticles of the as prepared catalyst (4.4 ± 1.5 nm) only slightly increase in size when used in aqueous solution in the presence of acetate (4.7 ± 1.6 nm), borate (5.2 ± 1.9 nm) and NaOH (5.4 ± 2.4 nm), but significantly increase in size in the presence of carbonate (7.6 ± 2.6 nm). In the solventless reaction, the gold particle size was maintained (5.1 ± 1.8 nm), but in cyclohexane particle's growth was noticed (10.8 ± 8.3 nm). There is not a clear correlation between the reactivity and the particle size changes in the catalytic reaction studied. None of the samples were attacked by the reaction media and all of them are comprised of supported gold nanoparticles after reaction.
Conclusions
The comparative study of weak and strong bases in the aqueous oxidation of benzyl alcohol by PVA-stabilized supported gold nanoparticles showed that conversion is related to the initial pH of the reaction, since the bases with higher pKa values (greater initial pH values) provided higher conversions, regardless the amount of base used. However, the increase in the [base]:[alcohol] molar ratio affects the reaction selectivity, favoring the oxidation of the aldehyde to benzoic acid. For the strong base, low amounts of base are not enough to keep the high pH during the reaction. The increase in the amount of base is beneficial for the catalyst activity until [base]:[alcohol] molar ratio is 1. In excess of base, the intermediate (H2O2) is degraded decreasing the conversion. These results suggest that weak bases are the most effective promoters for the selective oxidation of alcohols in aqueous media. The buffer formation: (1) allows better pH control over reaction; (2) provides lower [HO−] and consequently better selectivity to aldehyde, in sub-stoichiometric amount of base; (3) provides higher [HO−] and consequently better selectivity to benzoic acid, in excess of base; K2CO3 is a very effective base to produce benzaldehyde or benzoic acid in aqueous media, producing preferentially benzaldehyde in the [base]:[alcohol] = 0.1 and benzoic acid in the [base]:[alcohol] = 4. The study in non-aqueous media revealed that the presence of the base promoter increased conversion and prevented completely the formation of benzoic acid. Mainly in the solvent-free condition, the increase in the amount of base favored the formation of benzyl benzoate. In summary, benzaldehyde is produced with the best selectivity in cyclohexane at low amount of base, benzoic acid in water at high amount of base and benzyl benzoate in solvent-free condition also at high amount of base. These observations are quite important when the selective oxidation of substrates derived from biomass, such as HMF, glycerol and glucose are considered. These reactions are generally performed in water, so the nature and concentration of the base promoters evaluated here are of great importance for further developments in this field.
Experimental
General remarks
HAuCl4, NaBH4, TiO2 (anatase) and polyvinyl alcohol (PVA, MW ∼ 9000–10000 g mol−1, 80% hydrolyzed) were purchased from Sigma-Aldrich; NaOH, Na(CH3COO), K2CO3 and Na2B4O7 were purchased from Merck; benzyl alcohol was purchased from J.T. Baker and used as received. Standards for GC analysis (benzaldehyde, benzoic acid and benzyl benzoate) were obtained from Sigma-Aldrich.Instrumentation
FAAS analysis was performed on a Shimadzu AA-6300 spectrophotometer using an Au hollow cathode lamp (Photron). TEM analyses were performed at the LNNano Laboratory (CNPEM, Campinas, Brazil) using a JEOL-JEM 2100F. Catalyst samples for TEM were prepared by sonicating the catalyst powder in isopropanol. A drop of the resulting dispersion was placed on a thin carbon film, which was deposited on standard 400-mesh TEM copper grids and air-dried. GC analyses were carried out with a Shimadzu GC-2010 equipped with a RTx-Wax column (30 m × 0.25 mm × 0.25 mm) and a FID detector. Method: Ti = 50 °C, Tf = 200 °C, 30 min, T FID and SPLIT = 200 °C. Internal standard: biphenyl.Synthesis of AuPVA/TiO2
The Au nanoparticles were prepared based on literature.40,50 A 2% solution of PVA was added to an aqueous HAuCl4 solution (5.08 × 10−4 M) with vigorous stirring (PVA/Au (w/w) = 1.2); a 0.1 M freshly prepared solution of NaBH4 (NaBH4/Au (mol/mol) = 5) was then added to form a metallic sol; the color of the sol was light-red (Au). After 30 min of sol generation, the colloid was immobilized by adding commercial titania (anatase) under vigorous stirring. The amount of support was calculated to give a total final metal loading of 2 wt% (nominal). After 2 h the slurry was filtered, the solid washed with hot water (2 × 25 mL) and ethanol (2 × 25 mL) and dried at 100 °C for 1 h. The metal content in the AuPVA/TiO2 catalyst determined by FAAS was 1.7 wt% Au.Catalytic reactions
All reactions were performed using a modified Fischer–Porter 50 mL glass reactor. In a typical reaction, the glass reactor was loaded with MiliQ water or cyclohexane (9.7 mL), benzyl alcohol (0.3 mL, 2.9 mmol), the AuPVA/TiO2 catalyst (15.6 mg, 1.3 μmol Au) and the base ([base]:[alcohol] = 0.1, 0.5, 1, 2 or 4); where the bases are: K2CO3, NaOH, Na(CH3COO) and Na2B4O7. The reactor was purged five times with O2, leaving the vessel at 6 bar. The temperature was maintained at 100 °C with an oil bath on a hot stirring plate connected to a digital controller (ETS-D5 IKA). The reactions were constantly stirred using Teflon-coated magnetic stir bars for the desired amount of time (15, 30, 45, 60, 75, 150 min). The reaction mixture was collected and the catalyst was removed by centrifugation. The pH of the resulting solution was measured and adjusted to 5 and the products extracted with CH2Cl2. The aqueous phase was collected and the pH adjusted to 2. The products were extracted with CH2Cl2 and the organic phases were combined and analyzed by GC.
In a typical solvent-free reaction, the glass reactor was loaded with benzyl alcohol (1 mL, 9.7 mmol), the AuPVA/TiO2 catalyst (52 mg, 4.4 μmol Au) and the base ([K2CO3]:[alcohol] = 0.1 or 1 (133 mg or 1.33 g)). The reactor was purged five times with O2, leaving the vessel at 6 bar. The temperature was maintained at 100 °C with an oil bath on a hot stirring plate connected to a digital controller (ETS-D5 IKA). The reactions were constantly stirred using Teflon-coated magnetic stir bars for 60 min. To the reaction mixture, 9 mL of CH2Cl2 was added and the catalyst was removed by centrifugation. An aliquot of the solution was removed and analyzed by GC.
Quantification of hydrogen peroxide
The oxidation reactions were performed for 1 h and after cooling to room temperature, the catalyst was removed. Hydrogen peroxide was determined spectrophotometrically by the peroxidase assay.55 To estimate the amount of H2O2 present in the different conditions, an aliquot from the reaction (10 μL) was tested for the formation of triiodide (ε = 25500, λ = 350 nm) using the HRP enzyme (horseradish peroxidase). In a UV cell containing 2990 μL of KI in acetate buffer (pH = 3.8) and 10 μL of HRP enzyme solution (1.0 g mL−1), the aliquot from reaction was added and measured at 353 nm in duplicate. The blank was measured in a UV cell containing 2990 μL of KI in acetate buffer (pH = 3.0) and 10 μL of HRP enzyme solution (1.0 g mL−1).
Acknowledgements
The authors acknowledge support from FAPESP (Fellowship Process No. 2014/10824-3; Grant Process 2014/15159-8), CNPq and INCT-Catálise. We also thank LNNano-CNPEM (Campinas, Brazil) for the use of TEM facilities and the Laboratório de Quimiluminescência Orgânica (IQ-USP, Brazil) for the hydrogen peroxide quantification assay.References
- M. B. Smith and J. March, in March's Advanced Organic Chemistry, John Wiley & Sons, Inc., 2006, pp. 1703–1869 Search PubMed.
- Catalytic Oxidation: Principles and Applications, ed., R. A. Sheldon and R. A. V. Santen, World Scientific Pub Co Inc, Singapore, River Edge, NJ, 1996 Search PubMed.
- A. Corma and H. García, Chem. Soc. Rev., 2008, 37, 2096–2126 RSC.
- T. Mallat and A. Baiker, Chem. Rev., 2004, 104, 3037–3058 CrossRef CAS PubMed.
- C. Della Pina, E. Falletta, L. Prati and M. Rossi, Chem. Soc. Rev., 2008, 37, 2077–2095 RSC.
- C. W. Corti, R. J. Holliday and D. T. Thompson, Top. Catal., 2007, 44, 331–343 CrossRef CAS.
- J. S. McPherson and D. T. Thompson, Top. Catal., 2009, 52, 743–750 CrossRef CAS.
- O. Vaughan, Nat. Nanotechnol., 2010, 5, 5–7 CrossRef CAS PubMed.
- S. E. Davis, M. S. Ide and R. J. Davis, Green Chem., 2012, 15, 17–45 RSC.
- C. Della Pina, E. Falletta and M. Rossi, Green Chem., 2011, 13, 1624–1632 RSC.
- E. Falletta, C. Della Pina, M. Rossi, Q. He, C. J. Kiely and G. J. Hutchings, Faraday Discuss., 2011, 152, 367–379 RSC.
- J. C. Serrano-Ruiz and J. A. Dumesic, Energy Environ. Sci., 2010, 4, 83–99 Search PubMed.
- J. N. Chheda, G. W. Huber and J. A. Dumesic, Angew. Chem., Int. Ed., 2007, 46, 7164–7183 CrossRef CAS PubMed.
- L. Prati and M. Rossi, J. Catal., 1998, 176, 552–560 CrossRef CAS.
- M. Chen and D. W. Goodman, Acc. Chem. Res., 2006, 39, 739–746 CrossRef CAS PubMed.
- A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896–7936 CrossRef PubMed.
- B. K. Min and C. M. Friend, Chem. Rev., 2007, 107, 2709–2724 CrossRef CAS PubMed.
- M. Haruta, J. Vac. Soc. Jpn., 2008, 51, 721–726 CrossRef CAS.
- R. Meyer, C. Lemire, S. K. Shaikhutdinov and H.-J. Freund, Gold Bull., 2004, 37, 72–124 CrossRef CAS.
- G. Bond, Gold Bull., 2008, 41, 235–241 CrossRef.
- X. Yang, X. Wang, C. Liang, W. Su, C. Wang, Z. Feng, C. Li and J. Qiu, Catal. Commun., 2008, 9, 2278–2281 CrossRef CAS.
- S. K. Klitgaard, A. T. DeLa Riva, S. Helveg, R. M. Werchmeister and C. H. Christensen, Catal. Lett., 2008, 126, 213–217 CrossRef CAS.
- A. Villa, C. E. Chan-Thaw, G. M. Veith, K. L. More, D. Ferri and L. Prati, ChemCatChem, 2011, 3, 1612–1618 CrossRef CAS.
- V. R. Choudhary, A. Dhar, P. Jana, R. Jha and B. S. Uphade, Green Chem., 2005, 7, 768–770 RSC.
- A. Abad, A. Corma and H. García, Chem.–Eur. J., 2008, 14, 212–222 CrossRef CAS PubMed.
- R. L. Oliveira, P. K. Kiyohara and L. M. Rossi, Green Chem., 2009, 11, 1366–1370 RSC.
- H. Guo, A. Al-Hunaiti, M. Kemell, S. Rautiainen, M. Leskelä and T. Repo, ChemCatChem, 2011, 3, 1872–1875 CrossRef CAS.
- L. Prati, A. Villa, C. E. Chan-Thaw, R. Arrigo, D. Wang and D. S. Su, Faraday Discuss., 2011, 152, 353–365 RSC.
- A. Buonerba, C. Cuomo, S. Ortega Sánchez, P. Canton and A. Grassi, Chem.–Eur. J., 2012, 18, 709–715 CrossRef CAS PubMed.
- B. Karimi and F. K. Esfahani, Chem. Commun., 2009, 5555–5557 RSC.
- V. V. Costa, M. Estrada, Y. Demidova, I. Prosvirin, V. Kriventsov, R. F. Cotta, S. Fuentes, A. Simakov and E. V. Gusevskaya, J. Catal., 2012, 292, 148–156 CrossRef CAS.
- E. Skupien, R. J. Berger, V. P. Santos, J. Gascon, M. Makkee, M. T. Kreutzer, P. J. Kooyman, J. A. Moulijn and F. Kapteijn, Catalysts, 2014, 4, 89–115 CrossRef.
- L. Prati and F. Porta, Appl. Catal., A, 2005, 291, 199–203 CrossRef CAS.
- M. Comotti, C. Della Pina, R. Matarrese, M. Rossi and A. Siani, Appl. Catal., A, 2005, 291, 204–209 CrossRef CAS.
- Y. Yuan, N. Yan and P. J. Dyson, Inorg. Chem., 2011, 50, 11069–11074 CrossRef CAS PubMed.
- A. Thomas, Q. He and J. K. Edwards, Faraday Discuss., 2011, 152, 381–392 RSC.
- N. Zheng and G. D. Stucky, Chem. Commun., 2007, 3862–3864 RSC.
- H. Tsunoyama, H. Sakurai, Y. Negishi and T. Tsukuda, J. Am. Chem. Soc., 2005, 127, 9374–9375 CrossRef CAS PubMed.
- S. Rautiainen, O. Simakova, H. Guo, A.-R. Leino, K. Kordás, D. Murzin, M. Leskelä and T. Repo, Appl. Catal., A, 2014, 485, 202–206 CrossRef CAS.
- N. Dimitratos, J. A. Lopez-Sanchez, D. Morgan, A. Carley, L. Prati and G. J. Hutchings, Catal. Today, 2007, 122, 317–324 CrossRef CAS.
- S. Biella, L. Prati and M. Rossi, Inorg. Chim. Acta, 2003, 349, 253–257 CrossRef CAS.
- A. Abad, P. Concepción, A. Corma and H. García, Angew. Chem., Int. Ed., 2005, 44, 4066–4069 CrossRef CAS PubMed.
- A. Villa, N. Janjic, P. Spontoni, D. Wang, D. S. Su and L. Prati, Appl. Catal., A, 2009, 364, 221–228 CrossRef CAS.
- N. Dimitratos, A. Villa, D. Wang, F. Porta, D. Su and L. Prati, J. Catal., 2006, 244, 113–121 CrossRef CAS.
- J. A. Lopez-Sanchez, N. Dimitratos, P. Miedziak, E. Ntainjua, J. K. Edwards, D. Morgan, A. F. Carley, R. Tiruvalam, C. J. Kiely and G. J. Hutchings, Phys. Chem. Chem. Phys., 2008, 10, 1921–1930 RSC.
- D. I. Enache, J. K. Edwards, P. Landon, B. Solsona-Espriu, A. F. Carley, A. A. Herzing, M. Watanabe, C. J. Kiely, D. W. Knight and G. J. Hutchings, Science, 2006, 311, 362–365 CrossRef CAS PubMed.
- T. A. G. Silva, E. Teixeira-Neto, N. López and L. M. Rossi, Sci. Rep., 2014, 4, 5766 CAS.
- J. C. F. Rodríguez-Reyes, C. M. Friend and R. J. Madix, Surf. Sci., 2012, 606, 1129–1134 CrossRef.
- B. N. Zope, D. D. Hibbitts, M. Neurock and R. J. Davis, Science, 2010, 330, 74–78 CrossRef CAS PubMed.
- C. L. Bianchi, P. Canton, N. Dimitratos, F. Porta and L. Prati, Catal. Today, 2005, 102–103, 203–212 CrossRef CAS.
- A. Villa, D. Wang, G. M. Veith, F. Vindigni and L. Prati, Catal. Sci. Technol., 2013, 3, 3036–3041 CAS.
- J. A. Lopez-Sanchez, N. Dimitratos, C. Hammond, G. L. Brett, L. Kesavan, S. White, P. Miedziak, R. Tiruvalam, R. L. Jenkins, A. F. Carley, D. Knight, C. J. Kiely and G. J. Hutchings, Nat. Chem., 2011, 3, 551–556 CrossRef CAS PubMed.
- M. Conte, H. Miyamura, S. Kobayashi and V. Chechik, J. Am. Chem. Soc., 2009, 131, 7189–7196 CrossRef CAS PubMed.
- M. M. William and C. Ketchie, Top. Catal., 2007, 44, 307–317 CrossRef.
- M. L. Cotton and H. B. Dunford, Can. J. Chem., 1973, 51, 582–587 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra01795a |
| This journal is © The Royal Society of Chemistry 2016 |