
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly selective binding of methyl orange dye by cationic water-soluble pillar[5]arenes†
L. S.
Yakimova
,
D. N.
Shurpik
,
L. H.
Gilmanova
,
A. R.
Makhmutova
,
A.
Rakhimbekova
and
I. I.
Stoikov
*
Kazan Federal University, A.M. Butlerov Chemical Institute, 420008, Kremlevskaya Street, 18, Kazan, Russian Federation. E-mail: ivan.stoikov@mail.ru
First published on 4th April 2016
Abstract
A new water-soluble pillar[5]arene with an amide fragment and triethylammonium groups was synthesized by our original method of aminolysis of the ester groups. Using UV-spectroscopy, it is shown that cationic pillar[5]arenes are able to selectively form 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexes with some hydrophobic anions: the guests with bulky uncharged or negatively charged substituents hindering entry into the macrocycle cavity. Highly selective binding of the most lipophilic guest, methyl orange dye, in the form of organic anion salts by positively charged water-soluble pillar[5]arenes was detected. In the case of the azo dye the appropriate Kass values were 10–100-fold higher than those calculated for the other sulfonic acid derivatives studied. The 2D NMR NOESY 1H–1H spectroscopy confirms the formation of the inclusion complex: negative charge sulfonate head is outside the cavity of pillar[5]arenes and the hydrophobic fragment of the guest is located in the cavity.
:1 complexes with some hydrophobic anions: the guests with bulky uncharged or negatively charged substituents hindering entry into the macrocycle cavity. Highly selective binding of the most lipophilic guest, methyl orange dye, in the form of organic anion salts by positively charged water-soluble pillar[5]arenes was detected. In the case of the azo dye the appropriate Kass values were 10–100-fold higher than those calculated for the other sulfonic acid derivatives studied. The 2D NMR NOESY 1H–1H spectroscopy confirms the formation of the inclusion complex: negative charge sulfonate head is outside the cavity of pillar[5]arenes and the hydrophobic fragment of the guest is located in the cavity.
Introduction
Binding of the neutral and polar organic guest molecules in nonpolar solvents does not result in a significant increase of the energy due to the host–guest interactions.1 Such a binding is typically performed by dipole–dipole interactions and hydrogen bonding, often with the participation of ion–dipole interactions.2 Hydrophobic sites of a guest interact with the analogous sites of a host molecule. In water, the binding of an organic guest is greatly increased due to the hydrophobic effect.1 The association of a host and a guest during the complex formation reduces the deformation of the solvent structure.1Synthesis of water-soluble macrocycles3 is of significant interest because most of the biologically important compounds that can act as guests are soluble in water.4a–d The first water-soluble pillar[n]arene, i.e., carboxylatopillar[5]arene sodium salt, showed high water solubility and good selective binding ability toward basic amino acids, e.g., L-lysine, L-arginine and L-histidine.4e The decacarboxylic acid has a rigid spatial structure stabilized by hydrogen bonds, and its salt makes it water-soluble and improves complexation properties. In 2011, the synthesis of the first water-soluble cationic pillar[5]arene was proposed.4f The cationic pillar[5]arenes bind anionic guests mainly by hydrophobic and electrostatic interactions as in inclusion complexes with p-toluenesulfonate4g and 1-octanesulfonate.4f Methyl orange is commonly used as a pH-indicator due to its clear and distinct colour change.5 It is known that water-soluble macrocycles with negatively charged groups bind the protonated methyl orange5b whereas macrocycles with positively charged groups stabilize the non-protonated form of the azo dye.5c,d However, the binding of methyl orange with a large hydrophobic fragment requires pillar[5]arenes with higher hydrophobicity of the cavity. For this reason, besides well-known water-soluble pillar[5]arenes containing either carbonyl or ammonium groups, we first proposed to combine these structural fragments in the same molecule in order to increase the depth of the cavity and the selectivity of the binding. Moreover, we have expanded the family of cationic pillar[5]arenes (1–3) and sulfonic acid derivatives as guests (G1–G8) to estimate the influence of (a) the substituent at the p-position of the benzene ring in the guest molecule and (b) the length of the spacer and the alkyl substituent at the nitrogen atom in the host on the complexation properties and binding selectivity.
In our previous study, we have successfully synthesized two symmetric cationic water-soluble pillar[5]arenes 1 and 2 bearing trimethylammonium/methyldiethylammonium groups at both the rims and the formation of the 1:1 complexes with p-toluenesulfoacid G1 was shown.6 Now we have synthesized, in addition to macrocycles 1 and 2, a new water-soluble pillar[5]arene 3 with an amide fragment and triethylammonium groups by our original method of aminolysis of the ester groups.6 The structure of the compounds obtained was characterized by 1H and 13C NMR, IR spectroscopy and mass spectrometry (MALDI TOF) (ESI, Fig. S1, S2 and S4†). The spatial structure of the new functionalized pillar[5]arenes was established by 2D NMR NOESY 1H–1H spectroscopy (ESI, Fig. S3†).
Results and discussion
Macrocycles 1–3 possess both a hydrophobic cavity and ten positively charged substituents that allow recognizing guests by electrostatic, hydrophobic and π–π stacking interactions between the host and a guest as the driving forces.7 A series of organic sulfonic acid derivatives with various sizes and shapes were used, including aromatic guests without (G1, G2) and with alkyl linkers (G4–G7), linear aliphatic salt G3 and methyl orange G8 with two benzene units (Fig. 1). | ||
| Fig. 1 Structures of the ammonium pillar[5]arenes 1–3 and sulfonate salts as guests G1–G8. | ||
To quantify molecular recognition of the sulfonic acid derivatives by pillar[5]arenes 1–3, the stability constants and the stoichiometry of the host/guest complexes formed in water were estimated by UV-spectroscopy, which showed significant changes in the absorbance spectra of the macrocycles after the addition of the guest molecules. The hyperchromic effect was observed at 270–320 nm (ESI, Fig. S11†) in the case of the guest binding (Table 1). For methyl orange G8, changes in the absorbance were monitored at 350–600 nm, while the other hosts 1–3 did not absorb light waves (Fig. 2).
 | ||
| Fig. 2 Spectrophotometric titration of the system pillar[5]arene 3 and G8 in water. The molar ratio of the host and the guest was changed from 0.3:1 to 2:1 (0.3:1, 0.5:1, 0.8:1, 0.9:1, 1:1, 1.1:1, 1.3:1, 1.5:1, 2:1). | ||
Kass values of host/guest complexes formed by sulfonate salts as guests G1–G8 and pillar[5]arenes 1–3 as hosts at 298 K in D2O
| 1 | 2 | 3 | |
|---|---|---|---|
|
a Values for lgKass were determined as described in ref. 5.
b The guests G3, G5 and G6 form inclusion complexes with hosts 1–3 by very weak interactions (lgKass ≤ 0.5).
|
|||
| G1 | 1.43 ± 0.12a | 1.22 ± 0.08a | 1.94 ± 0.01 |
| G2 | 2.38 ± 0.10 | 2.52 ± 0.01 | 2.74 ± 0.02 |
| G3 | —b | —b | —b |
| G4 | 1.84 ± 0.15 | 2.25 ± 0.20 | 1.99 ± 0.01 |
| G5 | —b | —b | —b |
| G6 | —b | —b | —b |
| G7 | 2.04 ± 0.02 | 2.70 ± 0.07 | 3.06 ± 0.04 |
| G8 | 3.97 ± 0.03 | 3.84 ± 0.02 | 3.24 ± 0.01 |
A new adsorption band with the maximum near 426 nm was found for the system G8/3 with less than one equivalent of G8 added. At its higher concentration, its band overlapped with the adsorption maximum of free guest G8 (460 nm) in the visible spectral region. The new band probably corresponds to the complex formation in the system with methyl orange G8. The lgKass values of the complexes obtained varied from 1.22 to 3.97. The pillar[5]arenes 1–3 showed similar binding abilities toward G1, G2, G4, and G7.
In the case of the azo dye G8 the appropriate Kass values were 10–100-fold higher than those calculated for the other sulfonic acid derivatives studied. We can propose that the affinity of the guest toward a macrocycle cavity sharply increases with the lipophilicity of a guest. The logP value of methyl orange (2.0593) is more than two-fold higher than that of the other guests that formed an inclusion complex. Selectivity of complexation towards the guests studied decreases in the row of pillar[5]arenes 1–3.
The pillar[5]arene 1 with a propyl linker forms more stable inclusion complexes with methyl orange G8 than pillar[5]arenes 2 and 3 with an ethyl linker despite longer ethyl fragments at the ammonium group. Hence, pillar[5]arenes with a cavity depth larger due to alkyl linkers are able to highly selectively and efficiently bind methyl orange as compared with the other guests studied.
The guest G3 forms the inclusion complex with 3 in accordance with 1H NMR spectroscopy (ESI, Fig. S7†). The complex shows very weak interactions, so the association constant could not be quantified. No complexation of 1–3 with G5 and G6 was found. Despite the maximal lipophilicity (logP = 2.6096), G5 has a bulky tert-butyl substituent which hinders the substrate entering the macrocycle cavity. This suggests the complex formation by inclusion of an uncharged fragment of the guest into the macrocycle cavity and electrostatic association of the charged sulfonate moiety of a guest with the ammonium group of pillar[5]arene. A second negatively charged alkylsulfonate substituent at the p-position of G6 hinders its inclusion into the π-electron rich cavity. These phenomena offered evidence for the formation of an inclusion complex between the hosts 1–3 and the guests G1–G8 mainly driven by hydrophobic interactions and to a lesser extent by electrostatic interactions.
1H NMR data confirmed the host–guest complexation in solution.7c,8 As shown in Fig. 3, upon addition of one equiv. of the host 3, the signals of the guest G8 protons (A–E) exhibit an obvious upfield shift and a broadening effect against free guest signals. Strong upfield shifts (Δδ) of the aromatic and methyl signals indicate that guest G8 is located in the host cavity. These shifts appeared due to rather fast proton exchange observed for complexation in the 1H NMR timescale. It should be noted that Δδ (A) > Δδ (B) > Δδ (E) > Δδ (C) > Δδ (D). Thus, methyl groups of G8 should be deeper incorporated into the macrocycle cavity of 3 (Δδ = 0.71 ppm for A). Besides, the AB quadruplet of the proton signals H3 (diastereotopic protons) of the host merges into an unresolved multiplet in the inclusion complex. Also, significant chemical shifts (Δδ) from 0.11 to 0.25 ppm were observed for the inclusion complexes of the host 3 with G2, G3 and G7 (ESI, Fig. S6–S8†).
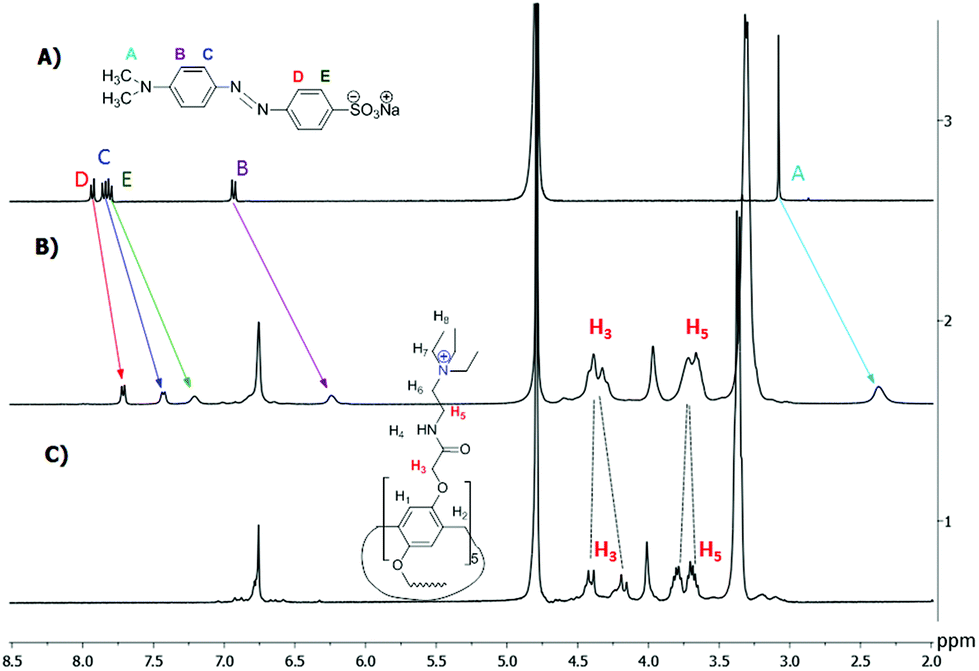 | ||
| Fig. 3 1H NMR spectra (D2O, 293 K, 400 MHz): (a) G8 (0.0112 mol l−1); (b) G8 (0.0112 mol l−1) + 3 (0.0112 mol l−1); (c) 3 (0.0112 mol l−1). | ||
The 2D NMR NOESY 1H–1H of the complexes confirms host–guest complexation (Fig. 4). The aromatic protons of G8 (B, C, D) have cross-peaks with H3 and H8 of the host 3. The methyl protons of the dimethylamino fragment (A) show cross-peaks with protons of the aromatic units (H1) and the methoxycarbonyl fragment (H3). Thus, we can conclude that the inclusion complex in which the guest threads the cavity of the pillar[5]arene 3 is formed. Its negative sulfonate head is close to the positive triethylammonium groups of 3, the fragment N(CH3)2-Ar– in methyl orange G8 is located in the cavity and the fragment –Ar-SO3Na is outside the cavity. From the UV spectroscopy results (Job's plot), the formation of the 1:1 inclusion complex between the host and the guest (see ESI, Fig. S23–S25†) can be proposed. The formation of the complex might be mainly driven by hydrophobic and electrostatic interactions, because the hydrophobic cavity of the host 3 meets hydrophobic alkyl and aryl chains of G8 and the cationic triethylammonium groups of the host 3 bind the anionic sulfonate group of G8 by electrostatic interactions.
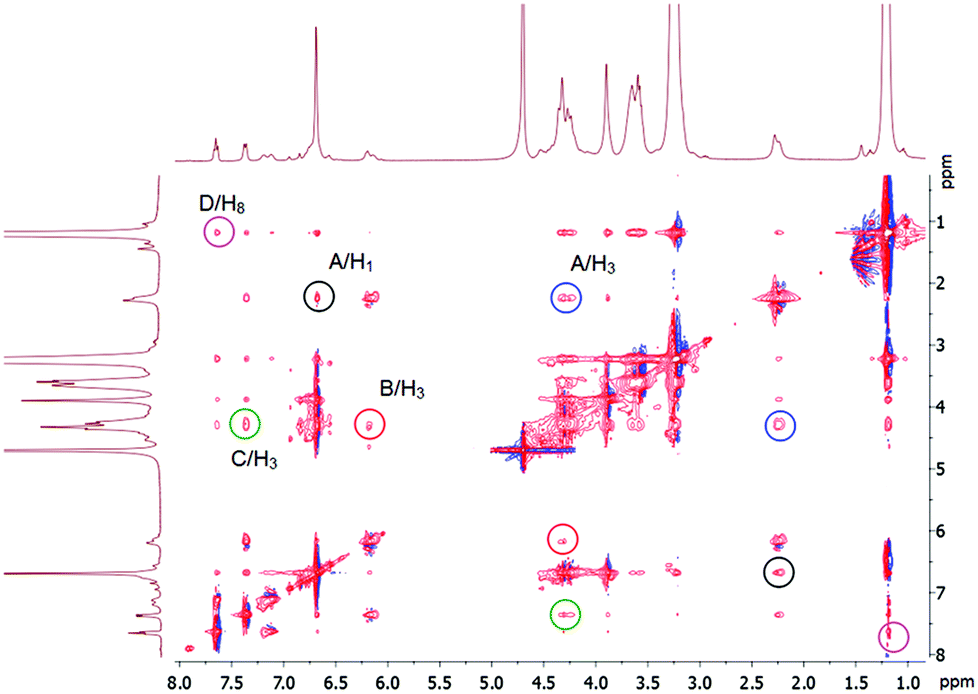 | ||
| Fig. 4 2D NMR 1H–1H NOESY (500 MHz) analysis of complex 3 with G8 in D2O. The concentrations of the host and the guest are 0.0112 M. | ||
The diffusion experiments with 1H NMR (DOSY) for the system 3/G8 offer information on the formation of the host–guest inclusion complex:9 the diffusion coefficient of a small guest molecule is large and decreases with complex formation. The DOSY spectrum (ESI, Fig. S5 and Table S1†) indicates a decrease of the mobility of the guest G8 after complexation (with diffusion coefficients of 3.43 × 10−10 m2 s−1 and 2.21 × 10−10 m2 s−1 for free and complexed G8, respectively). Also, in the DOSY spectrum the contour peaks lie on a horizontal line (ESI, Fig. S5†). All of these results confirm the formation of the inclusion complex between the host 3 and the guest G8.
Materials and methods
Instrumentation
1H NMR spectra were recorded on a Bruker Avance-400 (400 MHz) spectrometer and 13C and 2D NOESY NMR spectra were recorded on an impulse spectrometer Bruker Avance II (with 125 MHz and 500 MHz respectively). Chemical shifts were determined against the signals of residual protons of a deuterated solvent (D2O, CDCl3). The concentration of sample solutions was 3–5%. Attenuated total internal reflectance IR spectra were recorded with a Spectrum 400 (Perkin Elmer) Fourier spectrometer. Elemental analysis was performed with a Perkin Elmer 2400 Series II instrument. Mass spectra (ESI) were recorded on an AmaZonX mass spectrometer (Bruker Daltonik GmbH, Germany). The drying gas was nitrogen at 300° C. The capillary voltage was 4.5 kV. The samples were dissolved in acetonitrile (concentration ∼10−6 g ml−1). Melting points were determined using a Boetius Block apparatus. Additional control of the purity of compounds and monitoring of the reaction were carried out by thin-layer chromatography using Silica G, 200 μm plates, UV 254. 1H Diffusion Ordered Spectroscopy (DOSY): the spectra were recorded on a Bruker Avance 400 spectrometer, at 9.4 tesla, at the resonating frequency of 400.17 MHz for 1H, using a BBO Bruker 5 mm gradient probe. The temperature was regulated at 298 K and no spinning was applied to the NMR tube. DOSY experiments were performed using the STE bipolar gradient pulse pair (stebpgp1s) pulse sequence. 16 scans of 16 data points were collected. The maximum gradient strength produced in the z direction was 5.35 G mm−1. The duration of the magnetic field pulse gradients (δ) was optimized for each diffusion time (Δ) in order to obtain a 2% residual signal with the maximum gradient strength. The values of δ and Δ were 1.800 μs and 100 ms, respectively. The pulse gradients were incremented from 2 to 95% of the maximum gradient strength in a linear ramp.10–14Most chemicals were purchased from Aldrich and used as received without additional purification. Organic solvents were purified in accordance with standard procedures.
Synthesis of the compounds 3 and G3–G7
For the preparation of a new pillar[5]arene 3, initially, a four step literature procedure of 4,8,14,18,23,26,28,31,32,35-decakis-[N-(2′,2′-diethylaminoethyl)-carbamoylmethoxy]-pillar[5]arene was performed, starting from commercially available 1,4-dimethoxybenzene.11,12 )2), 2.41–2.52 (m, 60H, –CH2
)2), 2.41–2.52 (m, 60H, –CH2 –N(
–N( CH3)2), 3.24 (m, 20H, –
CH3)2), 3.24 (m, 20H, – CH2–N(CH2CH3)2), 3.79 (s, 10H, –
CH2–N(CH2CH3)2), 3.79 (s, 10H, – –), 4.32 (s, 20H, O–
–), 4.32 (s, 20H, O– C(O)–), 6.85 (s, 10H, ArH), 7.86 (t, 10H, 3JHH = 5.2, –C(O)NH). 13C NMR (CD3SOCD3) δC ppm: 167.64, 148.95, 127.97, 114.71, 67.71, 51.37, 46.47, 36.59, 28.80, 11.75. 1H–1H NOESY (NOE) (the major cross-peaks): H8/H4; H7/H4; H5/H4; H2/H4; H3/H4; H1/H4; H1/H8; H1/H7; H1/H5; H1/H5; H1/H2; H1/H3; H3/H8; H3/H7; H3/H5; H3/H1; H2/H8; H2/H7; H2/H5; H2/H3; H5/H6; H8/H7. IR ν cm−1: 3311.05 (N–H), 1661.33 (C
C(O)–), 6.85 (s, 10H, ArH), 7.86 (t, 10H, 3JHH = 5.2, –C(O)NH). 13C NMR (CD3SOCD3) δC ppm: 167.64, 148.95, 127.97, 114.71, 67.71, 51.37, 46.47, 36.59, 28.80, 11.75. 1H–1H NOESY (NOE) (the major cross-peaks): H8/H4; H7/H4; H5/H4; H2/H4; H3/H4; H1/H4; H1/H8; H1/H7; H1/H5; H1/H5; H1/H2; H1/H3; H3/H8; H3/H7; H3/H5; H3/H1; H2/H8; H2/H7; H2/H5; H2/H3; H5/H6; H8/H7. IR ν cm−1: 3311.05 (N–H), 1661.33 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). MALDI-TOF MS: calculated [M+] m/z = 2172.4, found [M + H]+m/z = 2173.4, [M + Na]+m/z = 2195.4. Found: C, 63.57; H, 8.81; N, 12.89. C115H190N20O20. Calculated for C115H190N20O20: C, 63.02; H, 8.55; N, 12.49.
O). MALDI-TOF MS: calculated [M+] m/z = 2172.4, found [M + H]+m/z = 2173.4, [M + Na]+m/z = 2195.4. Found: C, 63.57; H, 8.81; N, 12.89. C115H190N20O20. Calculated for C115H190N20O20: C, 63.02; H, 8.55; N, 12.49.
General procedure of the synthesis of compound 3
An equimolar amount of ethyl iodide was added to the solution of compound A (0.30 g, 0.14 mmol) in 10 ml acetonitrile. The reaction mixture was refluxed for 72 h and the solvent was removed under reduced pressure. The powder obtained was dried under reduced pressure (P2O5). )3), 3.36 (m, 80H, –CH2
)3), 3.36 (m, 80H, –CH2 –N(
–N( CH3)3), 3.63–3.85 (m, 20H, –
CH3)3), 3.63–3.85 (m, 20H, – CH2–N(CH2CH3)3), 3.98 (s, 10H, –
CH2–N(CH2CH3)3), 3.98 (s, 10H, – –), 4.08 (d, 10H, AB-system, 2JHH = 15.0, O–
–), 4.08 (d, 10H, AB-system, 2JHH = 15.0, O– C(O)NH–), 4.37 (d, 10H, AB-system, 2JHH = 15.0, O–
C(O)NH–), 4.37 (d, 10H, AB-system, 2JHH = 15.0, O– C(O)NH–), 6.73 (s, 10H, ArH). 13C NMR (CD3SOCD3) δC ppm: 168.95, 148.30, 127.45, 114.61, 66.94, 52.94, 52.94, 52.38, 32.28, 28.56, 7.21. IR ν cm−1: 3331.5 (–N+–(CH2CH3)3), 2975.3 (–CH2–CH3, –CH2–), 1665.9 (CO). ESI: calcd for [M − 4 I−]4+m/z = 806.2, [M − 5 I−]5+m/z = 619.6, [M − 6 I−]6+m/z = 496.8, found m/z = 806.1, 619.5, 495.2. Found: C, 57.4; H, 8.23; N, 9.65. C135H240N20O20. Calculated for C135H240Cl10N20O20: C, 57.54; H, 8.58; N, 9.94.
C(O)NH–), 6.73 (s, 10H, ArH). 13C NMR (CD3SOCD3) δC ppm: 168.95, 148.30, 127.45, 114.61, 66.94, 52.94, 52.94, 52.38, 32.28, 28.56, 7.21. IR ν cm−1: 3331.5 (–N+–(CH2CH3)3), 2975.3 (–CH2–CH3, –CH2–), 1665.9 (CO). ESI: calcd for [M − 4 I−]4+m/z = 806.2, [M − 5 I−]5+m/z = 619.6, [M − 6 I−]6+m/z = 496.8, found m/z = 806.1, 619.5, 495.2. Found: C, 57.4; H, 8.23; N, 9.65. C135H240N20O20. Calculated for C135H240Cl10N20O20: C, 57.54; H, 8.58; N, 9.94.
Pillar[5]arenes 1 and 2 were synthesized according our original method.6
NCH2 CH2NH–), 3.13 (s, 90H, (CH3)3N+–), 3.25 (m, 20H, NCH2CH2
CH2NH–), 3.13 (s, 90H, (CH3)3N+–), 3.25 (m, 20H, NCH2CH2 NH–), 3.39 (m, 20H, N
NH–), 3.39 (m, 20H, N CH2CH2NH–), 3.9 (s, 10H, –
CH2CH2NH–), 3.9 (s, 10H, – –), 4.41 (dd, 20H, O–
–), 4.41 (dd, 20H, O– C(O)–), 6.83 (s, 10H, ArH), 7.74 (t, 10H, 3JHH = 5.6, –C(O)NH). 13C NMR (CD3SOCD3) δC ppm: 170.89, 149.48, 129.88, 116.22, 68.30, 64.09, 53.09, 35.88, 29.12, 22.68. 1H–1H NOESY (NOE) (the major cross-peaks): H1/H3; H2/H1; H4/H8; H5/H6; H7/H8; H3/H2. IR ν cm−1: 3285.31, 3386.07 (N–H), 1662.39 (CO). MALDI-TOF MS: calculated [M − I−]+m/z = 3324.6, found [M − I−]+m/z = 3325.2. Found: C, 40.01; H, 5.84; N, 8.13. C115H200N20O20. Calculated for C115H200N20O20: C, 39.05; H, 5.43; N, 8.10.
C(O)–), 6.83 (s, 10H, ArH), 7.74 (t, 10H, 3JHH = 5.6, –C(O)NH). 13C NMR (CD3SOCD3) δC ppm: 170.89, 149.48, 129.88, 116.22, 68.30, 64.09, 53.09, 35.88, 29.12, 22.68. 1H–1H NOESY (NOE) (the major cross-peaks): H1/H3; H2/H1; H4/H8; H5/H6; H7/H8; H3/H2. IR ν cm−1: 3285.31, 3386.07 (N–H), 1662.39 (CO). MALDI-TOF MS: calculated [M − I−]+m/z = 3324.6, found [M − I−]+m/z = 3325.2. Found: C, 40.01; H, 5.84; N, 8.13. C115H200N20O20. Calculated for C115H200N20O20: C, 39.05; H, 5.43; N, 8.10.
 )2), 3.02 (s, 30H, –N–CH3), 3.41 (m, 40H, –CH2
)2), 3.02 (s, 30H, –N–CH3), 3.41 (m, 40H, –CH2 –N(
–N( CH3)2), 3.77 (m, 20H, –
CH3)2), 3.77 (m, 20H, – CH2–N(CH2CH3)2), 4.01 (s, 10H, –
CH2–N(CH2CH3)2), 4.01 (s, 10H, – –), 4.20 (d, 10H, AB-system, 2JHH = 15.1, O–
–), 4.20 (d, 10H, AB-system, 2JHH = 15.1, O– C(O)NH–), 4.40 (d, 10H, AB-system, 2JHH = 15.1, O–
C(O)NH–), 4.40 (d, 10H, AB-system, 2JHH = 15.1, O– C(O)NH–), 6.76 (s, 10H, ArH). 13C NMR (CD3SOCD3) δC, ppm: 171.06, 149.17, 128.90, 115.35, 67.88, 64.09, 57.74, 57.17, 47.48, 32.68, 30.36, 7.50. 1H–1H NOESY (NOE) (the major cross-peaks): H1/H8; H7/H1; H3/H8; H6/H7; H8/H9. IR ν cm−1: 3331.48 (N–H), 1665.92 (CO). MALDI-TOF MS: calculated [M]+m/z = 3591.73, found [M − I−]+m/z = 3463.7. Found: C, 41.79; H, 6.17; N, 7.80. C125H220N20O20. Calculated for C125H220N20O20: C, 40.53; H, 5.93; N, 7.45.
C(O)NH–), 6.76 (s, 10H, ArH). 13C NMR (CD3SOCD3) δC, ppm: 171.06, 149.17, 128.90, 115.35, 67.88, 64.09, 57.74, 57.17, 47.48, 32.68, 30.36, 7.50. 1H–1H NOESY (NOE) (the major cross-peaks): H1/H8; H7/H1; H3/H8; H6/H7; H8/H9. IR ν cm−1: 3331.48 (N–H), 1665.92 (CO). MALDI-TOF MS: calculated [M]+m/z = 3591.73, found [M − I−]+m/z = 3463.7. Found: C, 41.79; H, 6.17; N, 7.80. C125H220N20O20. Calculated for C125H220N20O20: C, 40.53; H, 5.93; N, 7.45.
Compounds G3–G7 were obtained from commercially available alcohols and phenols by literature methods.13, 14
Determination of the stability constant and stoichiometry of the complex by UV titration
The UV measurements were performed with a “Shimadzu UV-3600” instrument. A 1 × 10−3 M solution of G1, G2, G4 or G7 (0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9 and 1 ml) in water was added to 0.5 ml of the solution of receptors 1–3 (3 × 10−4 M) in water and diluted to a final volume of 3 ml with water. In the case of G8: a 3 × 10−5 M solution of hosts 1–3 (0.3, 0.5, 0.6, 0.8, 0.9, 1, 1.1, 1.3, 1.5 and 2 ml) in water was added to 0.03 ml of the solution of G8 (1 × 10−3 M) in water and diluted to a final volume of 3 ml with water. The UV spectra of the solutions were then recorded. The stability constant and stoichiometry of complexes were calculated as described in ESI.† Three independent experiments were carried out for each series. A Student's t-test was applied for statistical data processing.Job's plots
Series of solutions of pillar[5]arene derivatives 1–3 and sulfonic acid derivatives were prepared in water. The volume of the host and guest solutions varied from 0.6:2.4 to 2.4:0.6, respectively, with the total concentration of the host (H) and the guest (G) being constant and equal to 1 × 10−5 M. The solutions were used without further stirring. The absorbance Ai of the complexation systems was measured at the maximum absorbance wavelength of the complex. The absorbance values were used to plot a diagram from whose maximum the structures of the complexes were deduced. Three independent experiments were carried out for each system.
Conclusions
The recognition properties of pillar[5]arenes containing both carbonyl and ammonium groups at both the rims were confirmed regarding a series of charged organic sulfonate guests of various shapes and sizes. Cationic water-soluble pillar[5]arenes 1–3 are able to selectively form 1:1 complexes with hydrophobic anions (G1, G2, G4, and G7). The guests with bulky uncharged or negatively charged substituents (G5 and G6) hindering entry into the macrocycle cavity. Highly selective binding of the most lipophilic guest, i.e. methyl orange G8, in the form of organic anion salts by positively charged water-soluble pillar[5]arenes was detected.
Acknowledgements
The work was supported by the Russian Science Foundation (no. 14-13-00058).References
- J. Steed and J. Atwood, Supramolecular Chemistry, John Wiley & Sons, 2009, 998pp Search PubMed.
- (a) L. Li, R. Wu, S. Guang, X. Su and H. Xu, Phys. Chem. Chem. Phys., 2013, 15, 20753 RSC; (b) G. Yu, K. Jie and F. Huang, Chem. Rev., 2015, 115(15), 7240 CrossRef CAS PubMed; (c) K. Jie, Y. Zhou, Y. Yao and F. Huang, Chem. Soc. Rev., 2015, 44(11), 3568 RSC; (d) D. N. Shurpik, L. S. Yakimova, L. I. Makhmutova, A. R. Makhmutova, I. K. Rizvanov, V. V. Plemenkov and I. I. Stoikov, Macroheterocycles, 2014, 7(4), 351 CrossRef; (e) I. I. Stoikov, O. A. Mostovaya, L. S. Yakimova, A. A. Yantemirova, I. S. Antipin and A. I. Konovalov, Mendeleev Commun., 2010, 20(6), 359 CrossRef CAS; (f) V. A. Smolko, D. N. Shurpik, R. V. Shamagsumova, A. V. Porfireva, V. G. Evtugyn, L. S. Yakimova and G. A. Evtugyn, Electrochim. Acta, 2014, 147, 726 CrossRef CAS.
- (a) X. Chi, X. Ji, D. Xia and F. Huang, J. Am. Chem. Soc., 2015, 137(4), 1440 CrossRef CAS PubMed; (b) D. Xia, G. Yu, J. Li and F. Huang, Chem. Commun., 2014, 50, 3606 RSC; (c) Z. Li, J. Yang, G. Yu, J. He, Z. Abliz and F. Huang, Org. Lett., 2014, 16(7), 2066 CrossRef CAS.
- (a) F. Perret, A. N. Lazar and A. W. Coleman, Chem. Commun., 2006, 2425 RSC; (b) H. Zhang, B. Zhang, M. Zhu, S. M. Grayson, R. Schmehl and J. Jayawickramarajah, Chem. Commun., 2014, 50, 4853 RSC; (c) N. Manganaro, G. Lando, C. Gargiulli, I. Pisagatti, A. Notti, S. Pappalardo, M. F. Parisi and G. Gattuso, Chem. Commun., 2015, 51, 12657 RSC; (d) H. Ahmad, D. Ghosh and J. A. Thomas, Chem. Commun., 2014, 50, 3859 RSC; (e) S. Dasgupta, A. Chowdhury and P. S. Mukherjee, RSC Adv., 2015, 5, 85791 RSC; (f) Y. Ma, X. Ji, F. Xiang, X. Chi, C. Han, J. He, Z. Abliz, W. Chen and F. Huang, Chem. Commun., 2011, 47, 12340 RSC; (g) B. Gomez, V. Francisco, F. Fernandez-Nieto, L. Garcia-Rio, M. Martin-Pastor, M. Rita Paleo and F. Javier Sardina, Chem. – Eur. J., 2014, 20, 12123 CrossRef CAS PubMed.
- (a) G. Xiang, D. Wu, J. He and X. Wang, Chem. Commun., 2011, 47, 11456 RSC; (b) E. Kazakova, J. Morozova, D. Mironova, V. Syakaev, L. Muslinkina and A. Konovalov, Supramol. Chem., 2013, 25(12), 831 CrossRef CAS; (c) N. O. Mchedlov-Petrossyan, L. N. Vilkova, N. A. Vodolazkaya, A. G. Yakubovskaya, R. V. Rodik, V. I. Boyko and V. I. Kalchenko, Sensors, 2006, 6, 962 CrossRef CAS; (d) N. O. Mchedlov-Petrossyan, N. A. Vodolazkaya, L. N. Vilkova, O. Yu. Soboleva, L. V. Kutuzova, R. V. Rodik, S. I. Miroshnichenko and A. B. Drapaylo, J. Mol. Liq., 2009, 145, 197 CrossRef CAS.
- D. N. Shurpik, L. S. Yakimova, I. K. Rizvanov, V. V. Plemenkov and I. I. Stoikov, Macroheterocycles, 2015, 8(2), 128 CrossRef.
- (a) I. I. Stoikov, M. N. Agafonova, L. S. Yakimova, I. S. Antipin and A. I. Konovalov, Mol. Recognition: Biotechn., Chem. Eng. and Mat. Appl., 2011, 1–43 Search PubMed; (b) L. S. Yakimova, M. A. Ziganshin, V. A. Sidorov, V. V. Kovalev, E. A. Shokova, V. A. Tafeenko and V. V. Gorbatchuk, J. Phys. Chem. B, 2008, 112, 15569 CrossRef CAS PubMed; (c) M. A. Ziganshin, L. S. Yakimova, K. R. Khayarov, V. V. Gorbatchuk, M. O. Vysotsky and V. Böhmer, Chem. Commun., 2006, 389 Search PubMed; (d) V. V. Gorbatchuk, M. A. Ziganshin, L. S. Savelyeva, N. A. Mironov and W. D. Habicher, Macromol. Symp., 2004, 210, 263 CrossRef CAS; (e) V. V. Gorbatchuk, L. S. Savelyeva, M. A. Ziganshin, I. S. Antipin and V. A. Sidorov, Russ. Chem. Bull., 2004, 53(1), 60 CrossRef CAS.
- M.-S. Yuan, H. Chen, X. Du, J. Li, J. Wang, X. Jia and C. Li, Chem. Commun., 2015, 51, 16361 RSC.
- (a) M. A. Braga, M. F. Martini, M. Pickholz, F. Yokaichiya, M. K. D. Franco, L. F. Cabeç, V. A. Guilherme, C. M. G. Silva, C. E. G. Limia and E. de Paula, J. Pharm. Biomed. Anal., 2016, 119, 27 CrossRef CAS PubMed; (b) L. M. Arantes, C. Scarelli, A. J. Marsaioli, E. de Paula and S. A. Fernandesa, Magn. Reson. Chem., 2009, 47, 757 CrossRef CAS PubMed; (c) E. Hatzakis, P. Dais and M. Misiak, Anal. Methods, 2015, 7, 5226 RSC.
- E. Hatzakis, P. Dais and M. Misiak, Anal. Methods, 2015, 7, 5226 RSC.
- T. Boinski and A. Szumna, Tetrahedron, 2012, 68, 9419 CrossRef CAS.
- T. Ogoshi, T. Aoki, K. Kitajima, S. Fujinami, T. Yamagishi and Y. Nakamoto, J. Org. Chem., 2011, 76(1), 328 CrossRef CAS PubMed.
- W. Chen, A. G. Joly, J.-O. Malm, J.-O. Bovin and S. Wang, J. Phys. Chem. B, 2003, 107, 6544 CrossRef CAS.
- J. P. Canselier, J. L. Boyer, V. Castro, G. L. Gard, J. Mohtasham, D. H. Peyton and F. E. Behr, Magn. Reson. Chem., 1995, 33, 506 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Some synthetic details, determination of association constants, and other materials. See DOI: 10.1039/c6ob00539j |
| This journal is © The Royal Society of Chemistry 2016 |