DOI:
10.1039/C6NR03297D
(Paper)
Nanoscale, 2016,
8, 14925-14931
Formation of nanowires via single particle-triggered linear polymerization of solid-state aromatic molecules†
Received
22nd April 2016
, Accepted 16th June 2016
First published on 16th June 2016
Abstract
Nanowires occupy a prestigious place in nanoelectronics, nanomechanics, and biomimetics. Although there are notable methods to grow nanowires via self-assembly, there is a key drawback in the need to find out the specific conditions appropriate for each system. In this sense, universal techniques to fabricate such nanowires from various organic materials have been sought for the continued progress of the related research field. Here we report one of the promising and facile methodologies to quantitatively produce nanowires with controlled geometrical parameters. In this method, referred to as “Single Particle-Triggered Linear Polymerization (STLiP)”, organic thin films on a supporting substrate were irradiated with high-energy charged particles, accelerated by particle accelerators. Each particle penetrates from the top of the films to the substrate while gradually releasing kinetic energy along its trajectory (ion track), generating reactive intermediates such as radical species that eventually induce propagation reactions. The resulting polymerized products were integrated into nanowires with uniform diameter and length that can be isolated via development with appropriate organic solvents. Considering the widely applicable nature of STLiP to organic materials, the present technique opens a new door for access to a number of functional nanowires and their assembly.
Introduction
Since the first discovery of the radiation-induced polymerization reactions of acetylenes,1,2 ionizing radiation including β- and γ-rays has been a useful tool to initiate polymerization reactions in the condensed phases of monomeric materials, and grafting reactions in polymeric materials via radicals and/or ionic reactive intermediates produced in the primary processes induced by radiation. Crosslinking and main chain scission reactions caused by ionizing radiation also have major advantages in the functionalization of a variety of polymer materials,3 and are hitherto used in the industrial processing of these materials.4 Chain reactions in the propagation of polymer chains dramatically amplify the number of chemical reactions induced by incident ionizing radiation, which dissipates the energy discontinuously and leads to a dilute distribution of reactive intermediates in the matter in comparison with conventional bulk chemical reactions.5 The sophisticated application of this amplification through propagation chain reactions was developed, such as in polymer gel dosimeter systems designed for the detection of ultralow radiation doses, where considerable cloudiness of the matrix was induced by the polymer aggregates promoted by propagation and polymerization reactions.6–8 Radiation-induced graft polymerization onto the surface and/or in the bulk of organic media has also been a key technique for providing functional surfaces, and there has been a constant demand for grafting in industrial processing of plastics as rubbers, membranes, and so on.9–11
In the solid state, radiation-induced ionization of organic molecules often leads to quantitative production of neutral radical species after geminate/bulk recombination of ionic species and relaxation processes of excited states. However, propagation chain reactions in solid media have often been kinetically unfavourable because of the strictly limited molecular diffusion. To date, successful solid state polymerization reactions have been reported in a limited number of monomeric materials,12–14 rarely in their crystalline phases.15–17 Topochemical polymerization is one promising solution to overcome the above limitation and lead to single crystalline polymeric materials, where propagation reactions proceed with a minimum amount of atomic and molecular movement of diolefin,18 diacetylene,19,20etc.21 Prerequisite for the polymerization reactions was the orientation of the reactive monomers in the solid state, that should be pre-organized at a distance commensurate with the repeat distance in the final polymer products.22 Numerous studies have been carried out to understand the mechanisms of photo- and radiation-induced polymerization reactions, and to shed light on their practical applications.
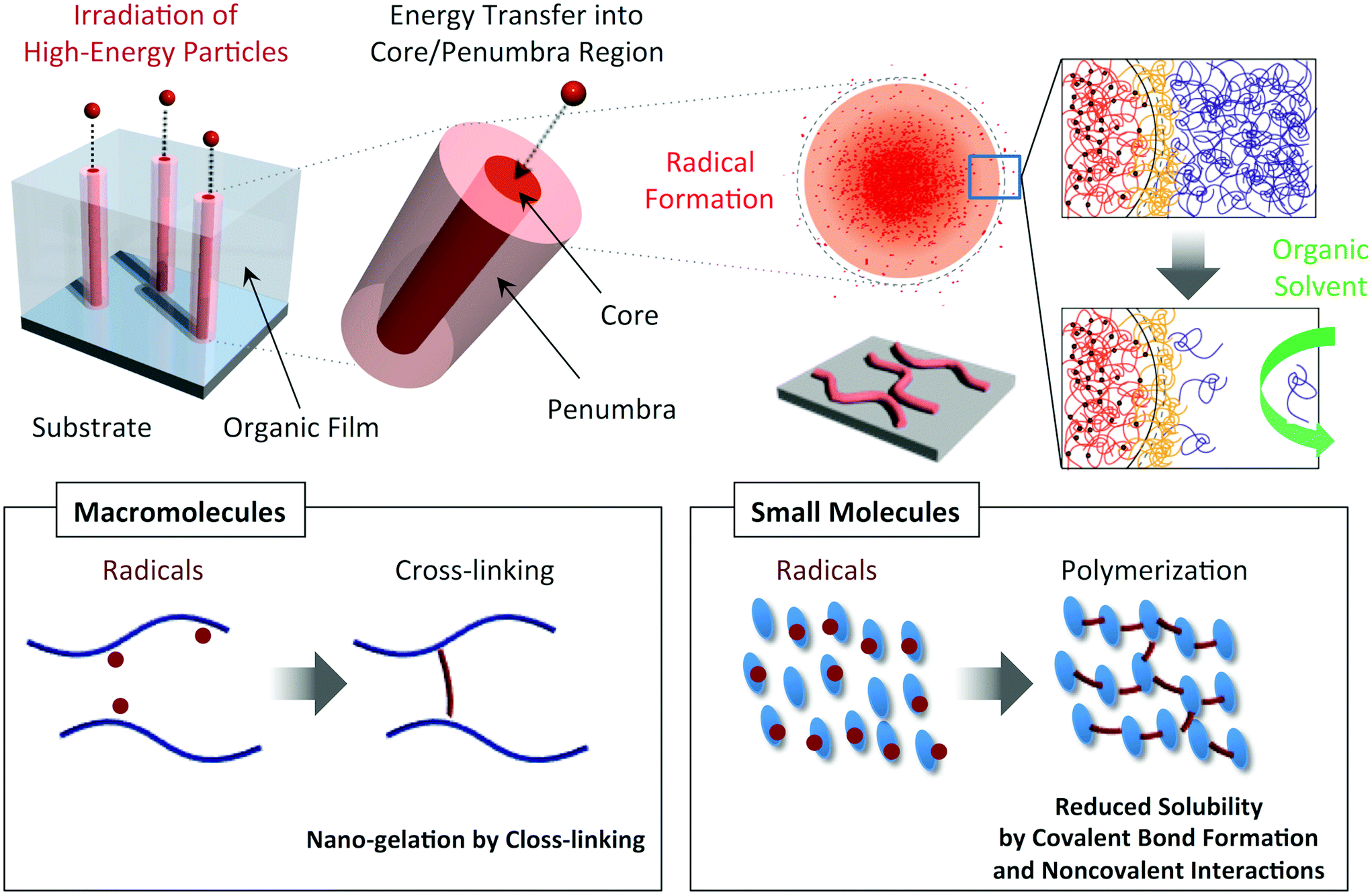
High-energy charged particles release their energy in condensed media with extremely high density, and induce non-homogeneous, cross-linking reactions in nanometer-scale cylindrical regions along their trajectories.23–25 By utilizing such radiation chemistry along the trajectories (penumbra regions) in polymer films, we have reported the formation of insoluble cylindrically shaped nanogels which are isolated as nanowires on substrates via a development process (Fig. 1).26–30 Although nanowire objects were successfully observed from various types of synthetic polymer26–29 and biomacromolecule,30 the application of this technique to small molecules was mostly limited to the fullerene derivatives.31–34 However, considering that the number density of active species (initiating radicals) is extremely high in the trajectories of the particles, we thought that the combination of solid-state polymerization reactions and high-energy charged particles paves the way for an extended variety of applicable monomers and for spatial limitation of the reactions. Here we demonstrate, using aromatic amine compounds, a potentially versatile technique of single particle triggered linear polymerization (STLiP) to give one-dimensional nanomaterials along the particle trajectories with confined sizes ranging over a few nm.
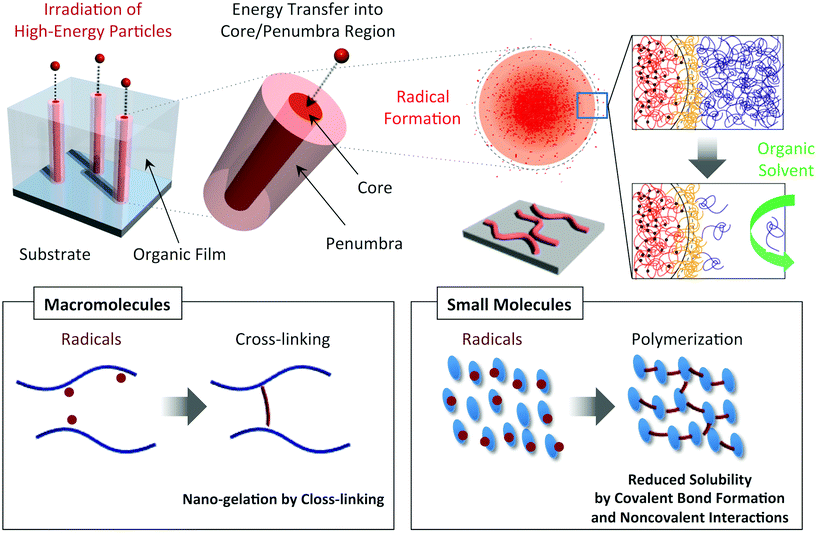 |
| | Fig. 1 Schematic illustrations of the nanowire fabrication processes utilizing high-energy ion particles that trigger cross-linking/polymerization reactions along ion tracks (penumbra regions). | |
Experimental section
Materials
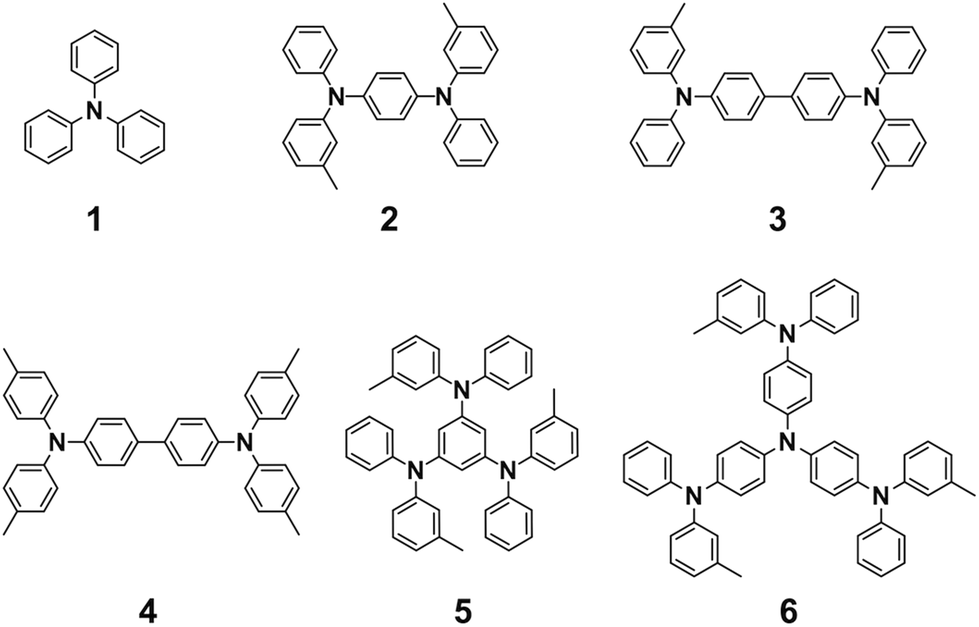
Triphenylamine derivatives 1, 3, 4, and 6 were purchased from TCI Co. and used without further purification. Compounds 2 and 5 were synthesized according to the literature.351H-NMR spectra were recorded in CDCl3 or acetone-d6 on a JEOL model AL-400 spectrometer, operating at 400 MHz, where chemical shifts were determined with respect to tetramethylsilane (TMS) as an internal reference.
Synthesis of N1,N4-diphenyl-N1,N4-di-m-tolylbenzene-1,4-diamine (compound 2).
Toluene (15 mL) was added to a mixture of 1,4-dibromobenzene (2.02 g, 8.49 mol), Pd2(dba)3 (467 mg, 0.51 mol), P(tBu)3·HBF4 (296 mg, 1.02 mol), tBuOK (3.26 g, 33.9 mol), and 3-methyl-N-phenylaniline (3.1 g, 17.0 mol) in a 2-neck flask, and the mixture was refluxed for 24 h under a N2 atmosphere. The reaction mixture was cooled down to room temperature and filtered from the insoluble fraction, and then evaporated to dryness under reduced pressure. The residue was subjected to column chromatography on silica gel using chloroform as an eluent, and evaporated to dryness under reduced pressure. The residue was recrystallized from chloroform/hexane to allow isolation of 2 (2.12 g, 54%) as white crystals. 1H NMR (400 MHz, acetone-d6, δ): 2.25 (s, 6H), 6.85–6.86 (m, 4H), 6.91 (s, 2H), 6.97–7.06 (m, 10H), 7.16–7.19 (t, J = 7.6 Hz, 2H), 7.25–7.30 (m, 4H).
Synthesis of N1,N3,N5-triphenyl-N1,N3,N5-tri-m-tolylenzene-1,3,5-triamine (compound 5).
Toluene (15 mL) was added to a mixture of 1,3,5-tribromobenzene (2.0 g, 6.35 mmol), Pd2(dba)3 (523 mg, 0.57 mmol), P(tBu)3·HBF4 (331 mg, 1.14 mmol), tBuOK (3.66 g, 38.1 mmol), and 3-methyl-N-phenylaniline (3.49 g, 19.06 mmol) in a 2-neck flask, and the mixture was refluxed for 24 h under a N2 atmosphere. The reaction mixture was cooled down to room temperature and filtered from the insoluble fraction, and then evaporated to dryness under reduced pressure. The residue was subjected to column chromatography on silica gel using chloroform as an eluent, and evaporated to dryness under reduced pressure. The residue was recrystallized from hexane to allow isolation of 5 (1.20 g, 30%) as pale gray crystals. 1H NMR (400 MHz, CDCl3, δ): 2.21 (s, 9H), 6.40 (s, 3H), 6.72–6.74 (d, J = 7.20 Hz, 2H), 6.80–6.84 (m, 6H), 6.89–6.93 (m, 3H), 7.00–7.07 (m, 9H), 7.13–7.17 (m, 6H).
Method
The Si substrate was cut into 1.5 cm2 squares, sonicated in 2-propanol, dried, and treated with UV-O3 prior to use. Triphenylamine derivatives were dissolved (5–10 wt%) in THF for 2 and toluene for 1 and 3–6, and the solution was spin-coated or drop-casted on the Si substrate to give thin films. The thickness of the films was evaluated using a Veeco Instruments Inc. model Dektak 150 surface profiler. 490 MeV 192Os30+ particles were generated using a cyclotron accelerator at National Institutes for Quantum and Radiological Science and Technology, Takasaki Advanced Radiation Research Institute. 150 MeV 107Ag11+, 100 MeV 58Ni7+, 60 MeV 28Si5+, and 60 MeV 16O5+ were generated using a pelletron accelerator at the Inter University Accelerator Centre. The prepared thin films of triphenylamine derivatives were exposed to the above ion beam in a vacuum chamber (<1 × 10−4 Pa). The number of incident particles was controlled at 108–1010 particles cm−2 to prevent severe overlapping of the ion tracks. Then, the irradiated films were further cut into small pieces and developed by immersing them into organic solvents for 10–60 s. The sizes and shapes of the isolated nanowires were observed using a Seiko Instruments Inc. model SPI-4000 atomic force microscope (AFM), Bruker Co. model Multimode 8 AFM, and JEOL Ltd model JSM-7001F scanning electron microscope (SEM). The loss of kinetic energy of the ions due to their traversal through the organic films was estimated using the SRIM 2010 simulation code.
Results and discussion
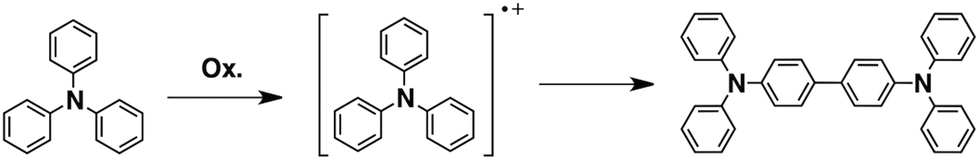
Triphenylamines are known to undergo dimerization reactions when oxidized to give radical cation species (Scheme 1).36,37 We consider that aromatic amine compounds provide radical species upon irradiation and efficient propagation reactions in the solid state. Compounds 1–5 with different molecular sizes were prepared in the present work (Fig. 2), and their thin films were prepared on Si substrates using conventional spin-coating or drop-cast methods. Ion-beam irradiation of the prepared films at a low fluence (∼108–1010 cm−2) hopefully results in propagation reactions, leading to the formation of one-dimensional organogels along the trajectories of the respective particles which are often called ion tracks. Because of the large difference between the linear energy transfers (LETs) of organic materials and the Si wafer, the primary charged particles and secondary electrons preferentially induce the formation of dense reactive species at the aromatic amines/Si interface, leading to heterogeneous covalent bond formation. Thus the termini of the obtained nanowires become covalently bound to the surface of the Si wafer via Si–C or Si–O–C bonds, preventing the nanowires from easily being washed away by solvents. The crucial requisite for this technique, named “single particle-triggered linear polymerization (STLiP)”, is the remarkable difference in solubility between irradiated and non-irradiated parts, which enables the easy isolation of the resultant nanowires via development solvents. In principle, the initially formed nanowires stand on a substrate, where the length of the nanowires corresponds to the thickness of the films. Nevertheless, due to the interfacial surface tension of organic solvents, observed nanowires usually lie flat on the substrate.
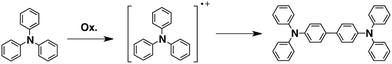 |
| | Scheme 1 Formation of tetraphenylbenzidine via dimerization of triphenylamine radical cations. | |
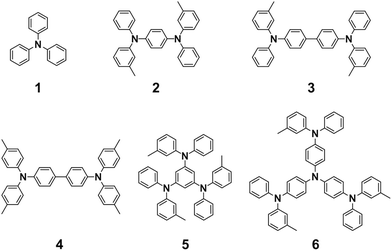 |
| | Fig. 2 Chemical structures of the aromatic amine derivatives. | |
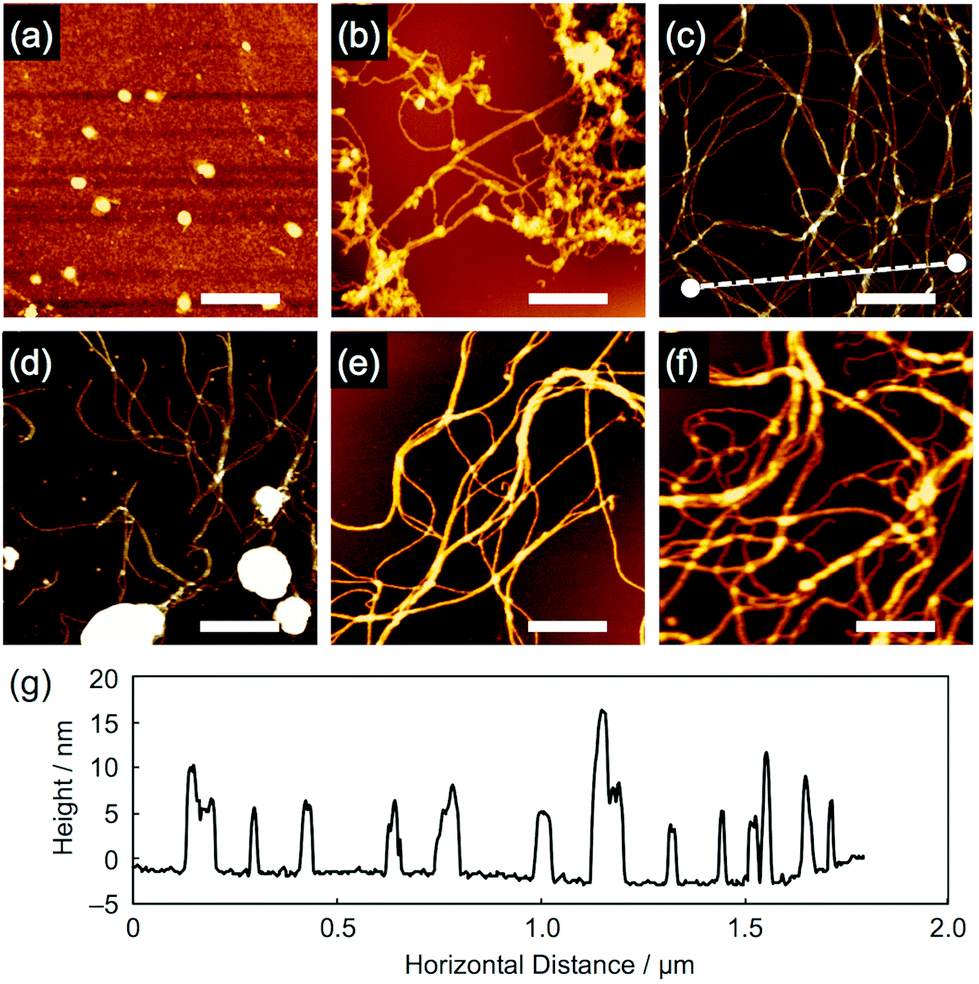
When the aromatic amine films were exposed to 490 MeV 192Os30+ particles (LET ∼13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 eV nm−1) at a low fluence, followed by development in hexane or toluene, nanowire objects consisting of aromatic derivatives were visualized via AFM, except for compound 1 (Fig. 3). We consider that the small molecular size of 1 caused a difficulty for its polymerization into high molecular-weight products upon radical generation and only gave, if any, fragile nanowires that are easily washed away during development. By carefully looking at the images for 2–6, partial fragmentation of the nanowires was confirmed, implying the bond-breakage or loss of the polymerized products. Nevertheless, even for the thick (>1 μm) films prepared using the drop-cast method, obvious nanowires with ultrahigh aspect ratios were observed. Compared to our previous reports based on polymer materials,26–30 the obtained nanowires appeared flexible, while their diameters appeared small. Most likely due to this flexibility, entangled, bundled, and networked structures of nanowires were formed, which is more obvious in the SEM images (Fig. 4). Moreover, cross-sectional profiles in AFM revealed the remarkable uniformity of nanowire diameter that is characterized by the single particle-triggered intra-track reactions. For example, when scanning along the dotted line drawn in Fig. 3c, we can visualize the height, which agrees well for the single nanowires as well as two or three overlapped nanowires (Fig. 3g).
800 eV nm−1) at a low fluence, followed by development in hexane or toluene, nanowire objects consisting of aromatic derivatives were visualized via AFM, except for compound 1 (Fig. 3). We consider that the small molecular size of 1 caused a difficulty for its polymerization into high molecular-weight products upon radical generation and only gave, if any, fragile nanowires that are easily washed away during development. By carefully looking at the images for 2–6, partial fragmentation of the nanowires was confirmed, implying the bond-breakage or loss of the polymerized products. Nevertheless, even for the thick (>1 μm) films prepared using the drop-cast method, obvious nanowires with ultrahigh aspect ratios were observed. Compared to our previous reports based on polymer materials,26–30 the obtained nanowires appeared flexible, while their diameters appeared small. Most likely due to this flexibility, entangled, bundled, and networked structures of nanowires were formed, which is more obvious in the SEM images (Fig. 4). Moreover, cross-sectional profiles in AFM revealed the remarkable uniformity of nanowire diameter that is characterized by the single particle-triggered intra-track reactions. For example, when scanning along the dotted line drawn in Fig. 3c, we can visualize the height, which agrees well for the single nanowires as well as two or three overlapped nanowires (Fig. 3g).
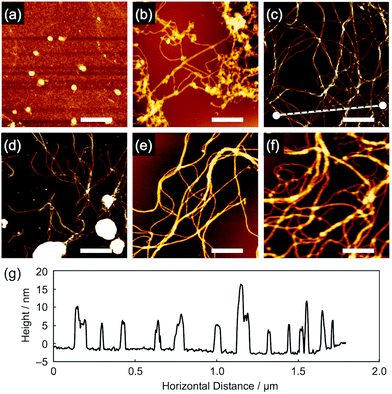 |
| | Fig. 3 AFM topographic images of the nanowires from drop-cast films of (a) 1, (b) 2, (c) 3, (d) 4, (e) 5 and (f) 6. The films were irradiated with 490 MeV 192Os30+ particles at the fluences of 1.0 × 109 and 2.0 × 108 ions cm−2 for (a–d, and f) and (e), respectively, and developed with n-hexane at r.t. for 1 and 60 °C for 2 and 4–6, and with n-hexane followed by toluene at r.t. for 3. Scale bars represent 500 nm. (g) Cross-sectional profile along the dotted line in (c). | |
 |
| | Fig. 4 SEM images of the nanowires from a drop-cast film of 5. The film was irradiated with 490 MeV 192Os30+ particles at the fluence of 1.0 × 109 ions cm−2 and developed with n-hexane followed by toluene. Scale bars represent 1 μm. | |
Then, we tried relatively lower LET ion beams to investigate the effect of given energy on nanowire formation. In a typical case, thin drop-cast films of 6 were irradiated with 150 MeV 107Ag11+ (LET: 9500 eV nm−1), 100 MeV 58Ni7+ (LET: 5500 eV nm−1), 60 MeV 28Si5+ (LET: 2100 eV nm−1), and 60 MeV 16O5+ (LET: 500 eV nm−1), and all of the respective films were developed in the same manner using n-hexane. As shown in Fig. 5, obvious differences were confirmed. We can recognize wavy nanowires in the case of Ag particle irradiation (Fig. 5a), while fragments of the nanowires were seen by means of Ni irradiation (Fig. 5b). In contrast, the lower-LET Si and O particle irradiations did not allow such objects, suggesting that high LET is required to obtain the stable nanowires.
 |
| | Fig. 5 AFM topographic images of the nanowires from drop-cast thin films of 6. The films were irradiated with (a) 150 MeV 107Ag11+, (b) 100 MeV 58Ni7+, (c) 60 MeV 28Si5+, and (d) 60 MeV 16O5+ particles at the fluence of 5.0 × 109 ions cm−2, and developed with n-hexane at room temperature. Scale bars represent 500 nm. | |
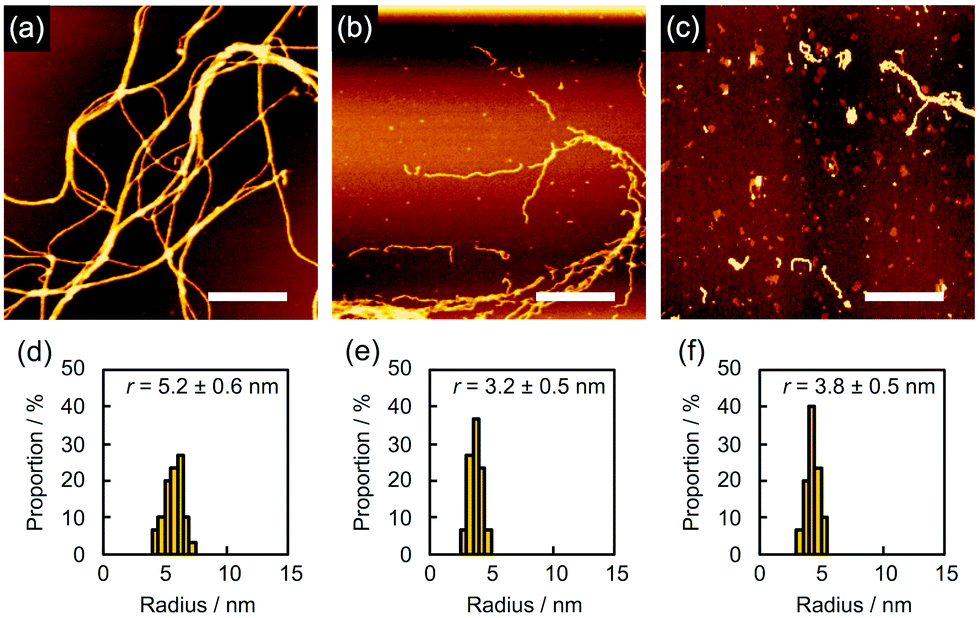
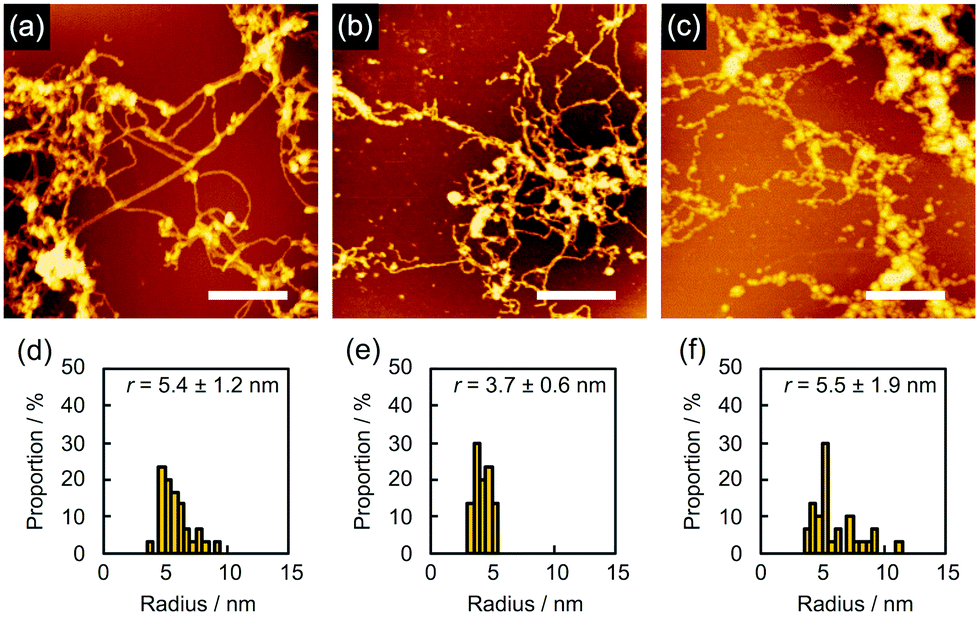
In order to find evidence that the nanowires were dominantly formed through not crosslinking but polymerization, we studied the effect of the solvents used for development on nanowire isolation. For example, as mentioned above, 5 gave bundled nanowires after treatment with n-hexane at 60 °C (Fig. 6a). When the identical sample was then immersed into CHCl3 for 10 s, dried in air, and subjected to AFM measurements, we discovered that the bundles of nanowires were mostly loosened and partially broken into wire-shaped fragments whose length was shorter than the initial thickness of the film (Fig. 6b). This observation clearly indicated that the nanowires were formed not only by crosslinking but also by entanglement of the polymerized products. On further development using CHCl3 for e.g. 2 min, we observed that the nanowires were partially washed away, suggesting the partial scission of the nanowire-substrate bonds (Fig. 6c). For quantitative analysis, the radius of the nanowires was evaluated via cross-sectional profiles in AFM. After the development process, the cross-section of the nanowires on the substrate was elliptically deformed as a consequence of adsorption forces. Taking this into account, the radius of nanowire is calculated by applying the ellipse model to the cross-section of the nanowire. The values of rx and ry are defined as the half-width and half-height of the cross-section of the nanowires, respectively. We defined the radius r as r = (rxry)1/2. Fig. 6d–f showed r distributions evaluated from cross-sectional profiles of the AFM images of the observed single nanowires. The averaged radius of n-hexane-treated nanowires was evaluated as 5.2 ± 0.6 nm, whereas those after development with CHCl3 for 10 s and an additional 2 min were 3.2 ± 0.5 and 3.8 ± 0.5 nm, respectively. The decrease in diameter implies the dissociation of polymerized products via solvation with CHCl3, which also resulted in the fragmentation of nanowires. In our previous reports of polymer-based nanowires, the nanowire diameter and morphology were not sensitive to the choice of solvents, and fragmentation behavior was not observed. Therefore, we concluded that the present nanowires from aromatic amine derivatives were mainly formed by the radiation-induced polymerized products. Another case of solvent-dependence was demonstrated for 2. Development with n-hexane afforded networked nanowires (Fig. 7a). The dispersed radius distribution of 5.4 ± 1.2 nm (Fig. 7d) indicated that non-developed small oligomers and monomers remained. Further development with toluene did not strongly affect the AFM images (Fig. 7b) but the averaged diameter of nanowires was decreased (Fig. 7e), suggesting the removal of the small oligomers and monomers. However, at the same time, the dissociation of polymeric products took place when immersing the nanowires into high solubility solvents. In fact, wavy and thin nanowires with considerable fragmentation were observed after further development with CHCl3 (Fig. 7c). In this case, the averaged radius was increased with dispersed distribution (Fig. 7f). We interpreted this phenomenon as a consequence of the dissociation of polymeric products by dissolution in solvent, which resulted in the observation of nanowires composed of loosely bound polymeric products.
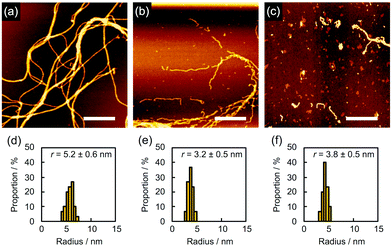 |
| | Fig. 6 (a–c) AFM topographic images and (d–f) the radius distributions of the nanowires from a drop-cast film of 5 isolated via sequential development. The film was irradiated with 490 MeV 192Os30+ particles at the fluence of 2.0 × 108 cm−2 and developed by (a and d) n-hexane at 60 °C, followed by (b and e) CHCl3 for 10 s and then (c and f) CHCl3 for 2 min. Scale bars represent 500 nm. | |
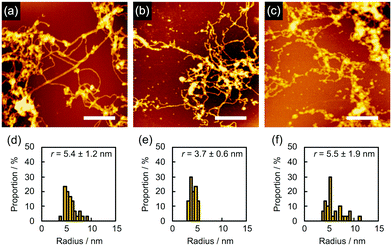 |
| | Fig. 7 (a–c) AFM topographic images and (d–f) the radius distributions of the nanowires from a drop-cast film of 2 isolated via sequential development. The film was irradiated with 490 MeV 192Os30+ particles at the fluence of 1.0 × 109 cm−2 and developed by (a and d) n-hexane, followed by (b and e) toluene and then (c and f) CHCl3 at room temperature. Scale bars represent 500 nm. | |
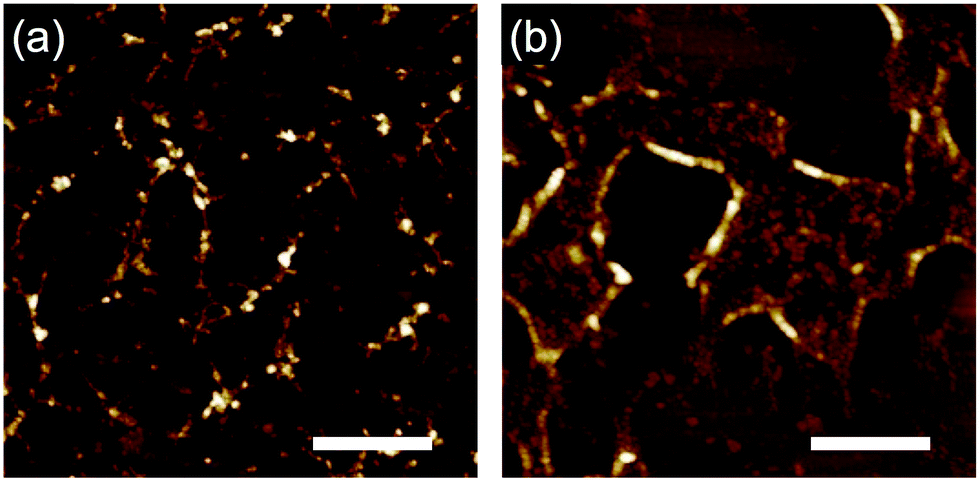
According to the observed range of r varying from 3 to 5 nm, the efficiency of the radiation induced polymerization reaction is assessed roughly based on the theoretical model of intra-track radial energy density.28,31,38 The derived G-value (number of propagation reaction steps) is G = 0.45 for r = 5 nm and LET ∼13800 eV nm−1 at the maximum, suggesting it is not in the range of efficient chain reactions.39 To gain insight into the mechanism of the polymerization reactions, electron paramagnetic resonance (EPR) spectroscopy of 3, 4 and 5 was measured (Fig. S1†) before and after the high-energy charged particle irradiation. As a result, we confirmed the presence of residual radical species for 3 and 5 even after irradiation and exposure to air. The g-values of the broad signal in the EPR spectra of 3 and 5 were around 2.0032, suggesting the presence of enough carbon centered neutral radicals and/or symmetric TPA radical cations. This is also confirmed by the analysis of the product using size exclusion chromatography (SEC), showing condensed (polymerized) products with longer wavelength absorption bands (Fig. S2†). The present results suggest that the generation of radical species and their coupling reactions initiate the polymerization reactions. From SEC charts, we can recognize that a large amount of monomeric compound remained in the nanowire filtrates (Fig. S2†). In addition, as exemplified in the SEC charts of 5 (Fig. S2†), some cleavage reactions also likely take place, giving smaller volume species than the monomeric compound. Dissociated phenyl groups and subsequent hydrogen extraction reactions are responsible for the fragmented molecules with absorption maxima at ∼270 nm. These results are suggestive of the major polymerization pathway; intermediate radical cations dimerize (propagate) with a neutral phenylamine molecule leading to benzidine structures and subsequent hydrogen dissociation.36,37,40 Now we recognize that aromatic amines are promising materials to afford uniform nanowires via the STLiP method. We are curious about the influence of the substitution patterns of methyl groups because triarylamines are known to react mostly at the para-position. In that sense, 3 and 4 have an identical tetraphenylbenzidine core but 3 is considered to be more reactive. As discussed above, 3 and 4 provided obvious nanowires by exposure to 490 MeV 192Os30+ particles followed by development with toluene (Fig. 3). On the other hand, when the samples were exposed to lower-LET 60 MeV 28Si5+ particles instead of 192Os30+, only 3 showed a sign of nanowire formation (Fig. 8). This implies the higher reactivity of 3 than 4. At the same time, since nanowire isolation is sensitive to the development processes, we did not obtain a clear answer at present.
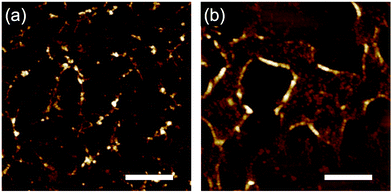 |
| | Fig. 8 AFM topographic images of the nanowires from drop-cast films of (a) 3 and (b) 4 isolated via development with toluene after irradiation with 60 MeV 28Si5+ particles at the fluence of 5.0 × 109 ions cm−2. Scale bars represent 500 nm. | |
Conclusions
We demonstrated a unique technique “Single Particle-Triggered Linear Polymerization (STLiP)” in the solid state, where high-energy ions deposit energy along their trajectories to generate active radicals for propagation reactions. The polymerized products were integrated into nanowires which were isolated via development with appropriate organic solvents. The experiments with a series of aromatic amine compounds suggested that a certain molecular size was required to obtain nanowire objects. By means of stepwise developments, we indirectly but certainly proved that the driving force for nanowire formation was not crosslinking but polymerization. Since the aromatic amines have been utilized for hole-transport materials in organic electronics,41,42 the evaluation of electrical conductivity along the single nanowires is an interesting subject worthy of investigation.
Acknowledgements
The authors are thankful to Ms. Manju Bala in IUAC for the help of ion irradiation experiments. This work was partly supported by a Grant-in-Aid for Scientific Research on Innovative Areas (no. 26102011) and Scientific Research (A) (no. 26249145) from the Japan Society for the Promotion of Science (JSPS), and a research grant from the Foundation for the Promotion of Ion Engineering.
Notes and references
- W. D. Coolidge, Science, 1925, 62, 441–442 CrossRef CAS PubMed.
- S. C. Lind and D. C. Bardwell, J. Am. Chem. Soc., 1926, 48, 1556–1575 CrossRef CAS.
-
D. Meisel, J. La Verne, Z. P. Zagorski, O. Gueven, J. M. Rosiak, T. Czvikovszky, M. Lavalle, A. J. Berejka and M. R. Cleland, Advances in radiation chemistry of polymers, IAEA, Vienna, 2004 Search PubMed.
-
O. Gueven, Advances in radiation chemistry of polymers, IAEA, Vienna, 2004, pp. 33–39 Search PubMed.
- P. Romiszowski and A. Kolinski, Polymer, 1982, 23, 1226–1229 CrossRef CAS.
- C. Baldock, Y. D. Deene, S. Doran, G. Ibbott, A. Jirasek, M. Lepage, K. B. McAuley, M. Oldham and L. J. Schreiner, Phys. Med. Biol., 2010, 55, R1–R63 CrossRef CAS PubMed.
- S. G. Vajapurkar and A. Bera, Indian J. Pure Appl. Phys., 2010, 48, 830–836 Search PubMed.
- A. Hiroki, S. Yamashita, Y. Sato, N. Nagasawa and M. Taguchi, J. Phys.: Conf. Ser., 2013, 444, 1–4 CrossRef.
- N. M. Claramma, N. M. Mathew and E. V. Thomas, Radiat. Phys. Chem., 1989, 33, 87–89 CrossRef CAS.
- M. M. Nasef and E. A. Hegazy, Prog. Polym. Sci., 2004, 29, 499–561 CrossRef CAS.
- V. Y. Kabanov, R. E. Aliev and N. Kudryavtsev, Radiat. Phys. Chem., 1990, 37, 175–192 CrossRef.
- A. Charlesby, Rep. Prog. Phys., 1965, 28, 463–518 CrossRef.
- M. J. Bowden, J. H. O'Donnell and R. D. Sothman, Macromolecules, 1971, 5, 269–274 CrossRef.
- K. Tajima and T. Aida, Chem. Commun., 2000, 24, 2399–2412 RSC.
- A. Usanmaz and E. Altürk, J. Macromol. Sci., Part A: Pure Appl. Chem., 2007, 39, 379–395 CrossRef.
- G. C. Eastmond, E. Haigh and B. Taylor, Trans. Faraday Soc., 1969, 65, 2497–2502 RSC.
- B. S. Elman, M. K. Thakur and R. J. Seymour, Radiat. Eff., 1986, 98, 139–149 CrossRef CAS.
- M. Hasegawa and Y. Suzuki, J. Polym. Sci., Part B: Polym. Chem., 1967, 5, 813–815 CrossRef CAS.
- G. Wegner, Z. Naturforsch., B: J. Chem. Sci., 1969, 24, 824–832 CAS.
- H. Bassler, Adv. Polym. Sci., 1984, 63, 1–48 CrossRef.
- L. Dou, Y. Zheng, X. Shen, G. Wu, K. Fields, W. Hsu, H. Zhou, Y. Yang and F. Wudl, Science, 2014, 347, 272–277 CrossRef PubMed.
- A. J. Melveger and R. H. Baughman, J. Polym. Sci., Polym. Phys. Ed., 1973, 11, 603–619 CrossRef CAS.
-
J. L. Magee and A. Chattarjee, in Kinetics of Nonhomogeneous Processes, ed G. R. Freeman, Wiley, New York, 1987, ch. 4, pp. 171–214 Search PubMed.
- J. A. LaVerne and R. H. Schuler, J. Phys. Chem., 1994, 98, 4043 CrossRef CAS.
- E. Kobetich and R. Katz, Phys. Rev., 1968, 170, 391–396 CrossRef CAS.
- S. Seki, K. Maeda, S. Tagawa, H. Kudoh, M. Sugimoto, Y. Morita and H. Shibata, Adv. Mater., 2001, 13, 1663–1665 CrossRef CAS.
- S. Seki, S. Tsukuda, K. Maeda, Y. Matsui, A. Saeki and S. Tagawa, Phys. Rev. B: Condens. Matter, 2004, 70, 144203 CrossRef.
- S. Seki, S. Tsukuda, K. Maeda, S. Tagawa, H. Shibata, M. Sugimoto, K. Jimbo, I. Hashitomi and A. Kohyama, Macromolecules, 2005, 38, 10164–10170 CrossRef CAS.
- M. Omichi, H. Marui, K. Takano, S. Tsukuda, M. Sugimoto, S. Kuwabata and S. Seki, ACS Appl. Mater. Interfaces, 2012, 4, 5492–5497 Search PubMed.
- M. Omichi, A. Asano, S. Tsukuda, K. Takano, M. Sugimoto, A. Saeki, D. Sakamaki, A. Onoda, T. Hayashi and S. Seki, Nat. Commun., 2014, 5, 3718 Search PubMed.
- Y. Maeyoshi, A. Saeki, S. Suwa, M. Omichi, H. Marui, A. Asano, S. Tsukuda, M. Sugimoto, A. Kishimura, K. Kataoka and S. Seki, Sci. Rep., 2012, 2, 600 CrossRef PubMed.
- A. Kumar, D. K. Avasthi, A. Tripathi, D. Kabiraj, F. Singh and J. C. Pivin, J. Appl. Phys., 2007, 101, 014308 CrossRef.
- R. Singhal, A. Tripathi and D. K. Avasthi, Adv. Mater. Lett., 2013, 4, 413–417 CrossRef.
- Y. Takeshita, T. Sakurai, A. Asano, K. Takano, M. Omichi, M. Sugimoto and S. Seki, Adv. Mater. Lett., 2015, 6, 99–103 CrossRef CAS.
- T. Yamamoto, M. Nishiyama and Y. Koie, Tetrahedron Lett., 1998, 39, 2367–2370 CrossRef CAS.
- K. Sreenath, C. V. Suneesh, V. K. R. Kumar and K. R. Gopidas, J. Org. Chem., 2008, 73, 3245–3251 CrossRef CAS PubMed.
- K. Sreenath, C. V. Suneesh, K. R. Gopidas and R. A. Flowers, J. Phys. Chem. A, 2009, 113, 6477–6483 CrossRef CAS PubMed.
- T. Washin, K. Enomoto, T. Sakurai, V. S. Padalkar, H. L. Cheng, M. T. Tang, A. Horio, D. Sakamaki, M. Omichi, A. Saeki, K. Kikuchi, Y. Hori, A. Chiba, Y. Saito, T. Kamiya, M. Sugimoto and S. Seki, ACS Sens., 2016, 1, 766–774 CrossRef.
- A. M. Funston and J. R. Miller, Radiat. Phys. Chem., 2005, 72, 601 CrossRef CAS.
- L. S. Marcoux, R. N. Adams and S. W. Feldberg, J. Phys. Chem., 1969, 73, 2611 CrossRef CAS.
- Y. Shirota and H. Kageyama, Chem. Rev., 2007, 107, 953–1010 CrossRef CAS PubMed.
- Z. Ning and H. Tian, Chem. Commun., 2009, 45, 5483–5495 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6nr03297d |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a
*a