
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew biphenyl iminium salt catalysts for highly enantioselective asymmetric epoxidation: role of additional substitution and dihedral angle†
Philip C.
Bulman Page
*a,
Christopher J.
Bartlett
a,
Yohan
Chan
a,
Steven M.
Allin
b,
Michael J.
McKenzie
c,
Jérôme
Lacour
d and
Garth A.
Jones
a
aSchool of Chemistry, University of East Anglia, Norwich Research Park, Norwich NR4 7TJ, UK. E-mail: p.page@uea.ac.uk
bDepartment of Chemistry, Nottingham Trent University, Clifton, Nottinghamshire NG11 8NS, UK
cCharnwood Molecular Ltd, 7 Beaumont Court, Prince William Road, Loughborough, Leicestershire LE11 5GA, UK
dDépartement de Chimie Organique, Université de Genève, Quai Ernest Ansermet 30, CH-1121 Genève 4, Switzerland
First published on 16th March 2016
Abstract
New biaryl iminium salt catalysts for enantioselective alkene epoxidation containing additional substitution in the heterocyclic ring are reported. The effects upon conformation and enantioselectivity of this additional substitution, and the influence of dihedral angle in these systems, has been investigated using a synthetic approach supported by density functional theory. Enantioselectivities of up to 97% ee were observed.
Introduction
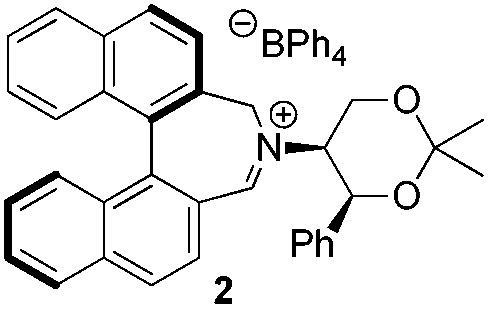
Asymmetric epoxidation of alkenes to generate non-racemic chiral epoxides is an extremely powerful synthetic tool,1 and the development of effective organocatalytic systems for epoxidation has received considerable attention.2 The most successful organocatalytic methods for epoxidation are those utilizing dioxiranes and those utilizing oxaziridinium salts. Chiral ketones used as precursors to dioxiranes, such as those of Yang,3 Denmark,4 Armstrong,5 and especially Shi,6 have achieved high enantioselectivities, with observed enantiomeric excesses exceeding 97% in the best cases. Oxaziridinium salts, first reported by Lusinchi in 1976,7 are also reactive reagents for oxygen transfer to nucleophilic substrates such as sulfides and alkenes, and may be generated catalytically by use of iminium salts in the presence of a stoicheiometric oxidant, typically Oxone.8 We have developed a range of catalysts based on biphenylazepinium (e.g.1), binaphthylazepinium (e.g.2) and dihydroisoquinolinium (e.g.3) moieties containing a chiral appendage on the nitrogen atom for epoxidation reactions, with the most successful to date containing the 1,3-dioxane motif of (S,S)-acetonamine 4 (Fig. 1).9 We have also shown that alternative oxidants may be used in iminium salt-catalysed epoxidation reactions such as hydrogen peroxide,10 sodium hypochlorite,11 and electrochemically-generated oxidants.12 Amines have also been used as iminium precursors in related epoxidation processes by us13 and others.14 | ||
| Fig. 1 Iminium salt catalysts. | ||
We reported the first very high enantioselectivities in the asymmetric epoxidation of alkenes using iminium salt catalysts, and we have developed non-aqueous conditions for these processes using tetraphenylphosphonium monoperoxysulfate (TPPP)15 as the oxidant.16 We have used the process in the syntheses of levcromakalim,17 (−)-lomatin, (+)-trans-khellactone,18 and scuteflorin19 utilizing catalysts 1 and 3.
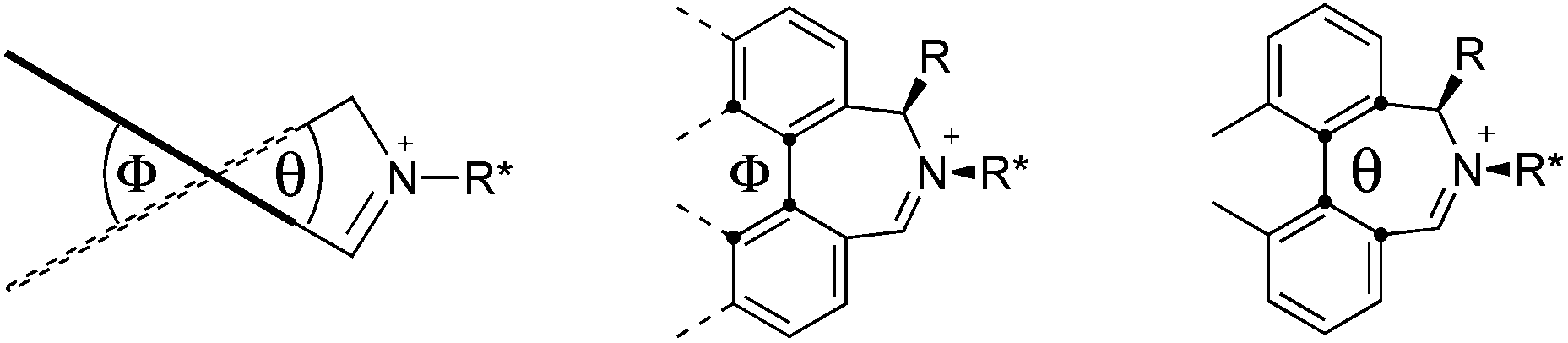
We have shown that incorporation of a pseudo-axial substituent at the prochiral carbon atom α- to the nitrogen atom in biaryl systems 1 and 2 affords higher enantioselectivities, with the addition of a methyl group found to have the greatest influence.20 It has also been reported that the level of enantiocontrol imparted by biaryl azepinium salt catalysts in the epoxidation of alkenes is in part correlated with the dihedral angle around the biphenyl axis in the iminium species,21 suggesting a parallel pattern of dihedral angles between the iminium parents and the putative derived oxaziridinium oxidative intermediates. It was further suggested that this angle is both larger and nearer to the optimum in the octahydrobinaphthyl series than in the parent binaphthyl series.
We were interested to investigate this potential correlation between dihedral angles in the iminium species and in the oxaziridinium oxidative intermediates, and if any correlation might also be observed between dihedral angles and induced enantioselectivities. Consequently, geometry optimizations were carried out on oxaziridinium intermediates, as well as the iminium species, at the B3LYP/6-31G* level of theory. These calculations indeed suggest that there is little change in dihedral angles between the iminium and oxaziridinium species in the lowest energy conformations (vide infra).
We were interested to learn if related structural modifications would also improve the enantioselectivity of our other biaryl-containing catalysts. We report herein the design and use of new iminium salt catalysts combining both of these modifications (Fig. 2).
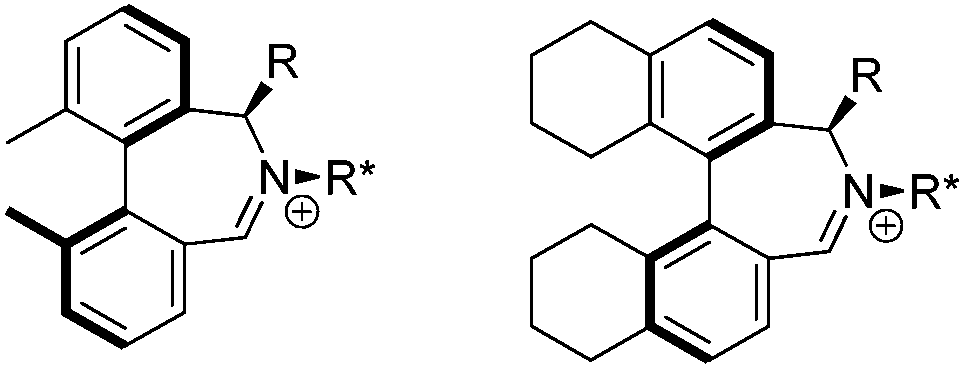 | ||
| Fig. 2 New biaryl catalysts. | ||
Results and discussion
Synthesis
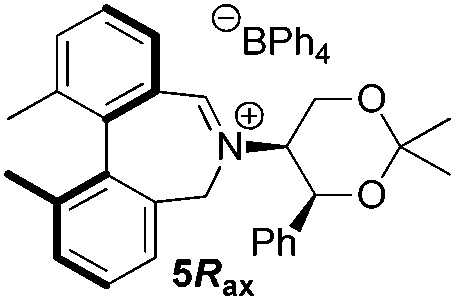
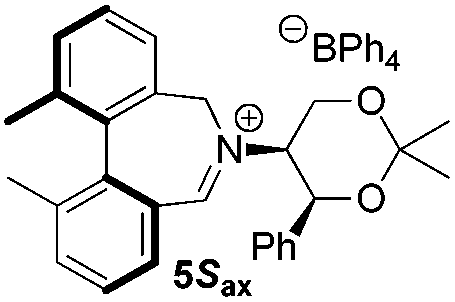
To begin with, we focused on synthesis of the simpler 6,6′-dimethylbiphenyl backbone, with iminium salts 5Rax, 5Sax and 6Rax, 6Sax as targets. The synthesis of racemic bis-carboxaldehyde (±)-7 was achieved following literature procedures.21,22 Formation of the diazonium salt from 3-methylanthranilic acid followed by coupling of the radical generated in situ gave bis-acid (±)-8 in 58% yield. High concentration of the diazonium salt and careful temperature control are key factors to obtain an acceptable yield of (±)-8, as formation of the corresponding diazo compound, a bright yellow solid, competes with the desired reaction. We first used the racemic bis-acid, ultimately to access both diastereoisomers 5Rax and 5Sax in order to assess separately their potential as catalysts, in the expectation of finding ‘matched’ and ‘mismatched’ systems, and reasoning that separation of the diastereoisomers would be more practical on sufficient scale than resolution of the bis-acid. Reduction of (±)-8 to the diol (±)-9 with lithium aluminium hydride proceeded in 96% yield. Oxidation of the two alcohol moieties of (±)-9 using PCC yielded the desired bis-carboxaldehyde (±)-7 in 87% yield. Reductive cycloamination of (±)-7 with enantiomerically pure (S,S)-acetonamine 4 gave diastereoisomers 10Rax, 10Sax, which were separated using silica gel column chromatography, in 49% and 26% yields respectively (Scheme 1).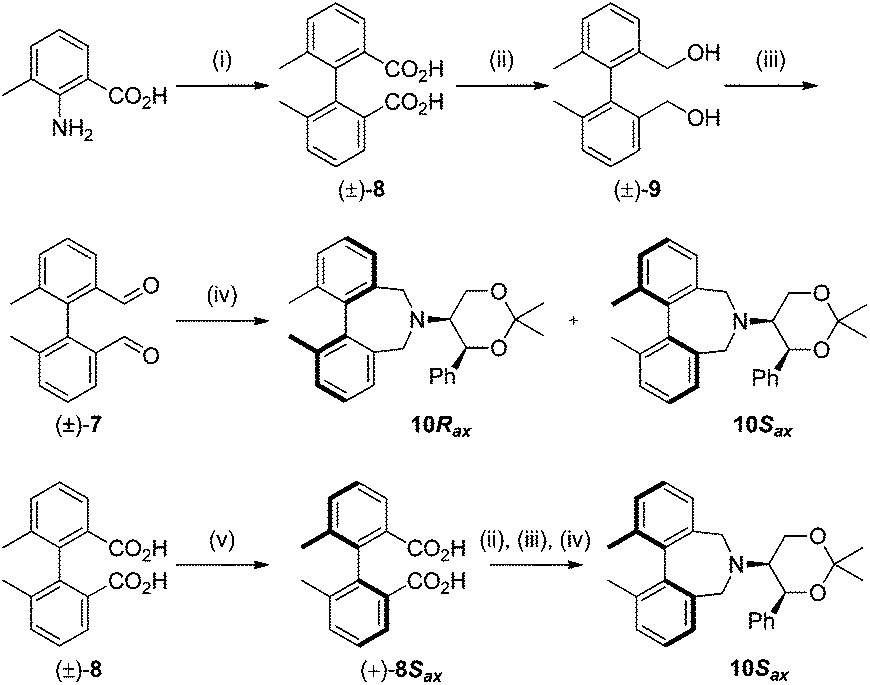 | ||
| Scheme 1 Synthesis of 6,6′-dimethylbiphenyl azepines. Reagents and conditions: (i) HCl, NaNO2; then Cu2SO4, (NH4OH)2·H2SO4, NH4OH, <5 °C, 58%; (ii) LiAlH4 (3 equiv.), Et2O, r.t., 96%; (iii) PCC (3 equiv.), CH2Cl2, r.t., 87%, (iv) Amine 4 (1 equiv.), NaBH3CN (2.2 equiv.), MeOH, AcOH, r.t., 24 h, [10Rax, 49%] and [10Sax, 26%]; (v) quinine, EtOH (90%), crystallization then (b) 3 M HCl, 70%. | ||
Resolution of bis-acid (±)-8 using quinine following a literature procedure provided us with the means to prepare a single diastereoisomer of 10.22 By comparison of spectroscopic data and optical rotation, the amine diastereoisomer obtained from (+)-8Sax was found to be identical to 10Sax, allowing us to attribute the absolute configurations of 10Rax and 10Sax.
Oxidation of 10Rax, 10Sax separately using NBS followed by counter-ion exchange gave the corresponding tetraphenylborate iminium salts 5Rax and 5Sax in 61% and 83% yields respectively. Diastereoselective addition of methyl magnesium bromide gave 11Rax and 11Sax as single diastereoisomers, the methyl groups being introduced into a pseudo-axial position in each case, as we have previously observed,20 controlled by the axial chirality of the biphenyl backbone, and appearing in the 1H NMR spectra at high field (11Rax: 0.21 ppm; 11Sax: 0.14 ppm).
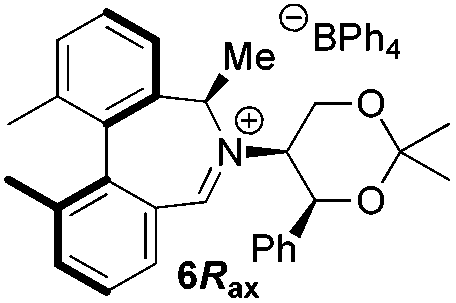
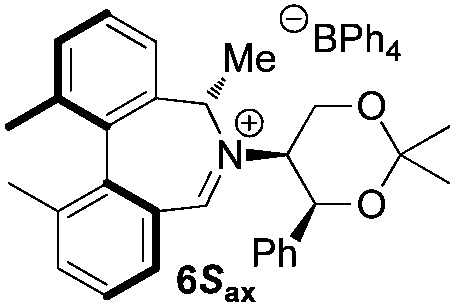
Oxidation of 11Rax, 11Sax and anion exchange gave α-methylated iminium salts 6Rax, 6Sax as single diastereo- and regio-isomers in 28% and 32% yields over the three steps, respectively (Scheme 2), resulting from loss of the more accessible single remaining axial hydrogen atom in each case.20,23 The yield of the oxidation step was moderated by competing bromination of the methyl groups at the 6,6′ positions.
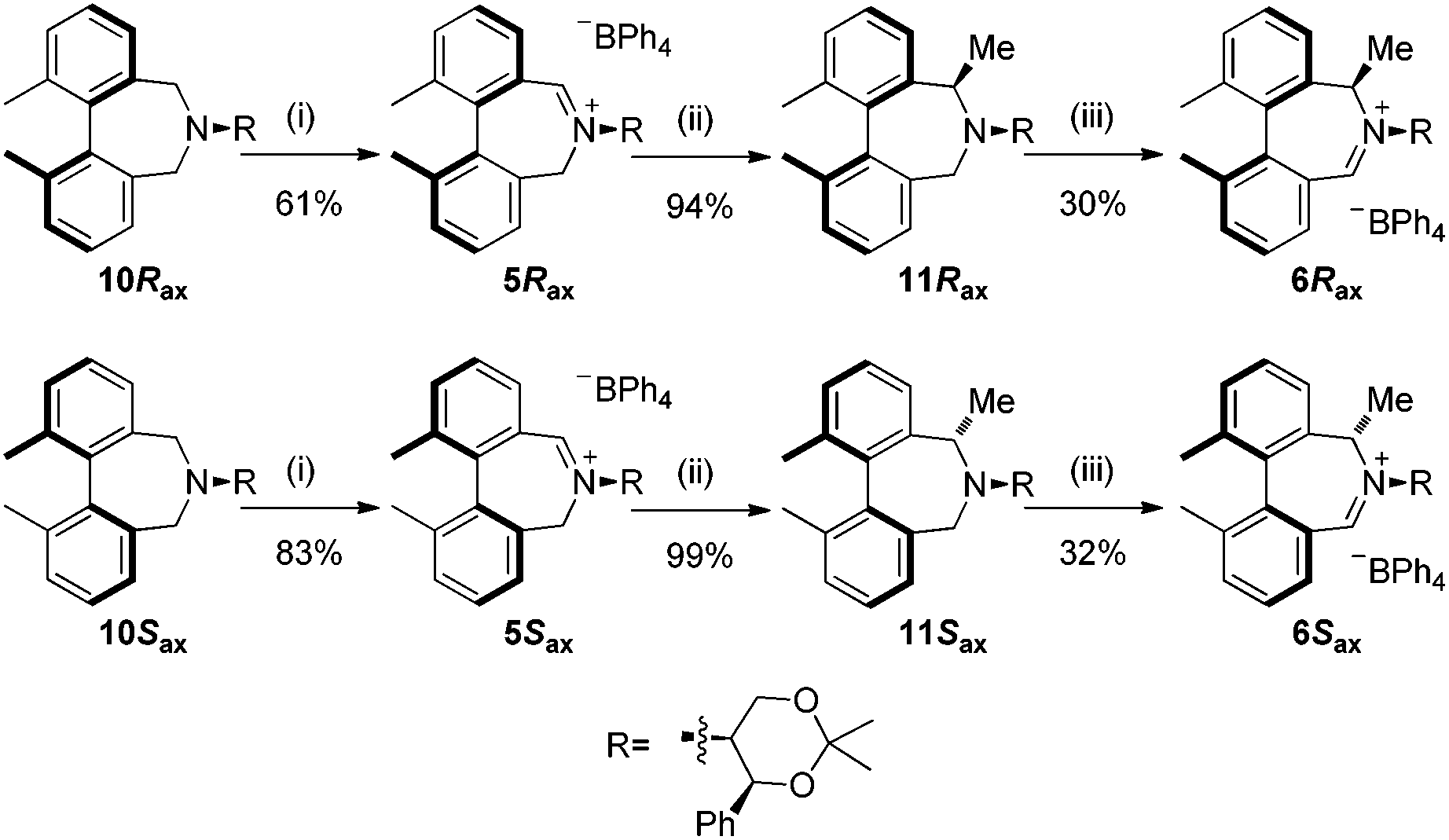 | ||
| Scheme 2 Synthesis of 6,6′-dimethylbiphenyl catalysts. Reagents and conditions: (i) NBS (1.1 equiv.), CH2Cl2, 0 °C then reflux, 2 h, then NaBPh4, EtOH, 5 min; (ii) MeMgBr (10 equiv.), THF, −78 °C to r.t.; (iii) NBS (1.1 equiv.), CH2Cl2, 0 °C then reflux, 2 h, then NaBPh4, EtOH, 5 min. | ||
We next turned our attention to the preparation of similar catalysts using the octahydrobinaphthyl backbone, where the iminium salt possesses large dihedral angles.21 Octahydrobinaphthyl-2,2′-bis-methanol 12 was obtained following literature procedures (Scheme 3).21,24 (R)-Binol was hydrogenated to (R)-octahydrobinol 13 using palladium on carbon under hydrogen pressure in 45% yield. Triflation of the phenolic hydroxyl moieties to give 14, followed by palladium-catalysed carbonylation to give 15,25 and reduction using lithium aluminium hydride gave (R)-octahydrobinaphthyl-1,1′-methanol 12 in 83% yield over the three steps.
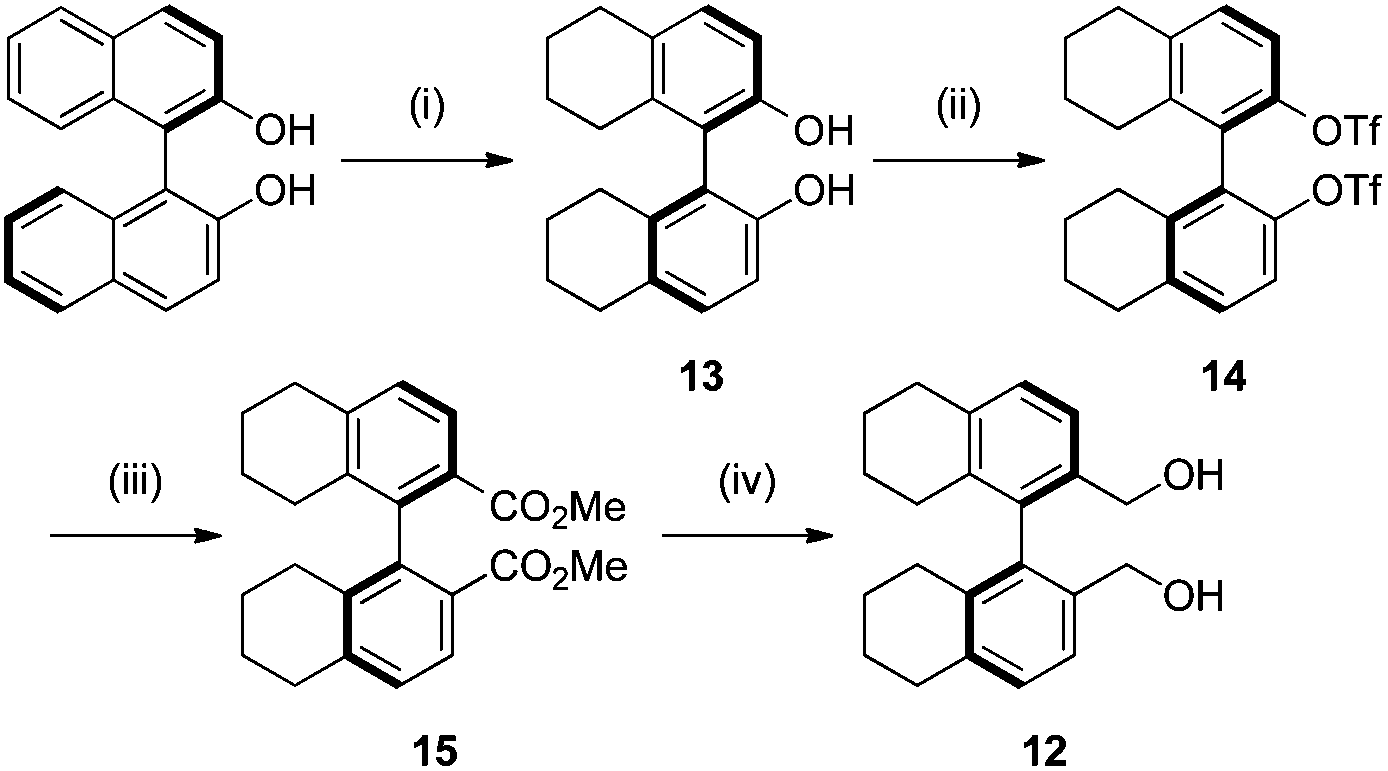 | ||
| Scheme 3 Synthesis of octahydrobinol derivatives. Reagents and conditions: (i) Pd/C, H2 (3.5 bar), AcOH, 80 °C, 10 d, 45%; (ii) Tf2O (3 equiv.), DMAP (0.4 equiv.), 2,6-lutidine (3 equiv.), −30 °C to r.t., 16 h, 99%; (iii) Pd(OAc)2 (0.15 equiv.), dppp (0.15 equiv.), MeOH (50 equiv.), DMSO, DIPEA, CO (2 bar), 80 °C, 48 h, 85%; (iv) LiAlH4 (2 equiv.), Et2O, 99%. | ||
Compound 12 was converted into the corresponding (R)-bis-carboxaldehyde 16 in quantitative yield. Tertiary amine 17 was, however, only obtained in 67% yield using the reductive cycloamination protocol. An alternative procedure consisting of displacing two bromide ions using (S,S)-acetonamine 4 was therefore used. Diol 12 was successfully bis-brominated to give 18 using phosphorus tribromide in 90% yield; double displacement with (S,S)-acetonamine 4 gave the desired amine 17 in an excellent 97% yield (Scheme 4).
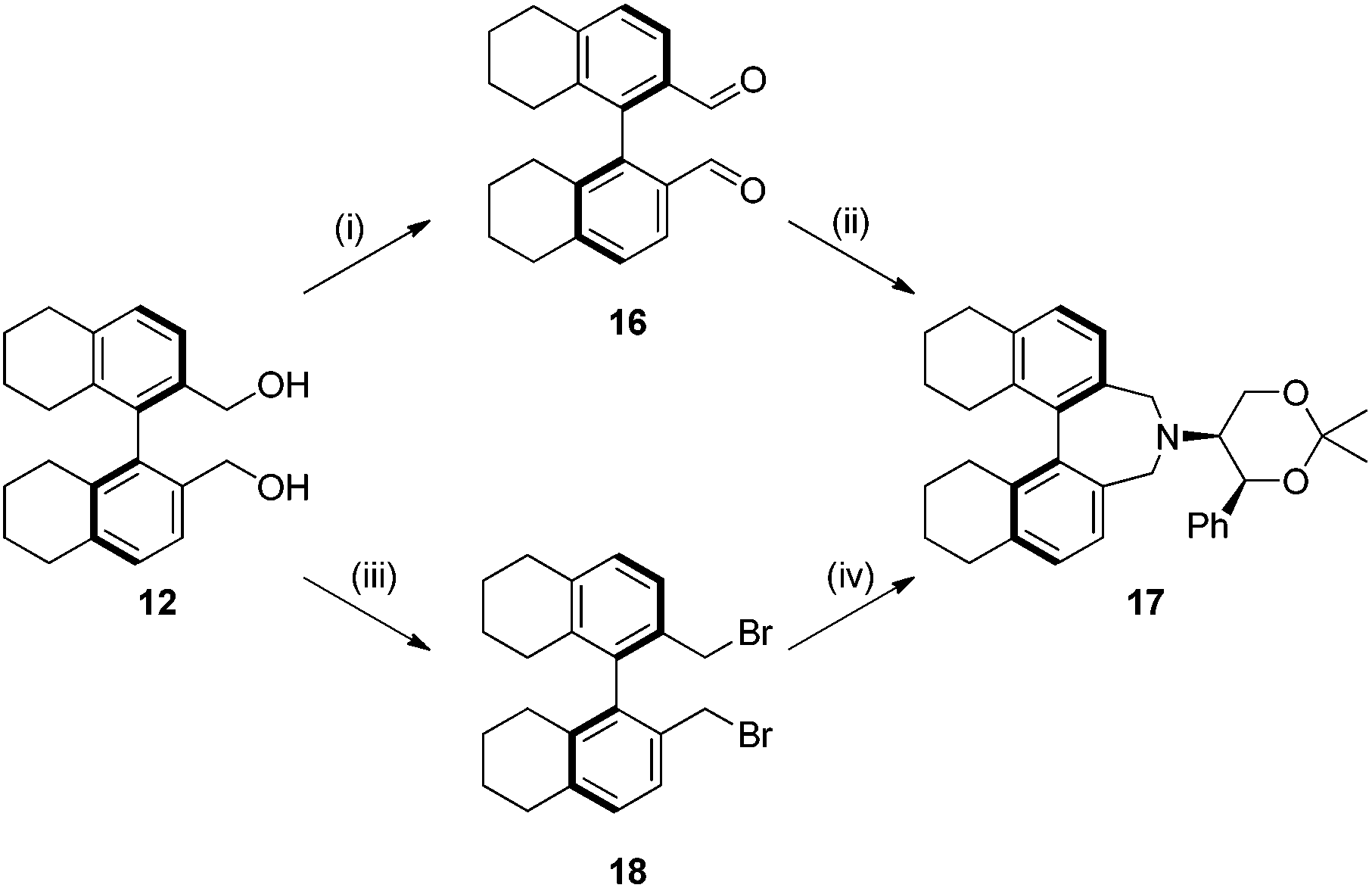 | ||
| Scheme 4 Synthesis of octahydrobinaphthyl azepine. Reagents and conditions: (i) PCC (1 equiv.), CH2Cl2, r.t., 2 h, 99%; (ii) 4 (1 equiv.), NaBH3CN (2.2 equiv.), AcOH, r.t., 24 h, 67%; (iii) PBr3 (3 equiv.), pyridine (0.1 equiv.), toluene, 60 °C, 3 h, 90%; (iv) Amine 4 (1 equiv.), K2CO3 (3 equiv.), MeCN, reflux, 16 h, 97%. | ||
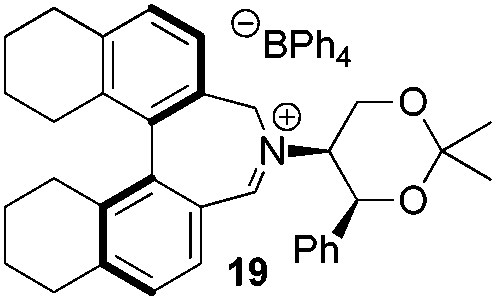
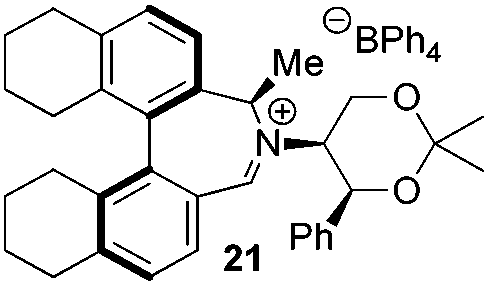
Amine 17 was oxidized using NBS to afford iminium salt 19 in 73% yield. The corresponding α-methylated azepinium salt was obtained through a similar sequence to that described above, involving diastereoselective Grignard reagent addition to give 20 followed by oxidation and anion exchange to give 21 in 56% yield over the two steps. Again, the α-methylated iminium salt 21 was obtained as a single regio- and diastereo-isomer with a pseudo-axial methyl group (Scheme 5).20
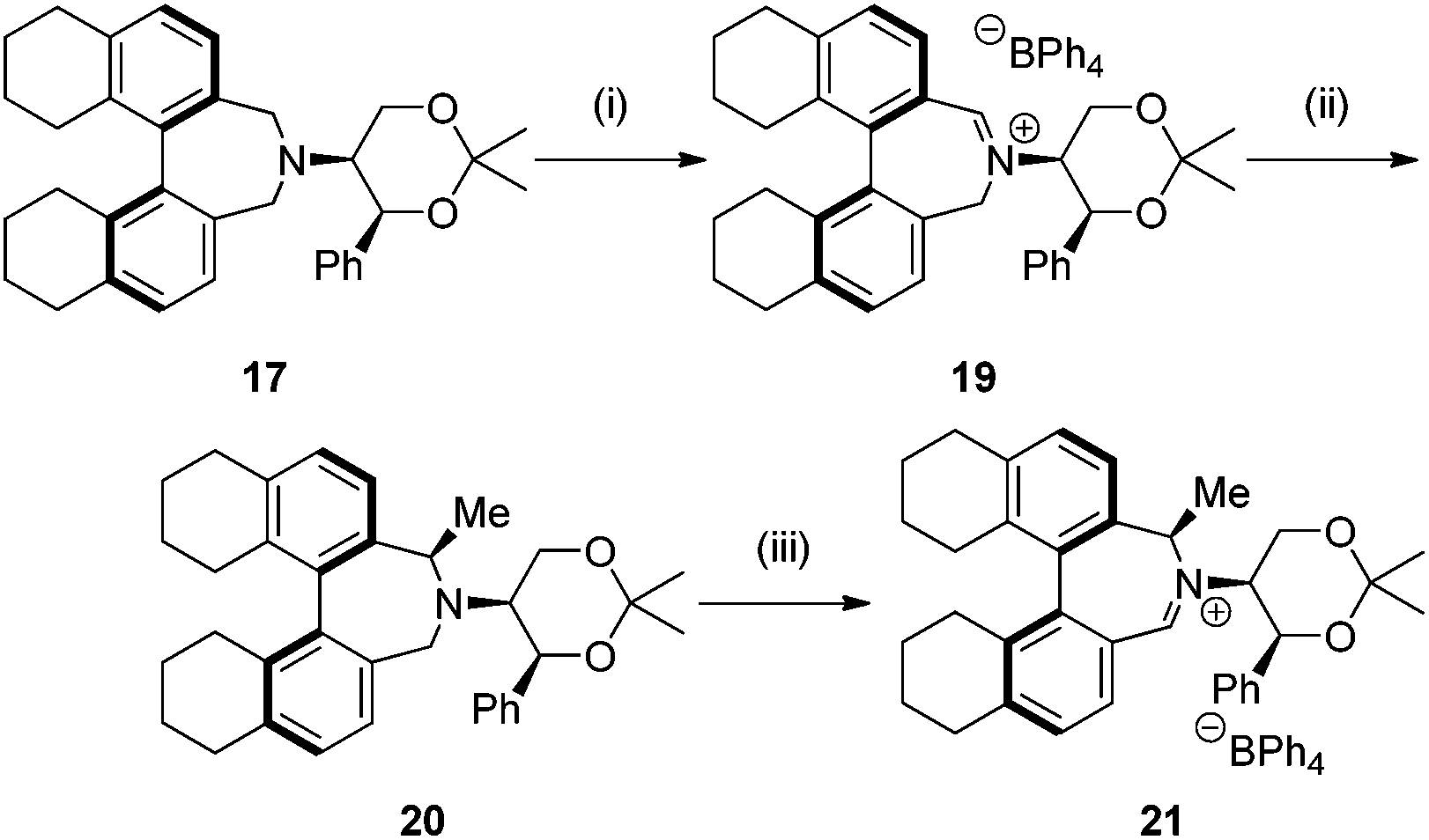 | ||
| Scheme 5 Synthesis of octahydrobinaphthyl catalysts. Reagents and conditions: (i) NBS (1.1 equiv.), CH2Cl2, r.t., 30 min then NaBPh4, EtOH, 73%; (ii) MeMgBr (10 equiv.), THF, −78 °C to r.t., 16 h, 79%; (iii) NBS (1.1 equiv.), CH2Cl2, r.t., 30 min then NaBPh4, EtOH, 71%. | ||
Calculations
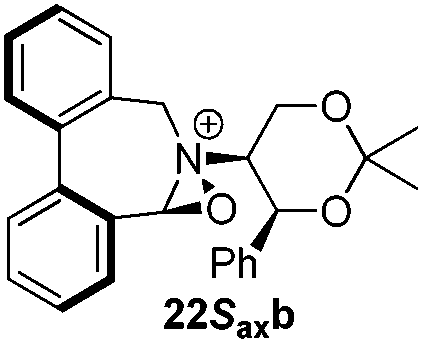
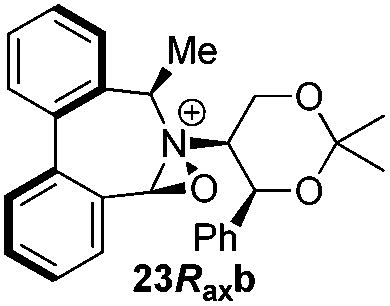
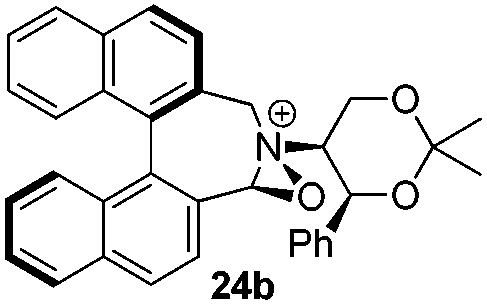
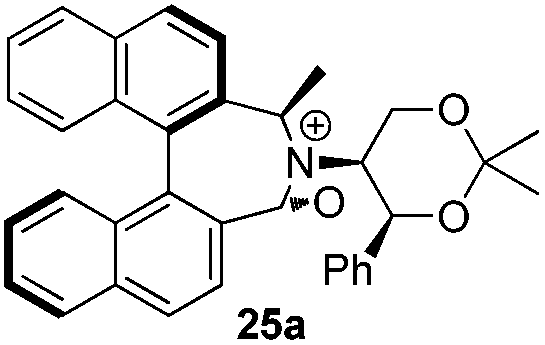
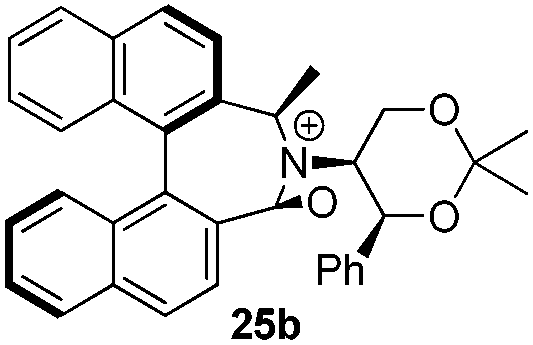
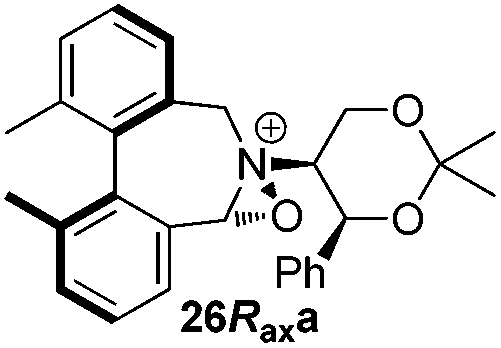
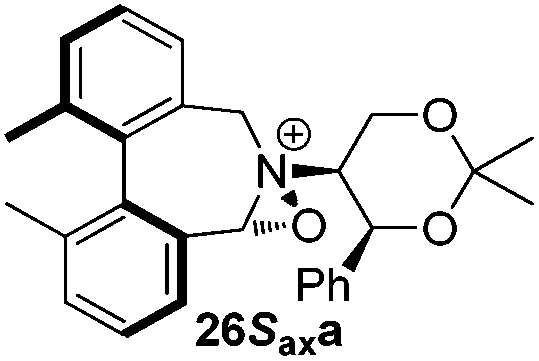
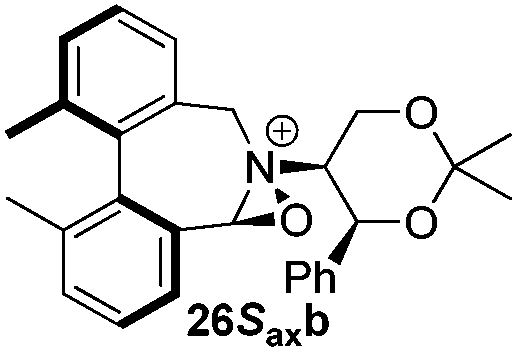
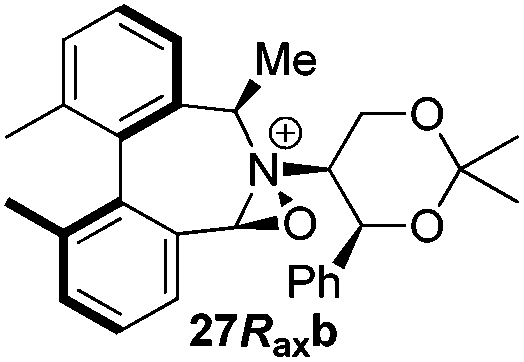
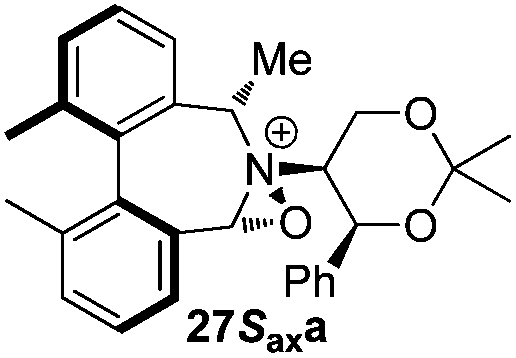
Geometry optimizations were carried out on a range of these iminium species and their putative derived oxaziridinium ions, at the B3LYP/6-31G* level using Gaussian 09,26 to determine the dihedral angles ϕ and θ in the lowest energy conformations. Both possible diastereoisomers were investigated in the case of each oxaziridinium ion. All stationary point structures were characterized by harmonic frequency analysis, and shown to be genuine energy minima (Tables 1 and 2). Cartesian co-ordinates of the optimized structures can be found in ESI.†|
|
|||
|---|---|---|---|
| Structure | ϕ/θ | Structure | ϕ/θ |
|
ϕ = −40.1 |

|
ϕ = −37.9 |
| θ = −41.3 | θ = −40.4 | ||
|
ϕ = 38.9 |
|
ϕ = 39.4 |
| θ = 41.7 | θ = 40.8 | ||

|
ϕ = −38.7 |
|
ϕ = −37.7 |
| θ = −39.2 | θ = −39.7 | ||

|
ϕ = −58.5 |
|
ϕ = −56.4 |
| θ = −54.8 | θ = −54.0 | ||
|
ϕ = −58.1 |
|
ϕ = −56.0 |
| θ = −54.0 | θ = −53.9 | ||
|
ϕ = −57.8 |

|
ϕ = −56.3 |
| θ = −54.7 | θ = −54.4 | ||
|
ϕ = 56.3 |
|
ϕ = 57.5 |
| θ = 55.0 | θ = 54.7 | ||
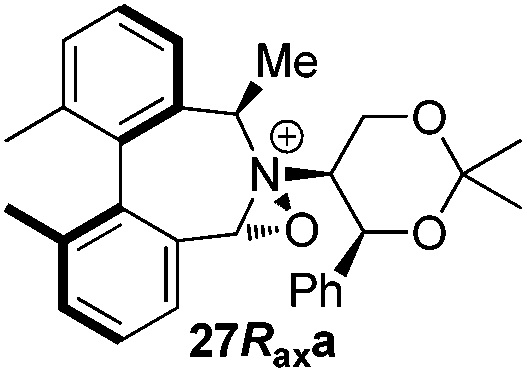
|
ϕ = −57.7 |
|
ϕ = −55.6 |
| θ = −53.9 | θ = −54.3 | ||
|
ϕ = 57.2 |
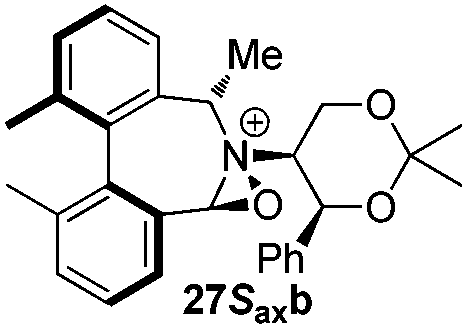
|
ϕ = 57.5 |
| θ = 54.9 | θ = 54.3 | ||

|
ϕ = −63.0 |
|
ϕ = −57.3 |
| θ = −58.3 | θ = −54.6 | ||

|
ϕ = −63.8 |
|
ϕ = −57.0 |
| θ = −57.6 | θ = −54.9 | ||
|
|
|||
|---|---|---|---|
| Structure | ϕ/θ | Structure | ϕ/θ |

|
ϕ = −34.9 |
|
ϕ = −33.8 |
| θ = −39.6 | θ = −37.6 | ||
|
ϕ = −52.2 |
|
ϕ = −52.2 |
| θ = −53.0 | θ = −52.2 | ||
|
ϕ = −53.1 |
|
ϕ = −52.7 |
| θ = −53.7 | θ = −52.5 | ||
|
ϕ = 52.4 |
|
ϕ = 52.7 |
| θ = 53.7 | θ = 53.3 | ||
|
ϕ = −59.7 |
|
ϕ = −60.0 |
| θ = −58.0 | θ = −57.3 | ||
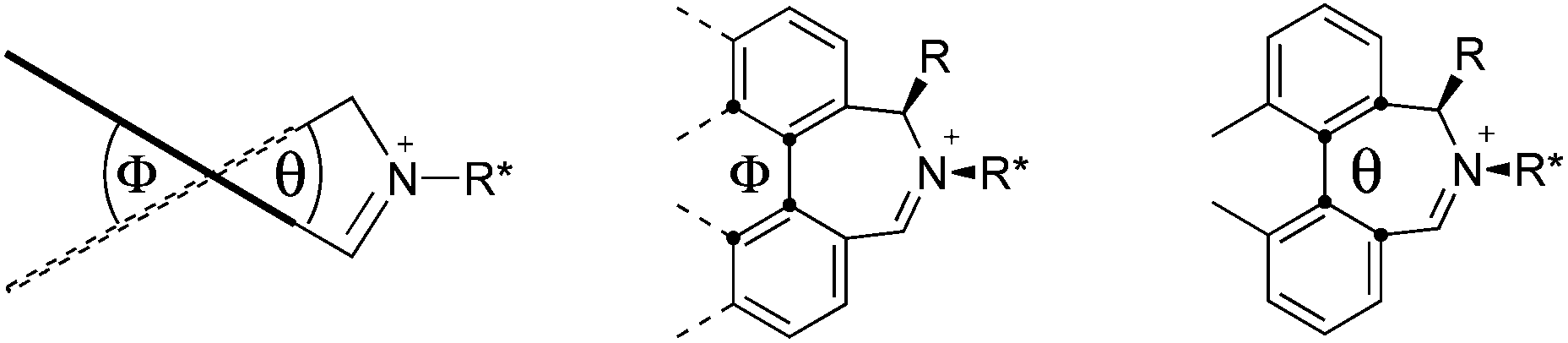
For the oxaziridinium ions (Table 1), the calculations demonstrate a clear increase in dihedral angles upon moving from the biphenyl species (around 38–42°) to the 6,6′-dimethylbiphenyl (around 54–58°) and the octahydrobinaphthyl (around 55–64°) species. Larger dihedral angles were also observed as expected in the binaphthyl species (around 54–59°).
It is interesting to note that the pair of diastereoisomeric oxaziridinium ions in each case display similar dihedral angles, with the differences varying from 0.3° to 6.8° for ϕ and 0.1° to 3.7° for θ, the larger figures being observed for the octahydro species. Similar calculations carried out on the derived iminium ions suggest that similar dihedral angles also obtain for the iminium and derived oxaziridinium species; the calculations suggest that the dihedral angles differ by no more than 6.3° from those of the corresponding iminium species.
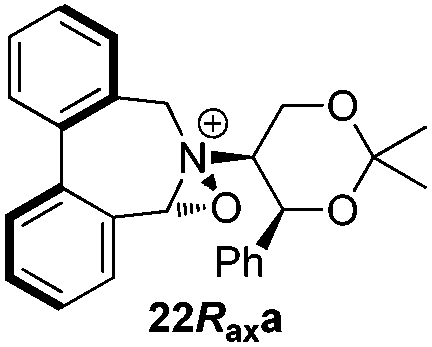
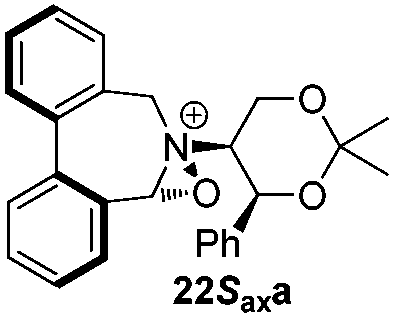
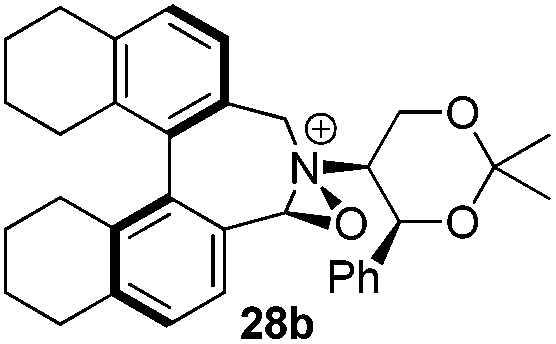
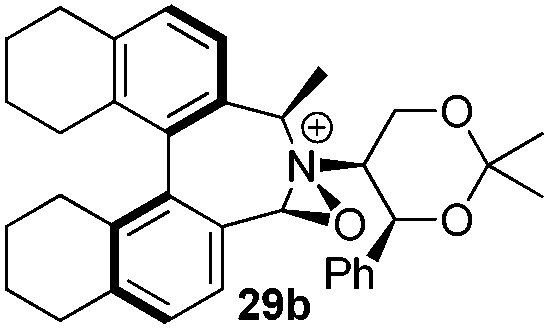
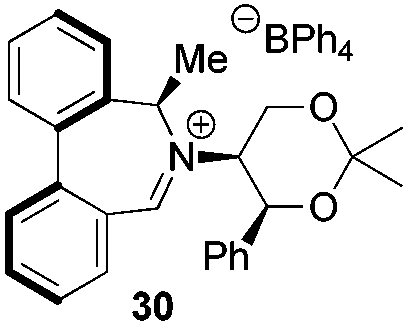
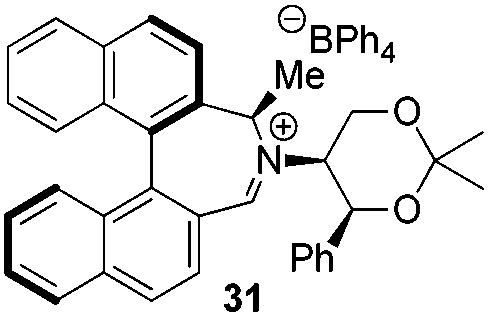
Incorporation of a methyl substituent at the prochiral carbon atom α- to the nitrogen atom in the heterocyclic ring does not, however, greatly affect the dihedral angles for either the iminium or oxaziridinium species (no more than about 2°). In the biphenyl cases 22 and 23, and the precursor iminium species 1 and 30, where interconversion between the atropoisomers is readily possible, the S,S-acetonamine unit induces a preference for the R-axial chirality in the biaryl unit. The methyl substituents in 23 and 30 occupy pseudo-axial positions, as is observed in the other methyl-substituted examples.
Epoxidation reactions
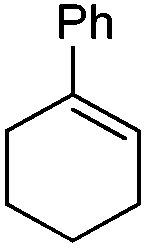
Iminium salts 5Rax, 5Sax, 6Rax, 6Sax, 19, and 21 were used in the asymmetric epoxidation of a number of alkenes under optimized reaction conditions,27 and the results compared with known results from species 1 and 30 (Table 3).20 We have previously observed that the related binaphthyl catalysts 2 and 31 display somewhat different profiles of reactivity and selectivity from those of the biphenyl series,9a,20 and these catalysts are therefore not included in this analysis.| Substrate | Cat. | Time | Conv.b/% | eec/% | Config.d |
|---|---|---|---|---|---|
Conditions: oxone® (2 equiv.), NaHCO3 (5 equiv.), catalyst (5 mol%), MeCN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O 10:1, 0 °C.a Conditions: oxone® (2 equiv.), NaHCO3 (5 equiv.), catalyst (5 mol%), MeCN:H2O 1:1, 0 °C.b Conversions were evaluated from the 1H NMR spectra by integration of the alkene and epoxide signals.c ee was determined using CSP HPLC using a Chiralcel OD-H column unless otherwise indicated.d Absolute configurations of the major enantiomers were determined by comparison of optical rotation with those reported in the literature.e ee was determined using CSP GC using a Chiraldex B-DM column.f ee was determined using 1H NMR spectroscopy in the presence of europium(III) tris[3-(heptafluoropropylhydroxymethylene)-(+)-camphorate] as chiral shift reagent. :H2O 10:1, 0 °C.a Conditions: oxone® (2 equiv.), NaHCO3 (5 equiv.), catalyst (5 mol%), MeCN:H2O 1:1, 0 °C.b Conversions were evaluated from the 1H NMR spectra by integration of the alkene and epoxide signals.c ee was determined using CSP HPLC using a Chiralcel OD-H column unless otherwise indicated.d Absolute configurations of the major enantiomers were determined by comparison of optical rotation with those reported in the literature.e ee was determined using CSP GC using a Chiraldex B-DM column.f ee was determined using 1H NMR spectroscopy in the presence of europium(III) tris[3-(heptafluoropropylhydroxymethylene)-(+)-camphorate] as chiral shift reagent. |
|||||
|
1 | 5 min | 100 | 60e | (−)-(1S,2S) |
| 30 | 0.5 h | 100 | 82e | (−)-(1S,2S) | |
| 5Rax | 0.8 h | 99 | 87e | (−)-(1S,2S) | |
| 6Rax | 1 h | 99 | 92e | (−)-(1S,2S) | |
| 5Sax | 1.8 h | 99 | 82e | (+)-(1R,2R) | |
| 6Sax | 3 h | 99 | 82e | (+)-(1R,2R) | |
| 19 | 0.6 h | 99 | 90f | (−)-(1S,2S) | |
| 19 | 1 h | 99 | 80f | (−)-(1S,2S) | |
| 21 | 1.5 h | 99 | 96f | (−)-(1S,2S) | |

|
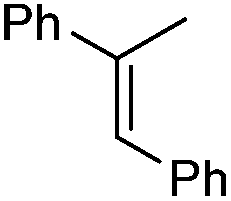
1 | 3 min | 90 | 41 | (+)-(1R,2S) |
| 30 | 0.5 h | 100 | 78 | (+)-(1R,2S) | |
| 5Rax | 0.5 h | 99 | 89 | (+)-(1R,2S) | |
| 5Rax | 0.3 h | 98 | 81 | (+)-(1R,2S) | |
| 6Rax | 1 h | 99 | 85 | (+)-(1R,2S) | |
| 5Sax | 2 h | 99 | 82 | (−)-(1S,2R) | |
| 6Sax | 6 h | 77 | 78 | (−)-(1S,2R) | |
| 19 | 0.7 h | 99 | 93 | (+)-(1R,2S) | |
| 19 | 1.3 h | 99 | 86 | (+)-(1R,2S) | |
| 21 | 3 h | 87 | 97 | (+)-(1R,2S) | |
|
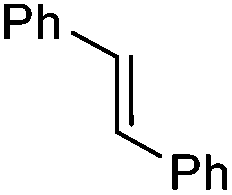
1 | 5 min | 95 | 37 | (−)-(1S,2S) |
| 5Rax | 3 h | 99 | 67 | (−)-(1S,2S) | |
| 6Rax | 3 h | 99 | 73 | (−)-(1S,2S) | |
| 5Sax | 6 h | 99 | 60 | (+)-(1R,2R) | |
| 6Sax | 6 h | 99 | 48 | (+)-(1R,2R) | |
| 19 | 1 h | 99 | 60 | (−)-(1S,2S) | |
| 19 | 2 h | 99 | 63 | (−)-(1S,2S) | |
| 21 | 18 h | 80 | 69 | (−)-(1S,2S) | |
|
1 | 5 min | 95 | 5 | (−)-(1S,2S) |
| 5Rax | 4.3 h | 99 | 15 | (−)-(1S,2S) | |
| 6Rax | 6 h | 99 | 39 | (−)-(1S,2S) | |
| 5Sax | 6 h | 99 | 8 | (+)-(1R,2R) | |
| 6Sax | 6 h | 99 | 10 | (+)-(1R,2R) | |
Aqueous conditions using Oxone as oxidant and a mixture of acetonitrile and water as solvent gave the highest reactivities and enantioselectivities. Non-aqueous conditions using TPPP as oxidant and acetonitrile as solvent were less successful, however; for example, use of chloroform as solvent gave an ee lower than 5%. The enantioselectivity observed when 18-crown-6 and a biphasic system28 were used was also poor.
From our results, it appears that observed enantioselectivities indeed increase for the trisubstituted alkenes from 37–60% ee for the simple biphenyl catalyst 1 (78–82% ee for the methylated derivative 30), with dihedral angles around 34–40°, to 60–93% ee for the matched 6,6′-dimethylbiphenyl catalyst 5R, with dihedral angles around 52–54°, and the octahydro binaphthyl catalyst 19, with dihedral angles around 57–60° (69–97% ee for the methylated derivatives 6R and 21 respectively).
For catalysts 5 and 6, the ‘matched’ diastereoisomers, inducing higher enantioselectivities, are the (Rax) 5Rax and 6Rax, and the ‘mismatched’ ones are the (Sax) 5Sax and 6Sax, which are also generally less reactive. It is interesting to note that the relative stereochemistry of diastereoisomers 5Rax and 6Rax corresponds to that seen in the lowest energy atropoisomer of the simple biphenyl analogue 1.
While the incorporation of a methyl substituent α to the nitrogen atom generally increases the enantiocontrol, it has a detrimental or no effect on the mismatched diastereoisomers 5Sax and 6Sax. The sense of stereochemical induction in the epoxidation process is controlled by the configuration of the biaryl backbone. Catalyst 6Rax, with the added methyl group, is in most cases superior to catalyst 5Rax. The octahydrobinaphthyl catalyst 21, with an additional methyl group, provides the highest enantioselectivities that we have seen for the epoxidation of 1-phenylcyclohexene, at 96% ee, and for 1-phenyl dihydronaphthalene, at 97% ee.
Conclusions
The results described herein provide further evidence that addition of additional methyl substituents adjacent to the nitrogen atom in the heterocyclic ring can provide improvements in enantioselectivities in catalysed epoxidation reactions, with the α-methylated octahydrobinaphthyl catalyst 21 giving the best results of this series; particularly noteworthy are the epoxidations of 1-phenylcyclohexene, at 96% ee, and of 1-phenyl dihydronaphthalene, at 97% ee.Calculations clearly suggest a parallel pattern of dihedral angles between the iminium parents and the putative derived oxaziridinium oxidative intermediates.
Experimental results also support the conjecture that some correlation may be observed between the dihedral angle found in the biaryl units of these iminium salt catalysts and the induced enantioselectivities in the catalysed epoxidation reactions. The 6,6′-dimethylbiphenyl catalysts 5 and 6 (dihedral angles around 52–54°) are superior to the simpler biphenyl species 1 and 30 (dihedral angles around 34–40°). The octahydrobinaphthyl catalysts 19 and 21 (dihedral angles around 57–60°) induce generally similar enantioselectivities to the 6,6′-dimethylbiphenyl species, perhaps suggesting optimum dihedral angles of around 50–60° as one factor in the induction of enantioselectivities in the catalysed epoxidation reactions.
Experimental section
General procedure for the addition of methyl Grignard reagent to iminium salts
The iminium salt was dissolved in Et2O (50 mL per g of iminium salt) under an atmosphere of N2. The solution was cooled to −78 °C. A 3 molar solution of MeMgCl in THF (10 equiv.) was added dropwise over 10 min. After 1 h, cooling was ceased, and the mixture allowed to reach ambient temperature overnight. Saturated aqueous NH4Cl (5 mL per g of iminium salt) and CH2Cl2 (30 mL per g of iminium salt) were added. The resulting mixture was transferred to a separating funnel, and brine (100 mL) added. The organic layer was collected and the aqueous layer washed with CH2Cl2 (2 × 50 mL per g of iminium salt). The combined organic layers were washed with water (2 × 100 mL per g of iminium salt) and brine (2 × 100 mL per g of iminium salt). The organic fraction was dried over MgSO4, and evaporated to dryness under reduced pressure to yield the desired methyl azepines.General procedure for the synthesis of iminium salts from the corresponding azepines
The azepine substrate was dissolved in CH2Cl2 (30 mL per g of azepine), and NBS (1.1 equiv.) added. The resulting bright yellow solution was stirred for 30 min at room temperature, after which time the solvent was switched to EtOH. To the ethanolic solution was added NaBPh4 (1.1 equiv.) in the minimum amount of MeCN with stirring, which caused the iminium salt to precipitate. The solvent was switched to CH2Cl2 and the solution transferred to a separating funnel. The organic layer was washed with water (2 × 60 mL per g of azepine) and brine (30 mL per g of azepine). The crude product isolated after the removal of solvents was recrystallized from EtOH. The crystalline product was filtered off and washed with cold EtOH followed by Et2O and hexane. The isolated iminium salt was left to dry under reduced pressure in an oven at 60 °C overnight.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) 4.
NaNO2 (7.8 g, 0.11 mol) was added in one portion to an ice cold solution of 2-amino-3-methylbenzoic acid (17.0 g, 0.11 mol) in 2.45 M NaOH (60 mL). The mixture was stirred at 0 °C until it became homogeneous. An ice-cooled 4 M aqueous solution of HCl (240 mL) was added to the solution at a rate such that the temperature remained below 5 °C. The resulting orange solution of the diazonium salt was stirred at 0 °C for 20 min. Cu2SO4·5H2O (24.0 g, 96 mmol) and water (75 mL) were added, and the resulting solution cooled in an ice-bath. When the solution had reached 5 °C, 30% w/v NH4OH solution (47.5 mL) was added in one portion. A freshly prepared solution of NH2OH, prepared from treatment of (NH2OH)·H2SO4 (8.8 g, 53.5 mmol) with a 3 M ice-cold aqueous solution of NaOH (38 mL, 0.11 mol), was added to the resulting deep-blue solution. The solution was stirred for 20 min to cool to 0 °C. The diazonium salt prepared above was added to the copper solution in three equal portions of 50 mL. The temperature was maintained below 7 °C throughout the procedure. After the addition was complete, the resulting orange/red solution was heated under reflux for 30 min, and allowed to cool. After the red solution had attained ambient temperature, conc. HCl (37.5 mL) was added slowly and the mixture was allowed to stand overnight to provide for the complete precipitation of the product. The yellow/brown precipitate was removed by filtration using a Büchner funnel, washed with water and air-dried. The crude material was stirred in boiling EtOH (200 mL); any remaining bright yellow solid was removed by filtration, leaving a clear brown filtrate. Crystallization of the bis(carboxylic acid) (±)-8 was induced by the addition of water to the filtrate to yield light brown crystals (8.4 g, 55%); m.p. 226–230 °C (dec.); νmax(neat)/cm−1 2982, 2907, 2667, 2579, 1680, 1583, 1436, 1377, 1316, 1159, 962, 799, 768, 722; δH (500 MHz; d6-DMSO) 1.81 (6 H, s), 7.31 (2 H, t, J = 7.5 Hz), 7.44 (2 H, d, J = 7.5 Hz), 7.71 (2 H, d, J = 7.5 Hz), 12.28 (2 H, broad); δC (125 MHz; d6-DMSO) 19.8, 126.6, 127.3, 132.9, 136.0, 140.7, 168.1.
4.
Compound (±)-8 (2.20 g, 8.2 mmol) was dissolved in Et2O (30 mL) under a nitrogen atmosphere. The resulting pale yellow solution was cooled to 0 °C, and LiAlH4 (0.93 g, 24.5 mmol) added slowly at a rate that just maintained the effervescence. The resulting suspension was heated under reflux for 1 h, after which time the reaction mixture was allowed to cool to room temperature. Solid Na2SO4 (6.0 g) was added to the suspension, which was cooled in an ice-bath and stirred for 5 min. Water (10 mL) was added dropwise to quench the excess LiAlH4. The mixture was stirred for 30 min, filtered through a layered pad of Celite and Na2SO4, and the colourless filtrate concentrated under reduced pressure to yield (±)-9 as a colourless crystalline solid (1.91 g, 96%); νmax(neat)/cm−1 3254, 3064, 3017, 2935, 2880, 1679, 1592, 1458, 1378, 1239, 1211, 1163, 1028, 994, 902, 783, 760, 623; δH (500 MHz; CDCl3) 1.86 (6 H, s), 3.19 (2 H, s), 4.08 (2 H, d, J = 11.5 Hz), 4.24 (2 H, d, J = 11.5 Hz), 7.24 (2 H, s, J = 7.5 Hz), 7.29 (2 H, t, J = 7.5 Hz), 7.34 (2 H, d, J = 7.5 Hz); δC (125 MHz; CDCl3) 20.2, 63.1, 127.6, 128.0, 129.9, 136.1, 138.3, 138.5; m/z (HNES) found for [C16H18O2 + Na]+ 265.1200; [M + Na]+ requires 265.1199.
29.
PCC (1.33 g, 62.0 mmol) was added in one portion to a solution of (±)-9 (0.50 g, 20.7 mmol) in CH2Cl2 (20 mL), producing a dark-orange solution. The solution was stirred vigorously for 3 h at room temperature. Et2O (20 mL) and a spatula of Celite was added to the resulting black mixture, which was stirred for a further 30 min. The reaction mixture was filtered through a layered pad of Celite and silica gel. Solvents were removed under reduced pressure to yield compound (±)-7 as a colourless crystalline solid (0.43 g, 87%); m.p. 108–110 °C; δH (500 MHz; CDCl3) 1.98 (6 H, s), 7.50 (2 H, t, J = 7.5 Hz), 7.59 (2 H, d, J = 7.5 Hz), 7.91 (2 H, d, J = 7.5 Hz), 9.60 (2 H, s); δC (125 MHz; CDCl3) 19.7, 126.4, 128.6, 134.5, 135.9, 137.6, 140.0, 191.4; m/z (HNES) found for [C16H14O2 + NH4]+: 256.1333; [M + NH4]+ requires 256.1332.
:1), buffered with 2% TEA, to give the desired compound as a mixture of diastereoisomers [First eluting 10Rax as a colourless crystalline solid (1.18 g, 49%); and second eluting 10Sax as a colourless foam (0.62 g, 26%)].
4.
NaNO2 (7.8 g, 0.11 mol) was added in one portion to an ice cold solution of 2-amino-3-methylbenzoic acid (17.0 g, 0.11 mol) in 2.45 M NaOH (60 mL). The mixture was stirred at 0 °C until it became homogeneous. An ice-cooled 4 M aqueous solution of HCl (240 mL) was added to the solution at a rate such that the temperature remained below 5 °C. The resulting orange solution of the diazonium salt was stirred at 0 °C for 20 min. Cu2SO4·5H2O (24.0 g, 96 mmol) and water (75 mL) were added, and the resulting solution cooled in an ice-bath. When the solution had reached 5 °C, 30% w/v NH4OH solution (47.5 mL) was added in one portion. A freshly prepared solution of NH2OH, prepared from treatment of (NH2OH)·H2SO4 (8.8 g, 53.5 mmol) with a 3 M ice-cold aqueous solution of NaOH (38 mL, 0.11 mol), was added to the resulting deep-blue solution. The solution was stirred for 20 min to cool to 0 °C. The diazonium salt prepared above was added to the copper solution in three equal portions of 50 mL. The temperature was maintained below 7 °C throughout the procedure. After the addition was complete, the resulting orange/red solution was heated under reflux for 30 min, and allowed to cool. After the red solution had attained ambient temperature, conc. HCl (37.5 mL) was added slowly and the mixture was allowed to stand overnight to provide for the complete precipitation of the product. The yellow/brown precipitate was removed by filtration using a Büchner funnel, washed with water and air-dried. The crude material was stirred in boiling EtOH (200 mL); any remaining bright yellow solid was removed by filtration, leaving a clear brown filtrate. Crystallization of the bis(carboxylic acid) (±)-8 was induced by the addition of water to the filtrate to yield light brown crystals (8.4 g, 55%); m.p. 226–230 °C (dec.); νmax(neat)/cm−1 2982, 2907, 2667, 2579, 1680, 1583, 1436, 1377, 1316, 1159, 962, 799, 768, 722; δH (500 MHz; d6-DMSO) 1.81 (6 H, s), 7.31 (2 H, t, J = 7.5 Hz), 7.44 (2 H, d, J = 7.5 Hz), 7.71 (2 H, d, J = 7.5 Hz), 12.28 (2 H, broad); δC (125 MHz; d6-DMSO) 19.8, 126.6, 127.3, 132.9, 136.0, 140.7, 168.1.
4.
Compound (±)-8 (2.20 g, 8.2 mmol) was dissolved in Et2O (30 mL) under a nitrogen atmosphere. The resulting pale yellow solution was cooled to 0 °C, and LiAlH4 (0.93 g, 24.5 mmol) added slowly at a rate that just maintained the effervescence. The resulting suspension was heated under reflux for 1 h, after which time the reaction mixture was allowed to cool to room temperature. Solid Na2SO4 (6.0 g) was added to the suspension, which was cooled in an ice-bath and stirred for 5 min. Water (10 mL) was added dropwise to quench the excess LiAlH4. The mixture was stirred for 30 min, filtered through a layered pad of Celite and Na2SO4, and the colourless filtrate concentrated under reduced pressure to yield (±)-9 as a colourless crystalline solid (1.91 g, 96%); νmax(neat)/cm−1 3254, 3064, 3017, 2935, 2880, 1679, 1592, 1458, 1378, 1239, 1211, 1163, 1028, 994, 902, 783, 760, 623; δH (500 MHz; CDCl3) 1.86 (6 H, s), 3.19 (2 H, s), 4.08 (2 H, d, J = 11.5 Hz), 4.24 (2 H, d, J = 11.5 Hz), 7.24 (2 H, s, J = 7.5 Hz), 7.29 (2 H, t, J = 7.5 Hz), 7.34 (2 H, d, J = 7.5 Hz); δC (125 MHz; CDCl3) 20.2, 63.1, 127.6, 128.0, 129.9, 136.1, 138.3, 138.5; m/z (HNES) found for [C16H18O2 + Na]+ 265.1200; [M + Na]+ requires 265.1199.
29.
PCC (1.33 g, 62.0 mmol) was added in one portion to a solution of (±)-9 (0.50 g, 20.7 mmol) in CH2Cl2 (20 mL), producing a dark-orange solution. The solution was stirred vigorously for 3 h at room temperature. Et2O (20 mL) and a spatula of Celite was added to the resulting black mixture, which was stirred for a further 30 min. The reaction mixture was filtered through a layered pad of Celite and silica gel. Solvents were removed under reduced pressure to yield compound (±)-7 as a colourless crystalline solid (0.43 g, 87%); m.p. 108–110 °C; δH (500 MHz; CDCl3) 1.98 (6 H, s), 7.50 (2 H, t, J = 7.5 Hz), 7.59 (2 H, d, J = 7.5 Hz), 7.91 (2 H, d, J = 7.5 Hz), 9.60 (2 H, s); δC (125 MHz; CDCl3) 19.7, 126.4, 128.6, 134.5, 135.9, 137.6, 140.0, 191.4; m/z (HNES) found for [C16H14O2 + NH4]+: 256.1333; [M + NH4]+ requires 256.1332.
:1), buffered with 2% TEA, to give the desired compound as a mixture of diastereoisomers [First eluting 10Rax as a colourless crystalline solid (1.18 g, 49%); and second eluting 10Sax as a colourless foam (0.62 g, 26%)].
First eluting amine 10Rax: [α]D +54.9 (c = 0.97, CDCl3); νmax(neat)/cm−1 2990, 2934, 2860, 1452, 1378, 1308, 1263, 1236, 1200, 1172, 1150, 1075, 1027 953, 852, 787, 749, 727, 698; δH (400 MHz; CDCl3) 1.58 (3 H, s), 1.64 (3 H, s), 2.11 (6 H, s), 2.65 (1 H, d, J = 1.9 Hz), 3.16 (2 H, d, J = 12.1 Hz), 3.67 (2 H, d, J = 12.1 Hz), 4.12–4.21 (2 H, m), 5.10 (1 H, d, J = 3.1 Hz), 6.98–7.02 (2 H, m), 7.14–7.18 (4 H, m), 7.24–7.36 (5 H, m); δC (100 MHz; CDCl3) 19.3, 20.0, 29.9, 53.2, 60.1, 61.9, 75.3, 99.3, 126.4, 126.7, 126.9, 127.5, 127.8, 129.0, 135.4, 136.7, 138.6, 140.5; m/z found for [C28H31NO2 + H]+ 414.2431; [M + H]+ requires 414.2428.
Second eluting amine 10Sax: [α]D +121.0 (c = 1.07, CDCl3); νmax(neat)/cm−1 2990, 2939, 2859, 1497, 1452, 1378, 1332, 1263, 1236, 1198, 1168, 1140, 1076 1024, 1001, 952, 853, 787, 750, 722, 697, 665; δH (400 MHz; CDCl3) 1.46 (3 H, s), 1.49 (3 H, s), 2.12 (6 H, s), 3.01 (1 H, br), 3.31 (2 H, d, J = 12.5 Hz), 3.46 (2 H, d, J = 12.4 Hz), 3.92 (1 H, d, J = 12.4 Hz), 4.13 (1 H, d, J = 12.4 Hz), 5.13 (1 H, d, J = 3.0 Hz), 6.80–7.02 (2 H, m), 7.16–7.27 (4 H, m), 7.33 (2 H, t, J = 7.6 Hz), 7.45 (2 H, d, J = 7.6 Hz); m/z found for [C28H31NO2 + H]+: 414.2425; [M + H]+ requires 414.2428.
![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2N), 5.82 (1 H, d, J = 2.5 Hz), 6.79 (4 H, t, J = 7.1 Hz), 6.92 (8 H, t, J = 7.3 Hz), 7.10–7.29 (14 H, m), 7.36 (1 H, d, J = 7.9 Hz), 7.41–7.48 (2 H, m), 7.53 (1 H, t, J = 7.8 Hz), 7.71 (1 H, d, J = 7.6 Hz), 8.92 (1 H, s); δC (100 MHz; DMSO-d6) 19.3, 20.2, 29.9, 56.2, 61.2, 66.5, 71.3, 100.8, 122.2, 125.5, 125.8, 125.9, 128.37, 128.38, 128.42, 129.1, 129.7, 132.2, 136.2, 137.1, 138.3, 139.0, 139.9, 164.0; m/z found for [C28H30NO2]+ 412.2273, iminium cation requires 412.2271.
29.
(Ra)-Binol (3.00 g, 11.5 mmol) was divided equally between three test tubes each containing Pd/C 10% w/w (0.20 g). Glacial acetic acid (4.0 mL) was added to each of the test tubes and mixed. The tubes were then placed in a hydrogenation apparatus, and were charged and evacuated with H2 twice. The pressure was increased to 50 bar and the reaction mixtures stirred at ambient temperature. The reactions were monitored over 10 days. The combined mixture was filtered through celite and washed through with CHCl3. The organic filtrate was washed with saturated aqueous NaHCO3 (1 × 50 mL), water (2 × 50 mL), and brine (1 × 50 mL). The organic layer was dried over MgSO4 and solvents were removed under reduced pressure to yield a colourless solid. Column chromatography was performed, eluting with light petroleum ether/EtOAc (4:1), to yield compound 13 as a colourless solid which was crystallized from hexane to give fine colourless needles (1.63 g, 52%); [α]20D −42.6 (c = 1.06, CHCl3), lit.29 [α]20D −42.5 (c = 1.00, CHCl3); νmax(neat)/cm−1 3475, 3381, 2929, 2856, 1587, 1472, 1333, 1287, 1248, 1195, 1152, 936, 828, 812, 726; δH (300 MHz; CDCl3) 1.63–1.78 (8 H, m), 2.16 (2 H, dt, J = 17.0 Hz, 6.0 Hz), 2.29 (2 H, dt, J = 17.0 Hz, 6.1 Hz), 2.75 (4 H, t, J = 6.2 Hz), 4.56 (2 H, br), 6.83 (2 H, d, J = 8.4 Hz), 7.07 (2 H, d, J = 8.3 Hz); δC (75 MHz; CDCl3) 22.8, 22.9, 27.0, 29.1, 113.0, 118.8, 130.2, 131.1, 137.2, 151.5.
28.
Compound 13 (10.0 g, 34 mmol) was dissolved in CH2Cl2 (250 mL) in a flame-dried flask under an atmosphere of N2. The vessel was cooled to −30 °C. DMAP (1.66 g, 13.6 mmol), 2,6-lutidine (11.9 mL, 102 mmol) and Tf2O (17.2 mL, 102 mmol) were added to the solution and the mixture stirred for 10 min, and allowed to reach room temperature overnight. Silica gel was added to the dark brown solution, and the mixture stirred for 20 min. The solvents were removed under reduced pressure. The residue was transferred to a sintered glass funnel containing a layer of silica gel and washed with hexane until the product had eluted. The solvent was removed under reduced pressure to yield compound 14 as a colourless crystalline solid (28.01 g, 99%); m.p. 119–121 °C; [α]20D −259.6 (c = 1.03, CHCl3), lit.28 [α]20D −260.3 (c = 1.00, CHCl3); νmax(neat)/cm−1; 2947, 2870, 2836, 1476, 1464, 1451, 1410, 1249, 1202, 1182, 1138, 1048, 926, 872, 926, 872, 853, 834, 806, 763, 699, 644; δH (300 MHz; CDCl3) 1.75 (8 H, m), 2.28 (2 H, dt, J = 6.0 Hz, 17.4 Hz), 2.43 (2 H, dt, J = 6.0, 17.7 Hz), 2.86 (4 H, t, J = 6.0 Hz), 7.16 (2 H, d, J = 8.6 Hz), 7.23 (2 H, d, J = 8.6 Hz); δC (75 MHz; CDCl3) 22.2, 22.3, 27.4, 29.3, 118.1, 118.2 (q, J = 322 Hz), 127.1, 130.9, 138.3, 139.3, 144.8.
28.
DMSO (90 mL), MeOH (36.0 mL, 1.56 mol) and Hünig's base (13.4 mL, 137 mmol) were added to 14 (1.50 g, 17.9 mmol), Pd(OAc)2 (0.61 g, 4.7 mmol) and dppp (1.11 g, 4.70 mmol) under an argon atmosphere. The vessel was sealed with a gas manifold and pressurised with CO (g) at 2 bar followed by evacuation of the atmosphere until the orange solution just began to boil. The cycle was repeated five times. During the cycles, the solution turned to opaque black. The vessel was heated to 80 °C and maintained at a pressure of 2.4 atm. of CO for 48 h. The reaction mixture was transferred to a round-bottomed flask, and solvents were removed under reduced pressure to yield an oily residue. This crude residue was purified by column chromatography, eluting with light petroleum/EtOAc (5:1). Compound 15 was obtained as a colourless crystalline solid (5.9 g, 88%); m.p. = 99–105 °C; [α]20D −2.1 (c = 0.96, CHCl3), lit.28 [α]20D −1.9 (c = 1.00, CHCl3); νmax(neat)/cm−1 2926, 2853, 1718, 1588, 1430, 1400, 1289, 1251, 1186, 1164, 1124, 1066, 1050, 878, 861, 832, 771, 749; δH (300 MHz; CDCl3) 1.69 (8 H, m), 1.99 (2 H, dt, J = 5.9 Hz, 17.0 Hz), 2.18 (2 H, dt, J = 6.4 Hz, 17.2 Hz), 2.86 (4 H, m), 3.57 (6 H, s), 7.13 (2 H, d, J = 8.1 Hz), 7.76 (2 H, d, J = 8.1 Hz); δC (75 MHz; CDCl3) 22.4, 23.0, 27.2, 30.2, 51.5, 126.6, 127.1, 128.0, 135.4, 141.7, 141.9, 167.6.
21.
A suspension of LiAlH4 (0.30 g, 7.94 mmol) in Et2O (50 mL) under a nitrogen atmosphere was cooled to 0 °C. Compound 15 (1.50 g, 3.97 mmol) was added slowly. After the effervescence had ceased, the reaction mixture was heated under reflux for 30 min. The mixture was cooled in an ice bath. Na2SO4 (1.50 g) and Celite were added in one portion and stirred for 10 min followed by the dropwise addition of water (2.0 mL). After 30 min, the mixture was filtered through a pad of Celite and Na2SO4, and washed though with Et2O. The solvents were removed under reduced pressure to yield compound 12 as a colourless foam (1.24 g, 99%); [α]20D −39.1 (c = 0.96, MeOH), lit.21 [α]20D −39.0 (c = 1.01, MeOH); νmax(neat)/cm−1 3257, 2923, 2853, 1594, 1433, 1231, 1061, 1005, 899, 815, 740; δH (300 MHz; CDCl3) 1.69 (8 H, m), 2.02 (4 H, m), 2.82 (4 H, t, J = 8.8 Hz), 2.92 (2 H, s), 4.02 (2 H, d, J = 12.0 Hz), 4.21 (2 H, d, J = 12.0 Hz), 7.11 (2 H, d, J = 8.8 Hz), 7.24 (2 H, d, J = 8.8 Hz); δC (75 MHz; CDCl3) 22.7, 23.2, 27.6, 29.8, 62.9, 127.2, 128.9, 134.8, 135.7, 137.6, 138.3.
21.
Compound 12 (0.11 g, 0.34 mmol) was dissolved in CH2Cl2 (20 mL), and PCC (0.22 g, 1.02 mmol) added in one portion. The resulting dark orange solution was stirred for 2 h, and celite added followed by Et2O (20 mL). The mixture was stirred for 30 min and filtered through a layered pad of silica gel and Celite. The desired compound was eluted with Et2O. Solvents were then removed to furnish compound 16 as a colourless crystalline solid (0.11 g, 99%); m.p. 129–131 °C; [α]20D +110.4 (c = 0.83, CHCl3), lit.21 [α]20D +108.5 (c = 1.00, CHCl3); νmax(neat)/cm−1 2931, 2851, 2747, 1688, 1672, 1582, 1441, 1383, 1315, 1292, 1268, 1246, 1230, 1162, 1125, 996, 968, 943, 913, 897, 836, 811, 760; δH (300 MHz; CDCl3) 1.73 (8 H, m), 2.15 (4 H, m), 2.91 (4 H, t, J = 6.1 Hz), 7.29 (2 H, d, J = 8.1 Hz), 7.82 (2 H, d, J = 8.1 Hz), 9.51 (2 H, s); δC (75 MHz; CDCl3) 22.2, 22.7, 27.4, 30.4, 125.1, 129.8, 132.4, 136.2, 140.7, 145.2, 191.5.
27.
Compound 12 (2.03 g, 6.3 mmol) was dissolved in toluene (60 mL) under an atmosphere of N2, and pyridine (0.05 mL, 0.7 mmol) added. Tribromophosphine (1.78 mL, 18.9 mmol) was added dropwise. After the addition was complete, the mixture was heated at 60 °C for 3 h. After cooling, water (50 mL) was added, and the resulting biphasic mixture transferred to a separating funnel. The organic layer was isolated and washed with saturated aqueous NaHCO3 (20 mL). The organic layers were dried over MgSO4 and decolourized with a spatula of carbon black, and filtered. The solvents were removed under reduced pressure to yield compound 18 as a colourless powder (2.79 g, 99%); [α]20D +34.3 (c = 2.05, CHCl3), lit.27 [α]20D +36.5 (c = 1.00, CH2Cl2); νmax(neat)/cm−1 2920, 2851, 1592, 1457, 1376, 1235, 1204, 928, 862, 831, 814, 759, 723, 623; δH (300 MHz; CDCl3) 1.71 (8 H, m), 2.07 (2 H, dt, J = 6.4 Hz, 17.6 Hz), 2.34 (2 H, dt, J = 5.5 Hz, 17.0 Hz), 2.83 (4 H, t, J = 5.9 Hz), 4.10 (2 H, d, J = 9.9 Hz), 4.15 (2 H, d, J = 10.0 Hz), 7.14 (2 H, d, J = 7.9 Hz), 7.33 (2 H, d, J = 7.9 Hz); δC (75 MHz; CDCl3) 22.6, 22.9, 27.5, 29.9, 32.9, 128.1, 129.6, 132.3, 135.6, 137.9, 138.6.
2N), 5.82 (1 H, d, J = 2.5 Hz), 6.79 (4 H, t, J = 7.1 Hz), 6.92 (8 H, t, J = 7.3 Hz), 7.10–7.29 (14 H, m), 7.36 (1 H, d, J = 7.9 Hz), 7.41–7.48 (2 H, m), 7.53 (1 H, t, J = 7.8 Hz), 7.71 (1 H, d, J = 7.6 Hz), 8.92 (1 H, s); δC (100 MHz; DMSO-d6) 19.3, 20.2, 29.9, 56.2, 61.2, 66.5, 71.3, 100.8, 122.2, 125.5, 125.8, 125.9, 128.37, 128.38, 128.42, 129.1, 129.7, 132.2, 136.2, 137.1, 138.3, 139.0, 139.9, 164.0; m/z found for [C28H30NO2]+ 412.2273, iminium cation requires 412.2271.
29.
(Ra)-Binol (3.00 g, 11.5 mmol) was divided equally between three test tubes each containing Pd/C 10% w/w (0.20 g). Glacial acetic acid (4.0 mL) was added to each of the test tubes and mixed. The tubes were then placed in a hydrogenation apparatus, and were charged and evacuated with H2 twice. The pressure was increased to 50 bar and the reaction mixtures stirred at ambient temperature. The reactions were monitored over 10 days. The combined mixture was filtered through celite and washed through with CHCl3. The organic filtrate was washed with saturated aqueous NaHCO3 (1 × 50 mL), water (2 × 50 mL), and brine (1 × 50 mL). The organic layer was dried over MgSO4 and solvents were removed under reduced pressure to yield a colourless solid. Column chromatography was performed, eluting with light petroleum ether/EtOAc (4:1), to yield compound 13 as a colourless solid which was crystallized from hexane to give fine colourless needles (1.63 g, 52%); [α]20D −42.6 (c = 1.06, CHCl3), lit.29 [α]20D −42.5 (c = 1.00, CHCl3); νmax(neat)/cm−1 3475, 3381, 2929, 2856, 1587, 1472, 1333, 1287, 1248, 1195, 1152, 936, 828, 812, 726; δH (300 MHz; CDCl3) 1.63–1.78 (8 H, m), 2.16 (2 H, dt, J = 17.0 Hz, 6.0 Hz), 2.29 (2 H, dt, J = 17.0 Hz, 6.1 Hz), 2.75 (4 H, t, J = 6.2 Hz), 4.56 (2 H, br), 6.83 (2 H, d, J = 8.4 Hz), 7.07 (2 H, d, J = 8.3 Hz); δC (75 MHz; CDCl3) 22.8, 22.9, 27.0, 29.1, 113.0, 118.8, 130.2, 131.1, 137.2, 151.5.
28.
Compound 13 (10.0 g, 34 mmol) was dissolved in CH2Cl2 (250 mL) in a flame-dried flask under an atmosphere of N2. The vessel was cooled to −30 °C. DMAP (1.66 g, 13.6 mmol), 2,6-lutidine (11.9 mL, 102 mmol) and Tf2O (17.2 mL, 102 mmol) were added to the solution and the mixture stirred for 10 min, and allowed to reach room temperature overnight. Silica gel was added to the dark brown solution, and the mixture stirred for 20 min. The solvents were removed under reduced pressure. The residue was transferred to a sintered glass funnel containing a layer of silica gel and washed with hexane until the product had eluted. The solvent was removed under reduced pressure to yield compound 14 as a colourless crystalline solid (28.01 g, 99%); m.p. 119–121 °C; [α]20D −259.6 (c = 1.03, CHCl3), lit.28 [α]20D −260.3 (c = 1.00, CHCl3); νmax(neat)/cm−1; 2947, 2870, 2836, 1476, 1464, 1451, 1410, 1249, 1202, 1182, 1138, 1048, 926, 872, 926, 872, 853, 834, 806, 763, 699, 644; δH (300 MHz; CDCl3) 1.75 (8 H, m), 2.28 (2 H, dt, J = 6.0 Hz, 17.4 Hz), 2.43 (2 H, dt, J = 6.0, 17.7 Hz), 2.86 (4 H, t, J = 6.0 Hz), 7.16 (2 H, d, J = 8.6 Hz), 7.23 (2 H, d, J = 8.6 Hz); δC (75 MHz; CDCl3) 22.2, 22.3, 27.4, 29.3, 118.1, 118.2 (q, J = 322 Hz), 127.1, 130.9, 138.3, 139.3, 144.8.
28.
DMSO (90 mL), MeOH (36.0 mL, 1.56 mol) and Hünig's base (13.4 mL, 137 mmol) were added to 14 (1.50 g, 17.9 mmol), Pd(OAc)2 (0.61 g, 4.7 mmol) and dppp (1.11 g, 4.70 mmol) under an argon atmosphere. The vessel was sealed with a gas manifold and pressurised with CO (g) at 2 bar followed by evacuation of the atmosphere until the orange solution just began to boil. The cycle was repeated five times. During the cycles, the solution turned to opaque black. The vessel was heated to 80 °C and maintained at a pressure of 2.4 atm. of CO for 48 h. The reaction mixture was transferred to a round-bottomed flask, and solvents were removed under reduced pressure to yield an oily residue. This crude residue was purified by column chromatography, eluting with light petroleum/EtOAc (5:1). Compound 15 was obtained as a colourless crystalline solid (5.9 g, 88%); m.p. = 99–105 °C; [α]20D −2.1 (c = 0.96, CHCl3), lit.28 [α]20D −1.9 (c = 1.00, CHCl3); νmax(neat)/cm−1 2926, 2853, 1718, 1588, 1430, 1400, 1289, 1251, 1186, 1164, 1124, 1066, 1050, 878, 861, 832, 771, 749; δH (300 MHz; CDCl3) 1.69 (8 H, m), 1.99 (2 H, dt, J = 5.9 Hz, 17.0 Hz), 2.18 (2 H, dt, J = 6.4 Hz, 17.2 Hz), 2.86 (4 H, m), 3.57 (6 H, s), 7.13 (2 H, d, J = 8.1 Hz), 7.76 (2 H, d, J = 8.1 Hz); δC (75 MHz; CDCl3) 22.4, 23.0, 27.2, 30.2, 51.5, 126.6, 127.1, 128.0, 135.4, 141.7, 141.9, 167.6.
21.
A suspension of LiAlH4 (0.30 g, 7.94 mmol) in Et2O (50 mL) under a nitrogen atmosphere was cooled to 0 °C. Compound 15 (1.50 g, 3.97 mmol) was added slowly. After the effervescence had ceased, the reaction mixture was heated under reflux for 30 min. The mixture was cooled in an ice bath. Na2SO4 (1.50 g) and Celite were added in one portion and stirred for 10 min followed by the dropwise addition of water (2.0 mL). After 30 min, the mixture was filtered through a pad of Celite and Na2SO4, and washed though with Et2O. The solvents were removed under reduced pressure to yield compound 12 as a colourless foam (1.24 g, 99%); [α]20D −39.1 (c = 0.96, MeOH), lit.21 [α]20D −39.0 (c = 1.01, MeOH); νmax(neat)/cm−1 3257, 2923, 2853, 1594, 1433, 1231, 1061, 1005, 899, 815, 740; δH (300 MHz; CDCl3) 1.69 (8 H, m), 2.02 (4 H, m), 2.82 (4 H, t, J = 8.8 Hz), 2.92 (2 H, s), 4.02 (2 H, d, J = 12.0 Hz), 4.21 (2 H, d, J = 12.0 Hz), 7.11 (2 H, d, J = 8.8 Hz), 7.24 (2 H, d, J = 8.8 Hz); δC (75 MHz; CDCl3) 22.7, 23.2, 27.6, 29.8, 62.9, 127.2, 128.9, 134.8, 135.7, 137.6, 138.3.
21.
Compound 12 (0.11 g, 0.34 mmol) was dissolved in CH2Cl2 (20 mL), and PCC (0.22 g, 1.02 mmol) added in one portion. The resulting dark orange solution was stirred for 2 h, and celite added followed by Et2O (20 mL). The mixture was stirred for 30 min and filtered through a layered pad of silica gel and Celite. The desired compound was eluted with Et2O. Solvents were then removed to furnish compound 16 as a colourless crystalline solid (0.11 g, 99%); m.p. 129–131 °C; [α]20D +110.4 (c = 0.83, CHCl3), lit.21 [α]20D +108.5 (c = 1.00, CHCl3); νmax(neat)/cm−1 2931, 2851, 2747, 1688, 1672, 1582, 1441, 1383, 1315, 1292, 1268, 1246, 1230, 1162, 1125, 996, 968, 943, 913, 897, 836, 811, 760; δH (300 MHz; CDCl3) 1.73 (8 H, m), 2.15 (4 H, m), 2.91 (4 H, t, J = 6.1 Hz), 7.29 (2 H, d, J = 8.1 Hz), 7.82 (2 H, d, J = 8.1 Hz), 9.51 (2 H, s); δC (75 MHz; CDCl3) 22.2, 22.7, 27.4, 30.4, 125.1, 129.8, 132.4, 136.2, 140.7, 145.2, 191.5.
27.
Compound 12 (2.03 g, 6.3 mmol) was dissolved in toluene (60 mL) under an atmosphere of N2, and pyridine (0.05 mL, 0.7 mmol) added. Tribromophosphine (1.78 mL, 18.9 mmol) was added dropwise. After the addition was complete, the mixture was heated at 60 °C for 3 h. After cooling, water (50 mL) was added, and the resulting biphasic mixture transferred to a separating funnel. The organic layer was isolated and washed with saturated aqueous NaHCO3 (20 mL). The organic layers were dried over MgSO4 and decolourized with a spatula of carbon black, and filtered. The solvents were removed under reduced pressure to yield compound 18 as a colourless powder (2.79 g, 99%); [α]20D +34.3 (c = 2.05, CHCl3), lit.27 [α]20D +36.5 (c = 1.00, CH2Cl2); νmax(neat)/cm−1 2920, 2851, 1592, 1457, 1376, 1235, 1204, 928, 862, 831, 814, 759, 723, 623; δH (300 MHz; CDCl3) 1.71 (8 H, m), 2.07 (2 H, dt, J = 6.4 Hz, 17.6 Hz), 2.34 (2 H, dt, J = 5.5 Hz, 17.0 Hz), 2.83 (4 H, t, J = 5.9 Hz), 4.10 (2 H, d, J = 9.9 Hz), 4.15 (2 H, d, J = 10.0 Hz), 7.14 (2 H, d, J = 7.9 Hz), 7.33 (2 H, d, J = 7.9 Hz); δC (75 MHz; CDCl3) 22.6, 22.9, 27.5, 29.9, 32.9, 128.1, 129.6, 132.3, 135.6, 137.9, 138.6.
Method 2: Compound 16 (0.11 g, 0.35 mmol) was suspended in MeOH (4 mL) under an atmosphere of N2. A solution of amine 4 (79 mg, 1.1 equiv., 0.38 mmol) in MeOH (1 mL) was added followed by NaCNBH3 (44 mg, 2 equiv., 0.70 mmol) and glacial acetic acid (0.2 mL). The mixture was stirred for 24 h at ambient temperature, and a 1 M aq. solution of NaOH (25 mL) added. The mixture was extracted with Et2O (3 × 20 mL). The combined organic layers were washed with brine (30 mL) and dried over MgSO4. Solvents were removed under reduced pressure to yield a pale yellow foam that was purified by column chromatography, eluting with light petroleum ether/EtOAc (5:1). Compound 17 was isolated as colourless crystals (116 mg, 67%). [α]20D −27.7 (c = 0.72, CHCl3); νmax(neat)/cm−1 2928, 2832, 1451, 1377, 1291, 1267, 1237, 1194, 1172, 1147, 1074, 1053, 1017, 939, 898, 852, 829, 810, 753, 723, 700, 654, 640, 628; δH (300 MHz; CDCl3) 1.46–1.57 (2 H, m), 1.57 (3 H, s), 1.62 (3 H, s), 1.67–1.82 (6 H, m), 2.13 (2 H, dt, J = 5.9 Hz, 11.5 Hz), 2.58–2.65 (3 H, m), 2.75–2.86 (4 H, m), 3.09 (2 H, d, J = 12.1 Hz), 3.57 (2 H, d, J = 12.2 Hz), 4.17 (2 H, m), 5.07 (1 H, d, J = 3.1 Hz), 6.89 (2 H, d, J = 7.6 Hz), 6.98 (2 H, d, J = 7.6 Hz), 7.30 (5 H, m); δC (75 MHz; CDCl3) 19.1, 22.7, 22.9, 27.6, 29.4, 29.6, 52.7, 59.8, 61.8, 75.0, 99.1, 126.0, 126.7, 127.6, 128.2, 133.6, 135.0, 136.0, 138.2, 140.4; m/z (HNES) found for [C34H39NO2 + H]+: 494.3039; [M + H]+: requires 494.3039.
:1), buffered with 2% TEA. The title compound 20 was isolated as a colourless oil (0.63 g, 79%). [α]20D −8.4 (c = 2.04, CHCl3); δH (300 MHz; CDCl3) 0.18 (3 H, d, J = 7.1 Hz), 1.42–1.63 (2 H, m), 1.55 (3 H, s), 1.63 (3 H, s), 1.68–1.81 (6 H, m), 2.09–2.19 (2 H, m), 2.54–2.69 (2 H, m), 2.77–2.82 (4 H, m), 2.91 (1 H, td, J = 4.2 Hz, 2.5 Hz), 3.31 (1 H, d, J = 11.2 Hz), 3.35 (1 H, d, J = 11.2 Hz), 4.09 (1 H, dd, J = 12.3 Hz, 2.4 Hz), 4.21 (1 H, dd, J = 12.3 Hz, 4.3 Hz), 4.34 (1 H, q, J = 7.0 Hz), 5.13 (1 H, d, J = 3.5 Hz), 6.81 (1 H, d, J = 7.6 Hz), 6.81 (1 H, d, J = 7.7 Hz), 6.94 (1 H, d, J = 7.6 Hz), 6.97 (1 H, d, J = 7.7 Hz), 7.22–7.38 (1 H, m); δC (75 MHz; CDCl3) 19.4, 21.0, 22.7, 22.8, 22.9, 27.3, 29.0, 29.3, 29.4, 54.3, 59.0, 60.1, 62.9, 74.1, 99.3, 125.9, 126.4, 126.5, 126.6, 127.6, 128.1, 128.2, 135.2, 135.5, 135.6, 135.7, 136.1, 136.6, 138.4, 138.8, 140.3; m/z (HNES) found for [C35H41NO2 + H]+: 508.3204; [M + H]+ requires 508.3210.
General procedure for the formation of racemic epoxides
The alkene was dissolved in CH2Cl2 (10 mL per g of alkene) and the solution cooled to 0 °C. A solution of m-CPBA (2 equiv.) in CH2Cl2 (10 mL per g of alkene), dried over MgSO4, was filtered into the solution of alkene. The reaction was allowed to achieve ambient temperature and stirred until complete conversion of the alkene was observed by TLC. Saturated aqueous NaHCO3 (10 mL per g of alkene) was added, and organic layer collected and washed with 1 M NaOH (10 mL per g of alkene), and dried over MgSO4. The solvents were removed under reduced pressure. Analytically pure samples of the epoxides were obtained by means of flash column chromatography, typically eluting with light petroleum/EtOAc (99:1), buffered with 2% TEA.
General procedures for catalytic asymmetric epoxidation of alkenes with Oxone® mediated by iminium salts under aqueous conditions
Method A: Oxone® (2 equiv.) and NaHCO3 (5 equiv.) were added with stirring to a solution of the catalyst (5 mol%) in MeCN (1 mL) and water (0.1 mL) at 0 °C. After 5 min, alkene (3 mmol) dissolved in MeCN (1 mL), was added. The mixture was stirred at 0 °C until complete conversion of the alkene was observed by TLC, or for up to 6 h. The reaction mixture was then diluted with Et2O (10 mL) and the resulting suspension filtered through a pad of mixed Na2SO3 and MgSO4. The solvents were removed under reduced pressure, and analytically pure samples of the epoxide were obtained by means of flash column chromatography or preparative TLC, typically eluting with light petroleum/EtOAc (99:1), containing 2% TEA. The major enantiomer was identified by [α]20D measurements and comparison to the literature; enantioselectivities were determined either by chiral HPLC, or by 1H NMR spectroscopy in the presence of a resolving agent.
Method B: A mixture of water (1 ml), MeCN (1 mL) and NaHCO3 (100 mg, 1.2 mmol) was cooled in an ice-bath at 0 °C. Oxone® (370 mg, 0.6 mmol) was added. After the effervescence had subsided, the catalyst (5 mol%) was added followed, after 1 min, by the alkene (0.3 mmol). The mixture was stirred at 0 °C until complete conversion of the alkene was observed by TLC, or for up to 6 h. The reaction mixture was diluted with Et2O (10 mL per 0.10 g of substrate), and the resulting suspension filtered through a pad of mixed Na2SO3 and MgSO4. The solvents were removed under reduced pressure, and analytically pure samples of the epoxide were obtained by either flash column chromatography or preparative TLC, eluting with light petroleum/EtOAc (99:1), containing 2% TEA. The major enantiomer was identified by [α]20D measurements and comparison with the literature; enantioselectivities were determined either by chiral HPLC, or by 1H NMR spectroscopy in the presence of a resolving agent.
Method C: A mixture of water (0.8 mL) and NaHCO3 (67 mg, 0.80 mmol) was cooled in an ice-bath at 0 °C. Oxone® (132.0 mg, 0.21 mmol) was added, and the solution stirred until the effervescence had ceased. The alkene (0.20 mmol) was dissolved in CH2Cl2 (0.5 mL) and added. A solution of the catalyst (5 mol%) and 18-crown-6 (1 mg, 2.5 mol%) in CH2Cl2 (0.7 mL) was cooled to 0 °C, and added with stirring. The reaction mixture was vigorously stirred, typically for 2 h at 0 °C. CH2Cl2 (10 mL) and water (10 mL), both cooled to around 0 °C, were added. The organic layer was collected and dried over MgSO4. The solvents were removed under reduced pressure, and analytically pure samples of the epoxide were obtained by either column chromatography or preparative TLC, eluting with light petroleum/EtOAc (99:1), containing 2% TEA. The major enantiomer was identified by [α]20D measurements; enantioselectivities were determined either by chiral HPLC, or by 1H NMR spectroscopy in the presence of a resolving agent.
Tetraphenylphosphonium monoperoxysulfate15
Tetraphenylphosphonium chloride (15.0 g, 40 mmol) was dissolved in CH2Cl2 (200 mL), and the solution cooled to 10 °C. A solution of Oxone® (15.0 g, 48 mmol) in deionised water (300 mL) was cooled to 10 °C, and added over a period of 5 min. The resulting biphasic mixture was stirred vigorously for 1 h. The organic layer was separated, and the solvents were removed under reduced pressure at room temperature. The crude colourless solid was washed with deionised water (3 × 80 mL). The solid was re-dissolved in CH2Cl2 (150 mL) and dried over MgSO4. Hexane was added until a solid precipitate just began to form, and the flask placed in a freezer overnight. The resulting colourless crystalline solid was collected by filtration (12.87 g, 71%); νmax(neat)/cm−1 3210, 3060, 1586, 1484, 1435, 1262, 1226, 1162, 1106, 1058, 1041, 996, 721; δH (400 MHz; CDCl3) 7.58–7.69 (8 H, m), 7.71–7.80 (8 H, m), 7.82–7.86 (4 H, m), 9.18 (1 H, s). The oxygen content, typically 94% peroxide, was estimated by comparing the integrals of the aromatic and hydroxyl signals.General procedure for catalytic asymmetric epoxidation of alkenes with TPPP mediated by iminium salts under non-aqueous conditions
A solution of alkene was cooled to the required temperature, and TPPP was added in one portion with stirring over 2 min. The catalyst was added as a solid in small portions over 1 min. The reaction was stirred until complete consumption of the starting alkene had been observed, or until it was judged that no further conversion was occurring, by TLC. Et2O, cooled to 0 °C, was added to precipitate out TPPP and its reduced by-products. The suspension was filtered through a thin pad of celite and silica gel to yield the desired epoxide. If column chromatography was required, it was typically performed eluting with light petroleum/EtOAc (99:1), containing 2% TEA. The major enantiomer was identified by [α]20D measurements and comparison to the literature; enantioselectivities were determined by either chiral HPLC, or by 1H NMR spectroscopy in the presence of a resolving agent.
:20), oven temp 20 °C, column Chiracel OD-H 01 250 × 4.6 mm, 5 μm particle size, flow rate 1 mL min−1, 254 nm: tr-4.00 min (−)-(1S,2S), 6.92 min (+)-(1R,2R).
:10), oven temp 20 °C, column Chiracel OD-H 01 250 × 4.6 mm, 5 μm particle size, flow rate 1 mL min−1, 254 nm: tr-4.51 min (−)-(1S,2R), 5.94 min (+)-(1R,2S).
:20), oven temp 20 °C, column Chiracel OD-H 01 250 × 4.6 mm, 5 μm particle size, flow rate 1 mL min−1, 254 nm: tr-4.98 min (−)-(1S,2S), 6.54 min (+)-(1R,2R).
:10), oven temp 20 °C, column Chiracel OD-H 01 250 × 4.6 mm, 5 μm particle size, flow rate 1 mL min−1, 254 nm: tr – 4.26 min (+)-(S), 7.47 min (−)-(R).
Acknowledgements
This investigation has enjoyed the support of the EPSRC, Loughborough University, the University of East Anglia, Charnwood Molecular Ltd, and the ERDF (ISCE-Chem & INTERREG IVa programme 4061). We acknowledge the support of The Royal Society. We are also indebted to the EPSRC Mass Spectrometry Unit, Swansea. The calculations presented in this paper were carried out on the High Performance Computing Cluster supported by the Research and Specialist Computing Support service at the University of East Anglia.References
- (a) C. H. Behrens and K. B. Sharpless, Aldrichimica Acta, 1983, 16, 67 CAS; (b) J. Gorzynski Smith, Synthesis, 1984, 629 CrossRef; (c) T. Katsuki, Coord. Chem. Rev., 1995, 140, 189 CrossRef CAS.
- (a) A. Armstrong, Angew. Chem., Int. Ed., 2004, 43, 1460 CrossRef CAS PubMed; (b) P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2001, 40, 3726 CrossRef CAS; (c) D. W. C. MacMillan, M. P. Brochu and S. P. Brown, J. Am. Chem. Soc., 2004, 126, 4108 CrossRef PubMed; (d) Y. Zhu, Q. Wang, R. G. Cornwall and Y. Shi, Chem. Rev., 2014, 114, 8199 CrossRef CAS PubMed.
- (a) D. Yang, Y.-C. Yip, M.-W. Tang, M.-K. Wong, J.-H. Zheng and K.-K. Cheung, J. Am. Chem. Soc., 1996, 118, 491 CrossRef CAS; (b) D. Yang, X. C. Wang, M.-K. Wong, Y.-C. Yip and M.-W. Tang, J. Am. Chem. Soc., 1996, 118, 11311 CrossRef CAS; (c) D. Yang, M.-K. Wong, Y.-C. Yip, X. C. Wang, M.-W. Tang, J.-H. Zheng and K.-K. Cheung, J. Am. Chem. Soc., 1998, 120, 5943 CrossRef CAS; (d) D. Yang, Y.-C. Yip, M.-W. Tang, M.-K. Wong and K. K. Cheung, J. Org. Chem., 1998, 63, 9888 CrossRef CAS.
- (a) S. E. Denmark, D. C. Forbes, D. S. Hays, J. S. DePue and R. G. Wilde, J. Org. Chem., 1995, 60, 1391 CrossRef CAS; (b) S. E. Denmark, H. Matsuhashi, C. M. Crudden and Z. Wu, J. Org. Chem., 1997, 62, 8288 CrossRef CAS PubMed; (c) S. E. Denmark and Z. Wu, J. Org. Chem., 1997, 62, 8964 CrossRef CAS; (d) S. E. Denmark and Z. Wu, J. Org. Chem., 1998, 63, 2810 CrossRef CAS; (e) S. E. Denmark and Z. Wu, Synlett, 1999, 847 CAS.
- (a) A. Armstrong and B. R. Hayter, Chem. Commun., 1998, 621 RSC; (b) A. Armstrong, G. Ahmed, B. Dominguez-Fernandez, B. R. Hayter and J. S. Wailes, J. Org. Chem., 2002, 67, 8610 CrossRef CAS PubMed.
- (a) Y. Tu, Z.-X. Wang and Y. Shi, J. Am. Chem. Soc., 1996, 118, 9806 CrossRef CAS; (b) Z.-X. Wang, Y. Tu, M. Frohn and Y. Shi, J. Org. Chem., 1997, 62, 2328 CrossRef CAS PubMed; (c) Z.-X. Wang and Y. Shi, J. Org. Chem., 1998, 63, 3099 CrossRef CAS; (d) Z.-X. Wang and Y. Shi, J. Org. Chem., 1997, 62, 8622 CrossRef CAS; (e) G. A. Cao, Z.-X. Wang, Y. Tu and Y. Shi, Tetrahedron Lett., 1998, 39, 4425 CrossRef CAS; (f) Y. Zhu, Y. Tu, H. Yu and Y. Shi, Tetrahedron Lett., 1998, 39, 7819 CrossRef CAS; (g) T. Yong, Z. X. Wang, M. Frohn, M. He, H. Yu, Y. Tang and Y. Shi, J. Org. Chem., 1998, 63, 8475 CrossRef; (h) J. D. Warren and Y. Shi, J. Org. Chem., 1999, 64, 7675 CrossRef CAS; (i) M. Frohn, X. Zhou, J.-R. Zhang, Y. Tang and Y. Shi, J. Am. Chem. Soc., 1999, 121, 7718 CAS; (j) L. Shu and Y. Shi, Tetrahedron Lett., 1999, 40, 8721 CrossRef CAS; (k) L. Shu and Y. Shi, J. Org. Chem., 2000, 65, 8807 CrossRef CAS PubMed; (l) H. Tian, X. She, J. Xu and Y. Shi, Org. Lett., 2001, 3, 1929 CrossRef CAS PubMed; (m) H. Tian, X. She, H. Yu, L. Shu and Y. Shi, J. Org. Chem., 2002, 67, 2435 CrossRef CAS PubMed; (n) X.-Y. Wu, X. She and Y. Shi, J. Am. Chem. Soc., 2002, 124, 8792 CrossRef CAS PubMed.
- (a) A. Picot, P. Milliet and X. Lusinchi, Tetrahedron Lett., 1976, 17, 1573 CrossRef; (b) P. Milliet, A. Picot and X. Lusinchi, Tetrahedron Lett., 1976, 17, 1577 CrossRef; (c) G. Hanquet, X. Lusinchi and P. Milliet, Tetrahedron Lett., 1987, 28, 6061 CrossRef CAS; (d) G. Hanquet, X. Lusinchi and P. Milliet, Tetrahedron Lett., 1988, 29, 3941 CrossRef CAS; (e) L. Bohé, G. Hanquet, M. Lusinchi and X. Lusinchi, Tetrahedron Lett., 1993, 34, 7271 CrossRef; (f) L. Bohé, M. Lusinchi and X. Lusinchi, Tetrahedron, 1999, 55, 141 CrossRef.
- (a) L. Bohé and M. Kammoun, Tetrahedron Lett., 2002, 43, 803 CrossRef; (b) L. Bohé and M. Kammoun, Tetrahedron Lett., 2004, 45, 747 CrossRef; (c) V. K. Aggarwal and M. F. Wang, Chem. Commun., 1996, 191 RSC; (d) A. Armstrong, G. Ahmed, I. Garnett and K. Gioacolou, Synlett, 1997, 1075 CrossRef CAS; (e) A. Armstrong, G. Ahmed, I. Garnett, K. Gioacolou and J. S. Wailes, Tetrahedron, 1999, 55, 2341 CrossRef CAS; (f) A. Gluszynska, I. Mackowska, M. D. Rozwadowska and W. Sienniak, Tetrahedron: Asymmetry, 2004, 15, 2499 CrossRef CAS; (g) M. R. Biscoe and R. Breslow, J. Am. Chem. Soc., 2005, 127, 10812 CrossRef CAS PubMed; (h) S. Minakata, A. Takemiya, K. Nakamura, I. Ryu and M. Komatsu, Synlett, 2000, 1810 CAS; (i) M.-K. Wong, L.-M. Ho, Y.-S. Zheng, C.-Y. Ho and D. Yang, Org. Lett., 2001, 16, 2587 CrossRef; (j) J. M. Crosthwaite, V. A. Farmer, J. P. Hallett and T. Welton, J. Mol. Catal. A: Chem., 2008, 279, 148 CrossRef CAS; (k) R. Novikov and J. Lacour, Tetrahedron: Asymmetry, 2010, 21, 1611 CrossRef CAS; (l) R. Novikov, J. Vachon and J. Lacour, Chimia, 2007, 61, 236 CrossRef CAS; (m) J. Vachon, C. Lauper, K. Ditrich and J. Lacour, Tetrahedron: Asymmetry, 2006, 17, 2334 CrossRef CAS; (n) J. Vachon, S. Rentsch, A. Martinez, C. Marsol and J. Lacour, Org. Biomol. Chem., 2007, 5, 501 RSC; (o) O. A. Wong and Y. Shi, Chem. Rev., 2008, 108, 3958 CrossRef CAS PubMed; (p) R. Novikov, G. Bernardinelli and J. Lacour, Adv. Synth. Catal., 2009, 351, 596 CrossRef CAS.
- (a) P. C. Bulman Page, B. R. Buckley, M. M. Farah and A. J. Blacker, Eur. J. Org. Chem., 2009, 3413 CrossRef; (b) P. C. Bulman Page, B. R. Buckley, L. F. Appleby and P. A. Alsters, Synthesis, 2005, 3405 CrossRef; (c) P. C. Bulman Page, B. R. Buckley, G. A. Rassias and A. J. Blacker, Eur. J. Org. Chem., 2006, 803 CrossRef.
- P. C. Bulman Page, P. Parker, G. A. Rassias, B. R. Buckley and D. Bethell, Adv. Synth. Catal., 2008, 350, 1867 CrossRef.
- P. C. Bulman Page, P. Parker, B. R. Buckley, G. A. Rassias and D. Bethell, Tetrahedron, 2009, 65, 2910 CrossRef.
- B. R. Buckley, Y. Chan, N. Dreyfus, C. E. Elliott, F. Marken and P. C. Bulman Page, Adv. Synth. Catal., 2008, 350, 1149 CrossRef.
- P. C. Bulman Page, M. M. Farah, B. R. Buckley, A. J. Blacker and J. Lacour, Synlett, 2008, 1381 CrossRef , and references therein.
- (a) M. F. A. Adamo, V. K. Aggarwal and M. A. Sage, J. Am. Chem. Soc., 2000, 122, 8317 CrossRef CAS; (b) M. F. A. Adamo, V. K. Aggarwal and M. A. Sage, J. Am. Chem. Soc., 2002, 124, 11223 CrossRef CAS; (c) V. K. Aggarwal, C. Lopin and F. Sandrinelli, J. Am. Chem. Soc., 2003, 125, 7596 CrossRef CAS PubMed; (d) C.-Y. Ho, Y.-C. Chen, M.-K. Wong and D. Yang, J. Org. Chem., 2005, 70, 898 CrossRef CAS PubMed.
- S. Campestrini, F. Di Furia, G. Labat and F. Novello, J. Chem. Soc., Perkin Trans. 2, 1994, 2175 RSC.
- (a) P. C. Bulman Page, B. R. Buckley, D. Barros, A. J. Blacker, H. Heaney and B. A. Marples, Tetrahedron, 2006, 62, 6607 CrossRef CAS; (b) P. C. Bulman Page, B. R. Buckley, D. Barros, A. J. Blacker, B. A. Marples and M. R. J. Elsegood, Tetrahedron, 2007, 63, 5386 CrossRef.
- P. C. Bulman Page, B. R. Buckley, H. Heaney and A. J. Blacker, Org. Lett., 2005, 7, 375 CrossRef CAS PubMed.
- P. C. Bulman Page, L. F. Appleby, D. Day, Y. Chan, B. R. Buckley, S. M. Allin and M. J. McKenzie, Org. Lett., 2009, 11, 1991 CrossRef CAS PubMed.
- C. J. Bartlett, D. P. Day, Y. Chan, S. M. Allin, M. J. McKenzie, A. M. Z. Slawin and P. C. Bulman Page, J. Org. Chem., 2012, 77, 772 CrossRef CAS PubMed.
- C. J. Bartlett, Y. Chan, D. Day, P. Parker, B. R. Buckley, G. A. Rassias, A. M. Z. Slawin, S. M. Allin, J. Lacour, A. Pinto and P. C. Bulman Page, J. Org. Chem., 2012, 77, 6128 CrossRef PubMed.
- R. Novikov, G. Bernardinelli and J. Lacour, Adv. Synth. Catal., 2008, 350, 1113 CrossRef CAS.
- S. E. Denmark and H. Matsuhashi, J. Org. Chem., 2002, 67, 3479 CrossRef CAS PubMed.
- S. L. Pira, T. W. Wallace and J. P. Graham, Org. Lett., 2009, 11, 1663 CrossRef CAS PubMed.
- J. Ruan, G. Lu, L. Xu, Y.-M. Li and A. S. C. Chan, Adv. Synth. Catal., 2008, 350, 76 CrossRef CAS.
- A. K. Unni, N. Takenaka, H. Yamamoto and V. H. Rawal, J. Am. Chem. Soc., 2005, 127, 1336 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, M. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- See Tables S1 and S2 in the ESI.†.
- (a) J. Lacour, D. Monchaud and C. Marsol, Tetrahedron Lett., 2002, 43, 8257 CrossRef CAS; (b) J. Vachon, C. Pérollier, D. Monchaud, C. Marsol, K. Ditrich and J. Lacour, J. Org. Chem., 2005, 70, 5903 CrossRef CAS PubMed.
- A. Korostylev, V. I. Tararov, C. Fischer, A. Monsees and A. Borner, J. Org. Chem., 2004, 69, 3220 CrossRef CAS PubMed.
- P. C. Bulman Page, G. A. Rassias, D. Bethell and M. B. Schilling, J. Org. Chem., 1998, 63, 2774 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ob00542j |
| This journal is © The Royal Society of Chemistry 2016 |