
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA two-dimensional conjugated polymer framework with fully sp2-bonded carbon skeleton†
Xiaodong
Zhuang‡
a,
Wuxue
Zhao‡
a,
Fan
Zhang
*a,
Yu
Cao
b,
Feng
Liu
 *b,
Shuai
Bi
a and
Xinliang
Feng
*ac
*b,
Shuai
Bi
a and
Xinliang
Feng
*ac
aShanghai Key Lab of Electrical Insulation and Thermal Ageing & Shanghai Electrochemical Energy Devices Research Center, School of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, Shanghai 200240, China. E-mail: fan-zhang@sjtu.edu.cn; xinliang.feng@tu-dresden.de
bState Key Laboratory for Mechanical Behavior of Materials, Xi'an Jiaotong University, Xi'an 710049, P. R. China. E-mail: feng.liu@mail.xjtu.edu.cn
cCenter for Advancing Electronics Dresden& Department of Chemistry and Food Chemistry, Technische Universitaet Dresden, 01062 Dresden, Germany
First published on 9th May 2016
Abstract
The synthesis of crystalline two-dimensional (2D) covalent organic frameworks (COFs) with fully unsaturated carbon–carbon backbones via a solution approach remains a great challenge. In this work, we report the first example of an olefin-linked 2D conjugated COF using a Knoevenagel polycondensation reaction of 1,4-phenylene diacetonitrile and three armed aromatic aldehyde. The resulting 2D poly(phenelyenevinylene) framework (2DPPV) possesses a sheet morphology, and a crystalline layered structure featuring a fully sp2-bonded carbon skeleton with pendant cyanide groups. Its unique alternating structure with a serrated configuration has been essentially evaluated using HR-TEM TEM analysis, nitrogen physisorption measurements, PXRD studies and theoretical simulations. Upon thermal and activation treatments, the as-prepared 2DPPV can be facilely converted into porous carbon nanosheets with large specific surface areas of up to 880 m2 g−1 which exhibit an excellent electrochemical performance as supercapacitor electrodes and electrocatalysts for the oxygen reduction reaction. This represents an economic non-template approach to 2D porous carbon materials for energy-related applications.
Introduction
In the family of two-dimensional (2D) soft materials,1 2D polymers2 have drawn increasing attention due to their laterally infinite, one-atom or one-monomer-unit thick, freestanding networks with topologically repeating units based on covalent/non-covalent connection bonds. These unique structural features of 2D polymers qualify their potential in optoelectronics, catalysis, membrane technology, etc.3 Most of the approaches to covalent 2D polymers have been focused on the “bottom-up” surface/interface synthesis4 or “top-down” delamination/exfoliation5 of synthesized laminar structures, in particular for crystalline covalent organic frameworks (COFs).6 Fully conjugated COFs and 2D polymers remain less explored by far, with only a few examples, limited to the imine-bond7 and metal–organic coordination derived frameworks.8 The synthesis of 2D conjugated COFs based on unsaturated C–C linkages remains a great challenge due to the lack of suitable synthetic protocols. Electrochemical property studies are one of the most important ways to get access to fundamental understanding of new conjugated materials. Unfortunately, very rare studies about the electrochemical properties of conjugated COFs have been reported.Herein, for the first time, we report the synthesis of a 2D conjugated COF based on olefin (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) linkages using the Knoevenagel condensation reaction. The Knoevenagel condensation9 is a widely used organic methodology that can convert an aldehyde or ketone (CO) into a cyano-substituted and cis-configuration preferred olefin using a base as catalyst (see model reaction in Scheme 1).10 The condensation has been previously used for synthesizing high molecular weight cyano-substituted linear poly(phenylenevinylenes).10 In this work, employing 1,4-phenylenediacetonitrile and three armed aromatic aldehyde as the key monomers, the Knoevenagel polycondensation reaction successfully leads to the formation of a 2D poly(phenylenevinylene) framework (2DPPV) with a crystalline layered structure and a surface area of 472 m2 g−1, as well as a defined optical band gap of 2.10 eV and a lowest unoccupied molecular orbital (LUMO) of −3.45 eV. Upon thermal and activation treatments, it can be facilely converted into porous carbon nanosheets with large specific surface areas of up to 880 m2 g−1, which exhibit an excellent electrochemical performance as supercapacitor electrodes and electrocatalysts for the oxygen reduction reaction (ORR), representing an economic non-template approach to 2D porous carbon materials for energy-related applications.
C) linkages using the Knoevenagel condensation reaction. The Knoevenagel condensation9 is a widely used organic methodology that can convert an aldehyde or ketone (CO) into a cyano-substituted and cis-configuration preferred olefin using a base as catalyst (see model reaction in Scheme 1).10 The condensation has been previously used for synthesizing high molecular weight cyano-substituted linear poly(phenylenevinylenes).10 In this work, employing 1,4-phenylenediacetonitrile and three armed aromatic aldehyde as the key monomers, the Knoevenagel polycondensation reaction successfully leads to the formation of a 2D poly(phenylenevinylene) framework (2DPPV) with a crystalline layered structure and a surface area of 472 m2 g−1, as well as a defined optical band gap of 2.10 eV and a lowest unoccupied molecular orbital (LUMO) of −3.45 eV. Upon thermal and activation treatments, it can be facilely converted into porous carbon nanosheets with large specific surface areas of up to 880 m2 g−1, which exhibit an excellent electrochemical performance as supercapacitor electrodes and electrocatalysts for the oxygen reduction reaction (ORR), representing an economic non-template approach to 2D porous carbon materials for energy-related applications.
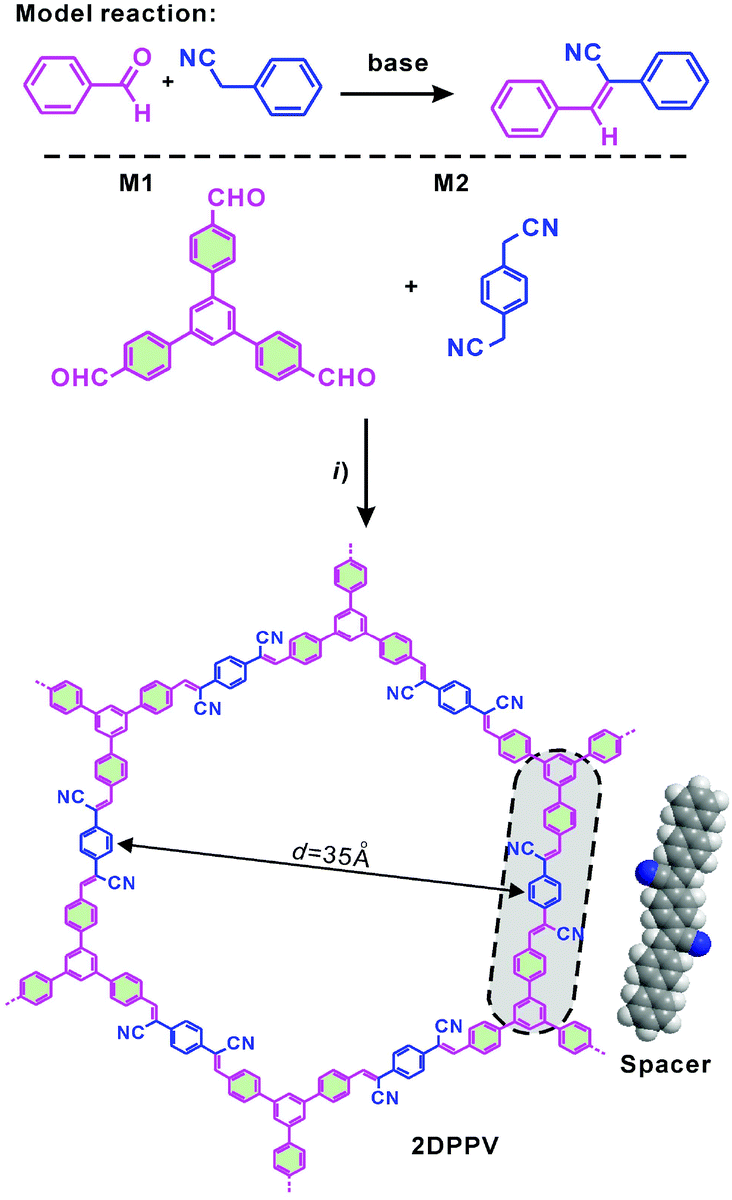 | ||
| Scheme 1 Synthesis and structures of the olefin-linked 2D conjugated polymer framework (2DPPV). (i) Argon, cesium carbonate, 1,2-dichlorobenzene, 150 °C, 3 days. | ||
Experimental section
All starting materials, unless otherwise noted, were purchased from Aladdin and TCI. All of the reactions were carried out under a nitrogen atmosphere and performed using Schlenk-line techniques. 1,3,5-tris-(4-formylphenyl)benzene (M1) was synthesized according to the reported procedures. Typically, the synthesis of 2DPPV was given as follows:To a 10 mL Pyrex tube, p-phenylenediacetonitrile (60 mg, 0.384 mmol), 1,3,5-tris(4-formylphenyl)benzene (100 mg, 0.256 mml), Cs2CO3 (250 mg, 1.15 mmol) and o-dichlorobenzene (4 ml) were added. This mixture was treated with freeze–pump–thaw cycles three times and sealed under vacuum. After heating at 150 °C for 3 days, the mixture was cooled to room temperature to collect the precipitate by centrifugation. After Soxhlet extraction using THF and ethanol for 3 days and further drying at 100 °C under vacuum for 10 h, a yellowish powder was produced (∼90%).
As a typical example, 2DPPV was pyrolyzed at 700, 800 and 900 °C under an argon atmosphere. The 800 °C pyrolyzed carbon was further treated at 800 °C under an ammonia atmosphere for 15 min to produce activated pores. The resulting porous carbons are denoted as 2DPPV-X (X = 700, 800 900 and 800a, respectively).
Characterization
Fourier transform infrared (FTIR) spectra were recorded with a Spectrum 100 spectrometer (Perkin Elmer, Spectrum 100). Elemental analysis was carried out using a Vario EL III/Isoprime. Solid-state 13C CPMAS NMR analysis was conducted using a Bruker AVANCE III 300 Spectrometer. Samples were spun at 5 kHz in a 4 mm zirconium rotor within a MAS probe. An acquisition time of 20 ms, a contact time of 1 ms and a 6.5 μs pre-scan delay were used. The recycle time was 2 s to obtain fully relaxed spectra. Chemical shifts were externally referenced to adamantane at 38.48 ppm. The morphology of the samples was investigated using a transmission electron microscope (TEM, JEOL, JEM-2010). Scanning electron microscopy (SEM) measurements were performed using a FEI Sirion-200 field emission microscope. X-ray photoelectron spectroscopy (XPS) was performed using a Kratos Axis X-ray photoelectron spectrometer. The binding energy was calibrated using the C 1s photoelectron peak at 284.6 eV as a reference. The elemental percentages were calculated from XPS spectra using the Casa XPS computer program with specific relative sensitivity factors for the Kratos Axis XPS (Ti 2p: 2.001, O 1s: 0.78, and N 1s: 0.47). Casa XPS was also used for peak fitting on the XPS spectra. Thermogravimetric analysis (TGA) was measured using a TAQ5000IR with a heating rate of 20 °C min−1 under a nitrogen flow. Nitrogen adsorption/desorption isotherm measurements were performed using a Micromeritics ASAP 2010 surface area and pore size analyzer at 77 K. Prior to the measurement, the samples were degassed under vacuum at 200 °C for 12 h.Results and discussion
The strategy of the synthesis of 2DPPV is presented in Scheme 1. Firstly, three-armed aromatic aldehyde (M1: 1,3,5-tri(4-formylphenyl)benzene) and 1,4-phenylenediacetonitrile (M2) were sealed with cesium carbonate and o-DCB which served as catalyst and solvent respectively after three freeze and thaw cycles. Then, the mixture was heated to 150 °C and kept for 3 days without stirring before termination of the reaction by exposing it to ambient atmosphere. After washing with water and tetrahydrofuran (THF) several times to remove the residual inorganic catalyst, a yellow, fluffy and reflective powder (yield > 90%) was produced after vacuum drying. The resulting polymer is insoluble in conventional organic solvents. Thermogravimetric analysis (TGA) reveals that as-prepared 2DPPV is thermally stable up to 400 °C (weight loss <10 wt%, see Fig. S1†). Further increasing the temperature to 800 °C, more than 60% of the weight is maintained, suggesting the carbon-rich nature of 2DPPV.The chemical identity of 2DPPV was first characterized using Fourier transform infrared (FTIR) spectroscopy. The FTIR spectra (see Fig. S2†) of 2DPPV clearly reveal the presence of cyano groups at 2250 cm−1. The peak at 1693 cm−1 which corresponds to the CO stretching vibration for aldehyde groups in monomers disappeared in the FT-IR spectrum of 2DPPV. Most importantly, the signal at 3020 cm−1 which is attributable to CCH, was found for the synthesized 2DPPV. These results strongly suggest the high polymerization efficiency of aromatic aldehydes and 1,4-phenylenediacetonitrile into polymeric structures. Unfortunately, a heavy overlap makes it difficult to clearly resolve the assignment of the individual carbon signals for 2DPPV using solid-state 13C nuclear magnetic resonance spectroscopy (Fig. S3a†) due to the carbon-rich skeleton. Therefore, X-ray photoelectron spectroscopy (XPS) was further used to disclose the structural information for the as-synthesized polymer framework (see Fig. S3b and S3c†). According to the C 1s XPS spectra, two kinds of carbon signals, namely sp2-hybridized and sp-hybridized carbon, can be found, which can be ascribed to the aromatic/olefin carbon (284.5 ± 0.1 eV) and the cyano carbon (286.0 ± 0.5 eV) respectively. In addition, 2DPPV shows a similar N1s binding energy at 398.0 ± 0.1 eV which originates from the nitrogen moiety in the cyano group. All of these results highlight that an olefin-linked conjugated structure has been successfully built up within 2DPPV.
An optical microscopy inspection of the powders of 2DPPV demonstrated their large-size (up to 100 μm), crystalline flake morphology and yellow semitransparent features (see Fig. 1a, b and S4†). The morphology and microstructure of the as-prepared 2DPPV were further investigated using scanning electron microscopy (SEM) and atomic force microscopy (AFM). As shown in Fig. 1e and S5,† many sheet-like flakes with sizes ranging from several to a hundred microns were observed. The thickness of 2DPPV measured using AFM ranged from 50 to 300 nm (Fig. S6†). All of these results indicate that the as-synthesized polymer framework may possess a crystalline lamellar structure. To gain insight into the thin layer morphology of the materials, steel ball milling exfoliation on as-synthesized 2DPPV was attempted to produce thinner nano-sheets for high resolution transmission electron microscopy (HR-TEM) studies. It was found that it was very difficult to achieve single layer sheets of 2DPPV using such an exfoliation method (Fig. S7†), while other solution exfoliation protocols also generated multilayer flakes.5d Nevertheless, it is notable that HR-TEM images of 2DPPV clearly demonstrate the local structural ordering with apparent layered textures (see Fig. 1c and S8†), which have an average layer distance of 0.35 ± 0.01 nm (see Fig. 1d), one of the most typical 2D structural characteristics for the COFs as disclosed in some recent reports.11
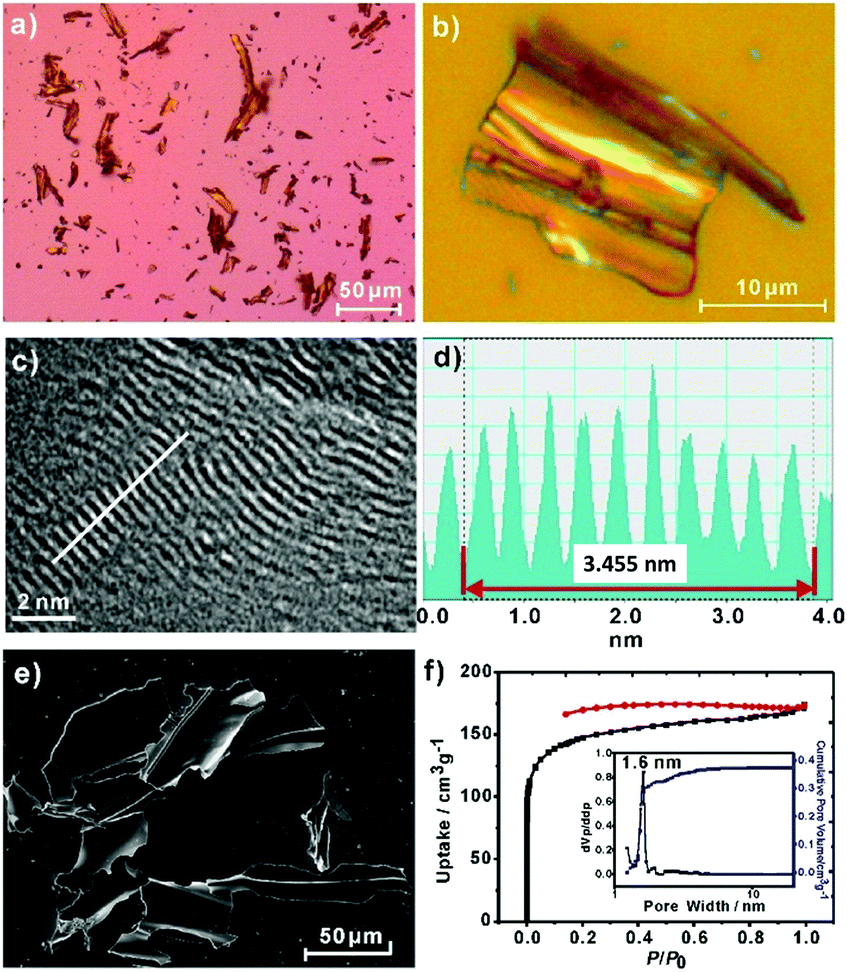 | ||
| Fig. 1 2DPPV: (a, b) optical microscope images; (c) HR-TEM image; (d) the white line trace from (c) shows the resolution of the interlayer distance at about ∼0.35 nm; (e) SEM image; (f) nitrogen adsorption–desorption isotherms, with the pore size distribution shown in the inset. | ||
The porosity of 2DPPV was investigated using nitrogen physisorption measurements at 77 K. As shown in Fig. 1f, 2DPPV exhibits type II isotherms according to IUPAC classification.13 The Brunauer–Emmett–Teller (BET) surface area and pore volume were found to be 472 m2 g−1 and 0.37 cm3 g−1, respectively. The pore size distribution was calculated on the basis of nonlocal density functional theory (NLDFT) to give a major pore diameter of around 1.6 nm. Such low surface area and small pore sizes are distinctly different from the values of the previously reported COFs comprising similar size building blocks,11b probably implying the unique porous structural natures of 2DPPV.
The crystallinity of 2DPPV was determined with powder X-ray diffraction (PXRD) experiments using a synchrotron radiation source at Shanghai Synchrotron Radiation Facility, SSRF. A series of high-resolution diffraction peaks were obtained in the PXRD pattern (see Fig. 2a and S9†). We did not find the diffraction data from the starting materials as carefully compared with their PXRD patterns (see Fig. S10†). It should also be noted that an important feature of the diffraction pattern is that the first peak exhibits the strongest diffraction intensity, indicating that a highly periodic structure and crystalline features exist in 2DPPV. In addition, a peak at q = 17.9 nm−1 correlating to the value of the interlayer distance was observed, and the d-spacing of the 2DPPV was calculated as being about 0.35 nm, which is consistent with the HR-TEM characterization (see Fig. 1c and S8†).
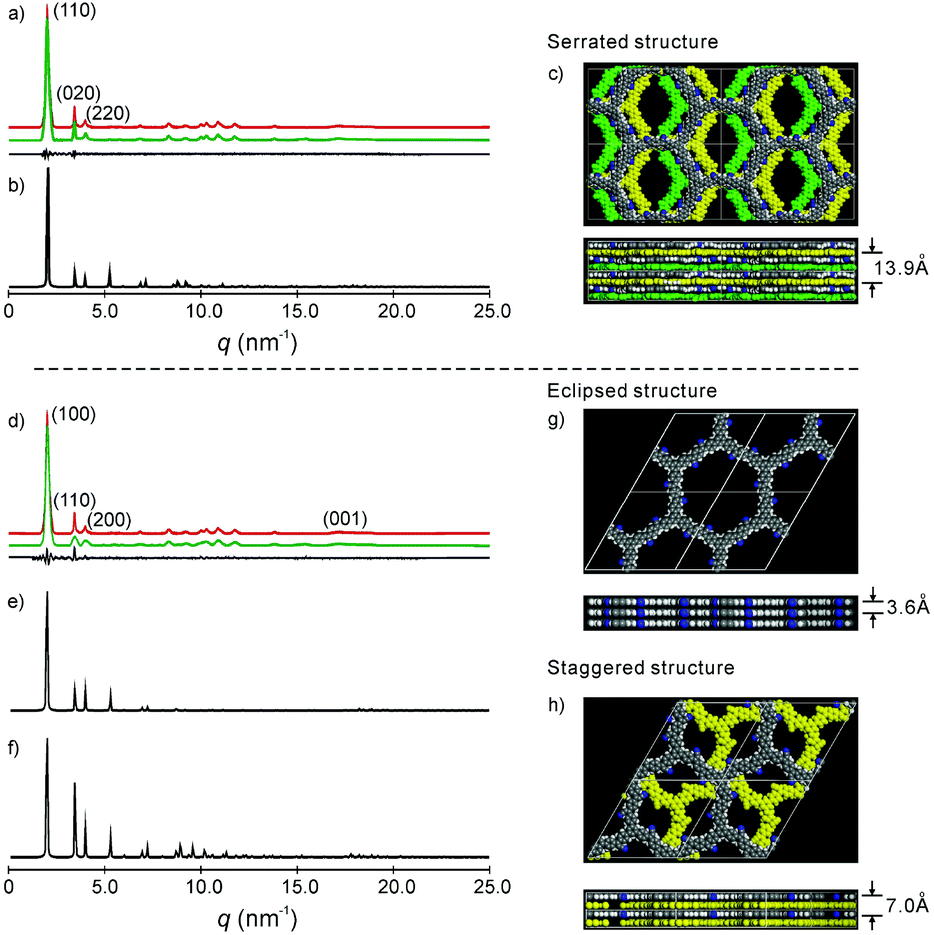 | ||
| Fig. 2 (a) and (d) Experimental (red) and refined (green) PXRD pattern of the serrated and eclipsed stacking mode of the as-prepared 2DPPV, with the difference plot between the experimental and refined patterns shown as a black line; (b), (e) and (f) simulated PXRD patterns of the serrated, eclipsed and staggered stacking mode of the 2DPPV; (c), (g) and (h) snapshots of the simulated structure of the serrated, eclipsed and staggered stacking modes, where the bottom row is the side view of the cell and for (c) and (h), the 2DPPV are colored according to their relative positions in the unit cell; please also see Fig. S13b.† | ||
To gain insight into the exact structure of 2DPPV, simulations were further conducted using Reflex in the Accelrys Material Studio software package and then compared with the experimental results. Considering the structure of 2DPPV and its low-pressure nitrogen adsorption uptake, an “A–B–A–B” type alternating structure based on a serrated stacking mode, in which adjacent sheets were slipped by 1/4 of the unit cell distance, was the first to be proposed and examined (see Fig. 2c, S11 and S13†). In this case, the first three diffractions can be assigned to the 110, 020 and 220 reflections of an orthorhombic lattice with the parameters a = 6.35 nm, b = 3.62 nm and c = 1.40 nm (see Fig. 2a and Table S1†). From Pawley refinement, using the space group of Cmcm with optimized parameters of a = 6.32 nm, b = 3.57 nm and c = 1.39 nm, a PXRD pattern in good agreement with the experimentally observed pattern was achieved (see Fig. 2a, b and S12†), as also evidenced by their negligible difference (black curve of Fig. 2a), with an Rwp of 3.40% and Rp of 7.12%, respectively. Furthermore, we also considered alternative structures wherein adjacent sheets are either eclipsed based on the space group of P6/mmm (see Fig. 2g and S14†) or staggered based on the space group of P63/mmc (see Fig. 2h and S16†). For the eclipsed stacking mode with a hexagonal periodic structure, a simulated PXRD pattern was also obtained in excellent agreement with the experimental ones, with an Rwp of 5.39% and Rp of 2.94%, respectively (see Fig. 2d, e and S15†). However, for such a model, 2DPPV should possess a pore size distribution with a theoretical value of 3.5 nm (see Scheme 1), much larger than that of the pore size (1.6 nm) calculated from NLDFT on the basis of the porosity analyses, and the gas uptake observed at low-pressure (472 m2 g−1) is less consistent with such a structure. For a staggered model, in which adjacent sheets were slipped by distances of a/2 and b/2 (see Fig. 2h and S16†), its simulated PXRD patterns did not match the observed data very well (see Fig. 2f and S17†), so the staggered model could be ruled out.
On the basis of the results discussed above, the exact architecture of 2DPPV is proposed to adopt the serrated stacking mode with neighboring layers slipped by 1/4 of the unit cell distance on average, separated by a distance of 0.35 nm (Fig. 2c, bottom row). Since the direction of the translation among those adjacent layers is random, calculation from rotating three in-plane orientations gives a pore size of about 1.6 nm (see Fig. S13†), which is well in line with the result predicted from NLDFT as shown above (Fig. 1f, inset). The detailed porosity analysis was summarized in Fig. S13 and Table S3.† Therefore, the results demonstrate that Knoevenagel polycondensation successfully leads to the formation of a 2D COF that consists of the serrated-type stacking of 2DPPV sheets with an “A–B–A–B” type alternating arrangement.6a
Encouraged by these interesting results, we can rationally consider that in the course of Knoevenagel condensation: (1) the resulting cyano stilbene motif with cis-conformation is favorable for the formation of hexagonal repeat units in the 2D sheet;12 (2) the generated carbon anion intermediate can be partly stabilized by the cyanide group, which contributes to the certain reversibility of the reaction under experimental conditions leading to eventual CC linkage formation (see Scheme S1†). Thus, here the Knoevenagel condensation can be considered as a quasi-dynamic covalent reaction which is essential for a high-degree polymerization efficiency and for the formation of a crystalline layered framework.
Given the sp2-bonded carbon skeleton of the resulting 2DPPV, we investigated its photophysical properties using UV-vis spectroscopy and cyclic voltammetry (CV). According to the UV-vis absorption spectrum of 2DPPV (see Fig. S18†), the absorption edge was found to be 587 nm. Therefore, the optical band gap for 2DPPV was calculated as 2.10 eV (see Table S4†), which is much lower than that of linear cyano-substituted PPV (2.5 eV).10 The electrochemical measurement was further carried out using cyclic voltammetry (CV) in deaerated acetonitrile containing recrystallized nBu4NPF6 (0.1 M) at room temperature. Due to the strong electron-withdrawing effect of cyano groups, 2DPPV exhibits quasi-reversible negative redox couples (see Fig. S19†). Based on this, the LUMO level of 2DPPV can be calculated as −3.45 eV. Thus with the optical band gap, the HOMO level of 2DPPV can be derived as −5.55 eV (see Table S4†).14
Polymer frameworks can serve as carbon-rich precursors by preserving their elemental composition and porous structure for the preparation of various porous carbon materials applicable for energy conversion and storage.15 Thanks to the high carbon yield (more than 60% even at 800 °C) for the as-prepared 2DPPV on the basis of TGA analysis (see Fig. S1†), 2DPPV was pyrolyzed at 700, 800 and 900 °C under an argon atmosphere. The resulting porous carbons are denoted as 2DPPV-X (X = 700, 800 and 900, respectively). 2DPPV-800 was further treated at 800 °C under an ammonia atmosphere for 15 min to produce activated pores. This sample is denoted as 2DPPV-800a. Nitrogen physisorption (see Fig. 3a, S20 and Table S5†) and SEM images (see Fig. 3b and S21†) reveal that 2DPPV-X maintained a nanosheet morphology with a remarkably improved specific surface area of up to 880 m2 g−1 (see Table S5†). To our knowledge, this is the first example of the preparation of large-sized (>10 μm) porous carbon nanosheets without using any 2D templates16 or inorganic porous templates.17 The N-doping character of the 2DPPV-X was further evaluated using XPS analysis (see Fig. S22 and Table S6†).
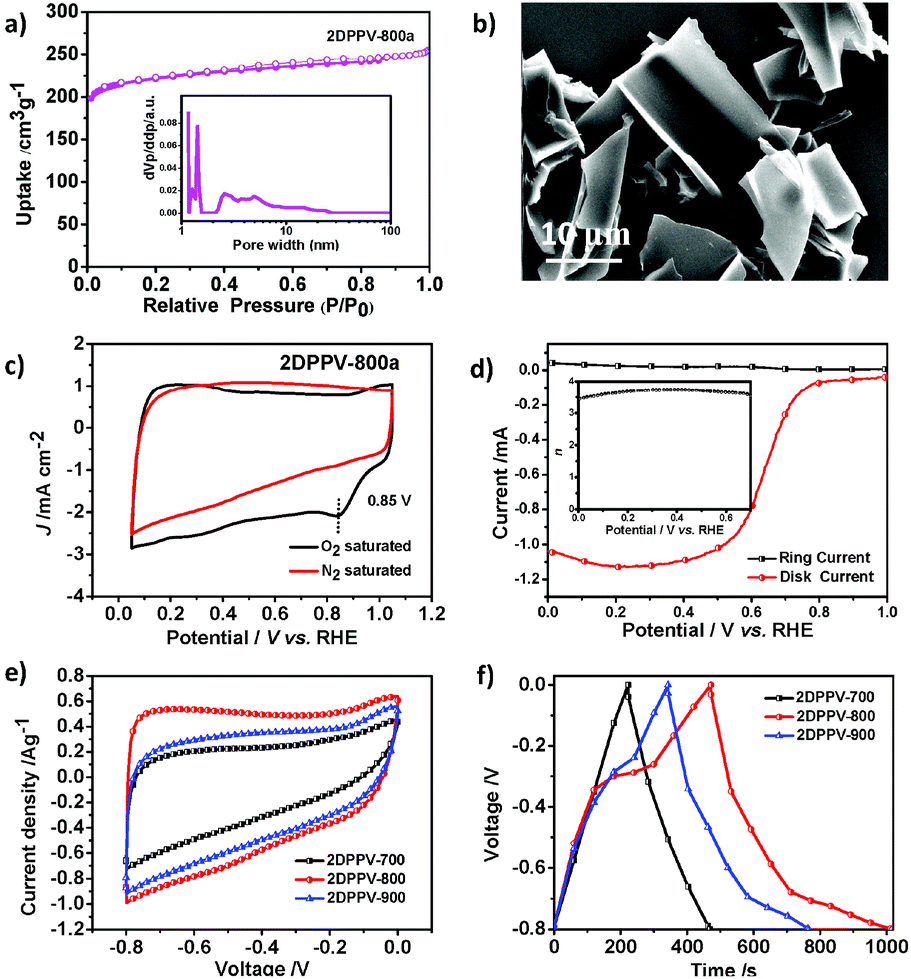 | ||
| Fig. 3 (a) Nitrogen adsorption–desorption isotherms of 2DPPV-800a (inset: pore size distribution); (b) SEM image of 2DPPV-700; (c) CV curves of 2DPPV-800a in N2-saturated and O2-saturated 0.1 M KOH at a scan rate of 50 mV s−1; (d) RRDE voltammogram for 2DPPV-800a in 0.1 M KOH solution saturated with O2 (the electrode rotation rate was 1600 rpm and the Pt ring electrode was held at 0.5 V, inset: electron transfer number of 2DPPV-800a as a function of potential); (e) CV curves of 2DPPV-X (X = 700, 800, 900) at 5 mV s−1 in 6 M KOH aqueous solution; and (f) galvanostatic charge/discharge curves for 2DPPV-X (X = 700, 800, 900) at a current density of 0.5 A g−1. | ||
One of the most important functions of N-doped porous carbons is as metal-free catalysts for the ORR. As a proof-of-concept, we examined the electrocatalytic activity of 2DPPV-800a, which possesses the highest specific surface area among 2DPPV-X, towards the ORR under alkaline conditions (0.1 M KOH) using both cyclic voltammetry (CV, Fig. 3c) and rotating ring-disk electrode voltammetry (RRDE, Fig. 3d). 2DPPV-800a exhibits a low onset potential at 0.85 V vs. RHE and primarily a four-electron pathway (n = 3.71 @ 0.4 V) based on CV and RRDE analysis respectively.18 These results indicate that as-prepared 2D porous carbon is a promising candidate for the electrochemically catalyzed ORR with a primary 4 e mechanism. These results are comparable to similar porous carbon-based materials and commercial precious metal Pt/C catalysts (Table S7†). The high surface area, N-doping features and 2D morphology of the as-prepared carbon nanosheets also render them applicable for use as electrochemical capacitors. As shown in Fig. 3e, the cyclic voltammetry profiles of 2DPPV-X (X = 700, 800, 900) in 6 M KOH show symmetric and horizontal characters, indicating their ideal capacitive behavior. In addition, an obviously higher current density was observed for 2DPPV-800 by comparison with others. The capacitive performance was further investigated with galvanostatic charge/discharge experiments (Fig. 3f). On the basis of the discharge curve, the specific capacitance of 2DPPV-800 was calculated to be 334 Fg−1 at 0.5 Ag−1, which is about 85% and 27% higher than those of 2DPPV-700 and 2DPPV-900, respectively (see Fig. S23†). This value is also among the highest performances of similar porous carbon materials (see Table S8†).19 The cycling stability of 2DPPV-800 (see Fig. S24†) was further evaluated using galvanostatic charge–discharge measurements at 0.5 Ag−1 in a 6 M KOH aqueous solution. Remarkably, almost no capacitance loss was observed after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles. Based on the Ragone plot, the power density and energy density of 2DPPV-800 were calculated at different rates (see Fig. S25†). Noticeably, a high energy density of 30 Wh kg−1 and power density of 6654 W kg−1 for 2DPPV-800 were attained. We consider that ultrathin graphitic layers with a high electrical conductivity for carbon nanosheets, combined with highly porous features, can promote the ion and electron transport in electrochemical processes. These excellent ORR and supercapacitor performances could be rationally attributed to the favorable long distance in-plane charge transporting features, associated with their unique porous structural characters.
000 cycles. Based on the Ragone plot, the power density and energy density of 2DPPV-800 were calculated at different rates (see Fig. S25†). Noticeably, a high energy density of 30 Wh kg−1 and power density of 6654 W kg−1 for 2DPPV-800 were attained. We consider that ultrathin graphitic layers with a high electrical conductivity for carbon nanosheets, combined with highly porous features, can promote the ion and electron transport in electrochemical processes. These excellent ORR and supercapacitor performances could be rationally attributed to the favorable long distance in-plane charge transporting features, associated with their unique porous structural characters.
Conclusions
In conclusion, for the first time, the olefin-linked 2D poly(phenelyenevinylene)-based COF (2DPPV) featuring a fully sp2 C–C skeleton has been successfully synthesized using a Knoevenagel polycondensation reaction. The as-prepared 2DPPV shows an intrinsic electronic structure related to its basic building blocks. The crystalline layered framework and excellent carbon yield at high temperatures mean that 2DPPV efficiently converted to large-sized porous carbon nanosheets with high surface areas through thermal treatment. These carbon nanosheets exhibit an excellent electrochemical performance as supercapacitor electrodes or as an electrocatalyst for the ORR. This work might pave a new way to 2D polymer and carbon materials with potential promising applications in a broad range involving optoelectronics, sensing and catalysis, as well as energy storage and conversion.Acknowledgements
The authors thank the financial support from the 973 Programs of China (2013CBA01602, 2012CB933400), the Natural Science Foundation of China (51403126, 21320102006, 21304057, 21574080, 21374086), and the ERC Grant on 2DMATER and EU Graphene Flagship. We also thank the Instrumental Analysis Center of Shanghai Jiao Tong University for some of the measurements. For help with the synchrotron experiments, we thank Beamline BL16B1 at SSRF (Shanghai Synchrotron Radiation Facility, China) for providing the beamtimes.References
- X. Zhuang, Y. Mai, D. Wu, F. Zhang and X. Feng, Adv. Mater., 2015, 27, 403 CrossRef CAS PubMed.
- (a) J. W. Colson and W. R. Dichtel, Nat. Chem., 2013, 5, 453 CrossRef CAS PubMed; (b) J. Sakamoto, J. van Heijst, O. Lukin and A. D. Schlüter, Angew. Chem., Int. Ed., 2009, 48, 1030 CrossRef CAS PubMed.
- (a) Y. Kou, Y. Xu, Z. Guo and D. Jiang, Angew. Chem., Int. Ed., 2011, 50, 8753 CrossRef CAS PubMed; (b) Z.-S. Wu, L. Chen, J. Liu, K. Parvez, H. Liang, J. Shu, H. Sachdev, R. Graf, X. Feng and K. Müllen, Adv. Mater., 2014, 26, 1450 CrossRef CAS PubMed.
- (a) L. Grill, M. Dyer, L. Lafferentz, M. Persson, M. V. Peters and S. Hecht, Nat. Nanotechnol., 2007, 2, 687 CrossRef CAS PubMed; (b) M. Bieri, M. Treier, J. Cai, K. Ait-Mansour, P. Ruffieux, O. Groning, P. Groning, M. Kastler, R. Rieger, X. Feng, K. Müllen and R. Fasel, Chem. Commun., 2009, 6919 RSC; (c) D. J. Murray, D. D. Patterson, P. Payamyar, R. Bhola, W. Song, M. Lackinger, A. D. Schluter and B. T. King, J. Am. Chem. Soc., 2015, 137, 3450 CrossRef CAS PubMed; (d) L. Xu, X. Zhou, Y. Yu, W. Q. Tian, J. Ma and S. Lei, ACS Nano, 2013, 7, 8066 CrossRef CAS PubMed.
- (a) P. Kissel, R. Erni, W. B. Schweizer, M. D. Rossell, B. T. King, T. Bauer, S. Goetzinger, A. D. Schlueter and J. Sakamoto, Nat. Chem., 2012, 4, 287 CrossRef CAS PubMed; (b) M. J. Kory, M. Worle, T. Weber, P. Payamyar, S. W. van de Poll, J. Dshemuchadse, N. Trapp and A. D. Schluter, Nat. Chem., 2014, 6, 779 CrossRef CAS PubMed; (c) P. Kissel, D. J. Murray, W. J. Wulftange, V. J. Catalano and B. T. King, Nat. Chem., 2014, 6, 774 CrossRef CAS PubMed; (d) D. N. Bunck and W. R. Dichtel, J. Am. Chem. Soc., 2013, 135, 14952 CrossRef CAS PubMed.
- (a) A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166 CrossRef PubMed; (b) Z. H. Xiang, D. P. Cao and L. M. Dai, Polym. Chem., 2015, 6, 1896 RSC; (c) S.-Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548 RSC; (d) X. Feng, X. S. Ding and D. L. Jiang, Chem. Soc. Rev., 2012, 41, 6010 RSC; (e) A. Thomas, Angew. Chem., Int. Ed., 2010, 49, 8328 CrossRef CAS PubMed.
- (a) S. Lin, C. S. Diercks, Y.-B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang, O. M. Yaghi and C. J. Chang, Science, 2015, 349, 1208 CrossRef CAS PubMed; (b) S. Kandambeth, V. Venkatesh, D. B. Shinde, S. Kumari, A. Halder, S. Verma and R. Banerjee, Nat. Commun., 2015, 6, 6786 CrossRef CAS PubMed; (c) S. Kandambeth, D. B. Shinde, M. K. Panda, B. Lukose, T. Heine and R. Banerjee, Angew. Chem., Int. Ed., 2013, 52, 13052 CrossRef CAS PubMed; (d) T. Y. Zhou, S. Q. Xu, Q. Wen, Z. F. Pang and X. Zhao, J. Am. Chem. Soc., 2014, 136, 15885 CrossRef CAS PubMed; (e) S.-Y. Ding, J. Gao, Q. Wang, Y. Zhang, W.-G. Song, C.-Y. Su and W. Wang, J. Am. Chem. Soc., 2011, 133, 19816 CrossRef CAS PubMed.
- (a) R. Dong, M. Pfeffermann, H. Liang, Z. Zheng, X. Zhu, J. Zhang and X. Feng, Angew. Chem., Int. Ed., 2015, 54, 12058 CrossRef CAS PubMed; (b) R. Sakamoto, K. Hoshiko, Q. Liu, T. Yagi, T. Nagayama, S. Kusaka, M. Tsuchiya, Y. Kitagawa, W. Y. Wong and H. Nishihara, Nat. Commun., 2015, 6, 6713 CrossRef CAS PubMed.
- E. Ber. Ber. Knoevenagel, Dtsch. Chem. Ges., 1898, 31, 2596 CrossRef.
- (a) N. C. Greenham, S. C. Moratti, D. D. C. Bradley, R. H. Friend and A. B. Holmes, Nature, 1993, 365, 628 CrossRef CAS; (b) M. Helbig and H.-H. Hörhold, Makromol. Chem., 1993, 194, 1607 CrossRef CAS; (c) A. C. Grimsdale, K. Leok Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897 CrossRef CAS PubMed.
- (a) M. Calik, F. Auras, L. M. Salonen, K. Bader, I. Grill, M. Handloser, D. D. Medina, M. Dogru, F. Lobermann, D. Trauner, A. Hartschuh and T. Bein, J. Am. Chem. Soc., 2014, 136, 17802 CrossRef CAS PubMed; (b) Y. Zhu, S. Wan, Y. Jin and W. Zhang, J. Am. Chem. Soc., 2015, 137, 13772 CrossRef CAS PubMed.
- S.-J. Yoon, J. W. Chung, J. Gierschner, K. S. Kim, M.-G. Choi, D. Kim and S. Y. Park, J. Am. Chem. Soc., 2010, 132, 13675 CrossRef CAS PubMed.
- K. S. W. Sing, Pure Appl. Chem., 1982, 54, 2201 CrossRef.
- X. Zhuang, Y. Chen, B. X. Li, D. G. Ma, B. Zhang and Y. X. Li, Chem. Mater., 2010, 22, 4455 CrossRef CAS.
- (a) S. Han, Y. Feng, F. Zhang, C. Yang, Z. Yao, W. Zhao, F. Qiu, L. Yang, Y. Yao, X. Zhuang and X. Feng, Adv. Funct. Mater., 2015, 25, 3899 CrossRef CAS; (b) N. Kang, J. H. Park, J. Choi, J. Jin, J. Chun, I. G. Jung, J. Jeong, J.-G. Park, S. M. Lee, H. J. Kim and S. U. Son, Angew. Chem., Int. Ed., 2012, 51, 6626 CrossRef CAS PubMed.
- (a) X. Zhuang, F. Zhang, D. Wu, N. Forler, H. Liang, M. Wagner, D. Gehrig, M. R. Hansen, F. Laquai and X. Feng, Angew. Chem., Int. Ed., 2013, 52, 9668 CrossRef CAS PubMed; (b) X. Zhuang, F. Zhang, D. Wu and X. Feng, Adv. Mater., 2014, 26, 3081 CrossRef CAS PubMed; (c) X. Zhuang, D. Gehrig, N. Forler, H. Liang, M. Wagner, M. R. Hansen, F. Laquai, F. Zhang and X. Feng, Adv. Mater., 2015, 27, 3789 CrossRef CAS PubMed.
- (a) W. Wei, H. Liang, K. Parvez, X. Zhuang, X. Feng and K. Müllen, Angew. Chem., Int. Ed., 2014, 126, 1596 CrossRef; (b) H. W. Liang, X. Zhuang, S. Bruller, X. Feng and K. Müllen, Nat. Commun., 2014, 5, 4973 CrossRef CAS PubMed.
- J. Zhang, Z. Zhao, Z. Xia and L. Dai, Nat. Nanotechnol., 2015, 10, 444 CrossRef CAS PubMed.
- (a) L. F. Chen, X. D. Zhang, H. W. Liang, M. G. Kong, Q. F. Guan, P. Chen, Z. Y. Wu and S. H. Yu, ACS Nano, 2012, 6, 7092 CrossRef CAS PubMed; (b) Z. Y. Jin, A. H. Lu, Y. Y. Xu, J. T. Zhang and W. C. Li, Adv. Mater., 2014, 26, 3700 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6py00561f |
| ‡ These two authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |