
Surface-enhanced Raman spectroscopy on porous silicon membranes decorated with Ag nanoparticles integrated in elastomeric microfluidic chips†

Abstract
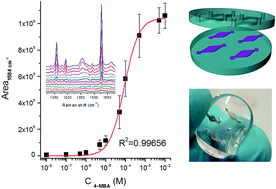
A microfluidic sensing device is fabricated by the integration of silver-coated porous silicon membranes as surface enhanced Raman scattering active nanostructures in a PDMS matrix showing plasmon resonances in the visible energy range. The membranes are shaped by defining a suitable pattern of the electrochemically etched silicon using the cover of the elastomeric chip. 4-Mercaptobenzoic acid, used as a probe molecule, shows Raman enhancement larger than 107 in electronic off-resonance conditions. The calibration of the SERS substrate is performed using a Langmuir isotherm fit, evidencing the analyte chemisorption. In order to check the potential of the chip as a multiplexed biosensing platform, model oligonucleotides (polyadenine, polyguanine, polythymine and polycytosine) are analyzed in a multichamber system.
- This article is part of the themed collection: Surface enhanced Raman Spectroscopy: Editors collection for RSC Advances
 Please wait while we load your content...
Please wait while we load your content...

