
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modulating the thermal properties of poly(hydroxybutyrate) by the copolymerization of rac-β-butyrolactone with lactide†
Jenny
Fagerland
a,
Anna
Finne-Wistrand
a and
Daniela
Pappalardo
*ab
aFiber and Polymer Technology, KTH Royal Institute of Technology, Stockholm, Sweden
bDipartimento di Scienze e Tecnologie, via dei Mulini 59/A, 82100 Benevento, Italy. E-mail: pappalardo@unisannio.it
First published on 7th July 2016
Abstract
Biobased poly(hydroxybutyrate) is produced by microorganisms under controlled conditions. It is a linear, high molecular weight, fully isotactic and highly crystalline polymer. However, it has poor mechanical and thermal properties. We have modulated the thermal properties of this material by ring-opening co-polymerization of rac-β-butyrolactone (BL) with lactide (LA) in the presence of salan-based yttrium and aluminum catalysts. The prepared poly(hydroxybutyrate-co-lactide) copolymers were characterized by proton and carbon nuclear magnetic resonance (1H and 13C NMR), size exclusion chromatography (SEC) and differential scanning calorimetry (DSC) analyses. The salan-yttrium compound was a more effective catalyst compared to the aluminum compound, affording high molecular weight copolymers with higher monomer conversion and a monomodal distribution of the molecular weights. The kinetic experiments showed a higher rate of polymerization for the LA with respect to the BL. The copolymers were amorphous and DSC showed unique transition temperatures for all of the samples. The formation of a gradient copolymer is proposed.
Introduction
Aliphatic polyesters are currently attracting growing scientific interest as biodegradable and biocompatible materials, and they are finding large applications in various fields, including agricultural, pharmaceutical and biomedical fields.Poly(hydroxybutyrate) (PHB) belongs to the family of thermoplastic polyesters, and the commercially available PHB is produced by bacteria through the fermentation of carbohydrates under controlled conditions. PHB is a linear, high molecular weight, fully isotactic and highly crystalline polymer, which has mechanical and physical properties similar to some commodity polymers, i.e., poly(ethylene terephthalate).1 Although poorly studied for biomedical applications because it is biodegradable, PHB has high potential to be integrated and processed within tissues.2,3 The major drawbacks of PHB are its poor mechanical properties, low solubility in some common solvents, slow degradation rate, high cost associated with the bacterial production, and melting temperature, which is too close to the thermal degradation temperature. As a consequence, recent efforts have been devoted to developing efficient chemical approaches, as alternatives to the bacterial approach, allowing for control of the thermal and mechanical properties of the material by modulating the chain microstructure. The ring-opening polymerization (ROP) of cyclic esters, such as lactone, lactides and glycolide, in the presence of metal-based initiators, represents the most convenient method for the preparation of aliphatic poly(ester)s with controlled and predictable molecular weights, stereochemistry, and end groups.4 The preparation of atactic, isotactic-enriched and syndiotactic-enriched PHBs has also been achieved by the ROP of commercially available racemic β-butyrolactone (BL) in the presence of the appropriate catalysts or initiators.5 In this regard, we described yttrium amido complexes bearing quadridentate binaphthyl-bridged Schiff-base salen-like and diamine bisphenolate salan-like ligands affording syndiotactic-enriched PHB, which showed modulated thermal properties depending on the degree of stereoregularity of the polymeric chains.6 Thomas et al. defined a straightforward synthesis of efficient syndiospecific yttrium salan-based catalysts by the in situ mixing of yttrium(III) isopropoxide and the salan ligand.7
The copolymerization of different monomers offers a way to match different polymeric materials. The synthesis of copolymers of various molecular weights, compositions and architectures has led to the ability to enlarge the library of new materials with unique properties. In addition to the control of the stereochemical microstructure, the copolymerization represents another way to control the physical properties of the polymeric materials. By varying the composition, monomer sequencing, and molecular weight, the polymer microstructure can be tailored for specific applications, where a proper balance between the mechanical properties, thermal properties and degradation rate is needed. For example, the properties of biodegradable and biocompatible poly(lactide) (PLA), which has found several applications in the pharmaceutical, medical, and agricultural fields,1 have been modulated by the copolymerization of the monomer, LA, with several cyclic esters, such as caprolactone, valerolactone and glycolide.4b Because PLA has good mechanical and thermal properties as well as higher degradation rates with respect to PHB, the ring-opening co-polymerization of BL with LA may be a suitable and promising approach to modulate the poly(hydroxybutyrate) microstructure and, as a consequence, the thermal properties.
Examples of the copolymerization of LA with BL are rare.8 Copolymerization of LA with BL has been described in the presence of catalysts based on main group metals. The ring-opening copolymerization of BL with (S,S)-lactide was reported in the presence of stannoxane catalysts.8a Aluminum-based catalysts, bearing salan and salen ligands, have also been described for BL/L-LA copolymerization.8b,c The synthesis of related block-copolymers by one-pot cationic polymerization has been reported.8d Star-shaped and triblock PHB/PLA copolymers were synthetized with dinuclear indium catalysts.8e
An interesting alternative for the BL/LA copolymerization could be the use of yttrium complexes, which are among the most efficient catalysts for the ROP of cyclic esters as well as for the polymerization of BL to high molecular weight, syndiotactic-enriched PHB.5a–c,6,7 Herein, we describe the use of a salan-based yttrium isopropoxide catalyst, previously synthetized by Thomas et al.7 and the analogous aluminum complex bearing the same tetradentate ligand in the copolymerization of BL and LA. Interestingly, yttrium complexes stabilized by tetradentate salan-type ligands have never been used in the copolymerization of BL with LA. The synthesis and characterization of copolymers of BL with LA in the presence of these catalysts are described and discussed.
Experimental
Materials
Unless otherwise stated, all reactions were performed under standard Schlenk conditions, using a Schlenk line or a glovebox.The salan ligand N,N′-dimethyl-N,N′-bis[(3,5-di-t-butyl-2-hydroxyphenyl)methylene]-1,2-diaminoethane9 and the yttrium-salan compound7 have been synthesized as previously described.
Aluminum isopropoxide was purchased from Sigma-Aldrich and used as received. rac-β-Butyrolactone (BL) (98%, Sigma Aldrich) was stirred above calcium hydride for at least 12 hours, then distilled in vacuo and stored above 3 Å molecular sieves inside a glovebox. Toluene (HPLC grade, Fisher) was stirred above calcium hydride for at least 48 hours, distilled from calcium hydride under a nitrogen atmosphere, and then stored above 3 Å molecular sieves. The NMR-solvent chloroform-d (Cambridge Isotope Laboratories) was used as received. Methanol (technical, VWR), n-heptane (99%, Fisher) and dichloromethane (HPLC grade, Fisher) were also used as received.
Methods
Nuclear magnetic resonance (NMR) spectroscopy was performed using a BRUKER 400 Ultrashield™ instrument with an autosampler.For analysis of the tacticity, 13C NMR spectroscopy was carried out with either the precipitated or the crude product and the characteristic peaks for the triads of the carbonyl group (∼169 ppm) and the methylene group (∼40 ppm) as well as for the diad of the methyl group (∼20 ppm) were studied.
Size exclusion chromatography (SEC) was performed using a Polymer Laboratory PL-GPC 50 Plus. Chloroform with toluene as an internal standard was used as the solvent.
Thermal analysis of the polymers was performed using a METTLER TOLEDO Differential scanning calorimeter (DSC) with an autosampler under a nitrogen flow. The analysis program is as follows: heating from −20 °C to 220 °C at a heating rate of 10 K min−1, holding at 220 °C for two minutes, cooling to −20 °C at a cooling rate of 10 K min−1, holding at −20 °C for two minutes and reheating to 220 °C at a heating rate of 10 K min−1. Due to the degradation of PHB at temperatures above 180 °C, only results from first heating were considered.
Elemental analysis of the aluminum compound was performed on a Thermo Finningan Flash EA 1112 series C, H, N analyzer in the microanalytical laboratory of the Chemistry and Biology Department of the University of Salerno.
The MALDI mass spectra were recorded in reflector mode using a 4800 MALDI TOF/TOF™ Analyzer (Applied Biosystem, Framingham, MA, USA), equipped with a Nd:YAG laser (λ = 355 nm) and working in positive ion mode. This MALDI TOF instrument is equipped with a laser with a wavelength of <500 ps pulse and 200 Hz repetition rate. The laser irradiance was set slightly above the threshold. 2-(4-Hydroxyphenylazo)benzoic acid (HABA) (0.1 M in THF) was used as a matrix. Appropriate volumes of polymer solution (5–10 mg mL−1 THF) and matrix solution were mixed in order to obtain 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1:2 and 1:3 ratios (sample/matrix v/v). 2 μL of each sample/matrix mixture was spotted on the MALDI sample holder and slowly dried to allow matrix crystallization. The resolution of the MALDI spectra is about 8000 (FWHM), and the mass accuracy was 100–150 ppm for masses in the range of 1000–2000 Da.
:1, 1:2 and 1:3 ratios (sample/matrix v/v). 2 μL of each sample/matrix mixture was spotted on the MALDI sample holder and slowly dried to allow matrix crystallization. The resolution of the MALDI spectra is about 8000 (FWHM), and the mass accuracy was 100–150 ppm for masses in the range of 1000–2000 Da.
Synthesis of the aluminum-salan compound
The salan ligand, N,N′-dimethyl-N,N′-bis[(3,5-di-t-butyl-2-hydroxyphenyl)methylene]-1,2-diaminoethane, was synthesized by a Mannich condensation from N,N′-dimethylethylenediamine (99%), formaldehyde (37 wt% in water) and 2,4-ditertbutyl phenol as previously described (99%).9In a glovebox, a Schlenk flask was charged with 524.8 mg of the salan ligand (1 mmol, 1 eq.), 204.3 mg of aluminum(III) isopropoxide (1 mmol, 1 eq.) and 10 mL of dry toluene. The reaction mixture was stirred at 80 °C for six days. After cooling to room temperature, the solvent was evaporated. The product was dried in vacuo and stored inside the glovebox. The product was characterized by proton and carbon nuclear magnetic resonance (1H and 13C NMR) and elemental analyses.
1H NMR (400 MHz, chloroform-d) δ 7.24 (d, J = 2.4 Hz, 2H), 6.84 (d, J = 2.6 Hz, 2H), 6.74 (d, J = 2.6 Hz, 2H), 6.72 (d, J = 2.6 Hz, 2H) Ar-H; 4.85 (d, J = 2.6 Hz, 2H, ArCH2N); 4.44 (m, J = 5.8 Hz, 2H, OCH(CH3)2); 4.26 (d, J = 12.9 Hz, 2H, ArCH2N); 3.98 (m, J = 5.8 Hz, 1H, OCH(CH3)2); 3.95 (d, J = 12.4 Hz, 1H), 3.54 (d, J = 12.3 Hz, 1H), 3.48 (d, J = 13.0 Hz, 1H), 3.41 (d, J = 13.4 Hz, 2H) ArCH2N; 3.04–2.73 (complex m, broad, 4 H) CH2CH2; 2.54 (s, 6H), 2.53 (s, 3H), 2.38 (s, 3H), NCH3; 1.53 (s, 9H), 1.51 (s, 9 H), 1.40 (s, 9 H), 1.31 (s, 9 H)1.30 (s, 18H), 1.28 (s, 18H), C(CH3)3; 1.05 (d, J = 5.9 Hz, 12H), 0.95 (d, J = 5.9 Hz, 3H), 0.83 (d, J = 5.9 Hz, 3H), OCH(CH3)2.
13C NMR (75 MHz, chloroform-d) δ 157.04, 156.47, 156.11 (ArC-O); 138.31, 138.22, 138.02, 137.76, 137.33, 136.49 (ArC-CMe3); 123.80, 123.65, 123.58, 123.37, 123.13, 122.78 (ArCH); 121.26, 119.80, 118, 48 (ArCipso); 64.79, 64.29, 62.77 (br) (CH2); 62.24, 62.07 (OCH(CH3)2); 60.84, 56.47, 54.17 (br), 52.22 (CH2); 46.10, 45.14, 44.25 (NCH3); 35.33, 35.06, 34.86, 34.03, 33.95, 33.93 (C(CH3)3); 31.83, 31.78, 29.96, 29.92, 29.87, 29.72 (C(CH3)3); 28.15, 27.91, 27.30 (OCH(CH3)2).
Anal. calcd for C37H61AlN2O3 C, 72,99; H, 10,1; N, 4,60. Found C 73,45; H, 10,67; N, 4,43.
Polymerization tests
In a glove box, selected amounts of BL, LA, toluene and catalyst were added into a glass vial equipped with a magnetic stirrer and sealed with a screw-on lid. The vial was then poured into an oil bath and stirred at the desired temperature. After the desired time, the reaction was quenched by the addition of dichloromethane. The product was precipitated in heptane, filtered and dried in a vacuum oven.Kinetics analysis
The BL, L-lactide (L-LA), toluene and the catalyst were added into a screw vial inside the glove box. The mixture was stirred at 25 °C. A single drop of the crude product was taken at definite time intervals, poured into CDCl3 and analyzed by 1H NMR. The reaction conditions are as follows: [Y] = 3 M, [LA]:[BL]:[Y] = 90:90:1, toluene as solvent, and T = 25 °C. The same sets of experiments were performed in the presence of D-lactide (D-LA).
Results and discussion
Synthesis of the catalysts
Aluminum alkyl or alkoxide derivatives stabilized by salen- or salan-type ligands have been largely described for the stereocontrolled polymerization of rac-lactide.10 The catalytic behavior of the aluminum complexes bearing tetradentate ligands strongly depended on the architecture of the ancillary ligand. In detail, besides isotactic PLA obtained from enantiomerically pure L- or D-LA, the preparations of heterotactic,10a syndiotactic,10b stereo-block isotactic10c–e and stereo-gradient isotactic10f,g PLAs have been described.Notably, Gibson et al. noticed a remarkable range of microstructures achievable by changing the steric bulk at the ortho position of the phenolate ligands. Moreover, when the size of the phenoxide alkyl substituent increases (from H to Me to tBu), the rate of polymerization of the LA decreases.10
Given the generally higher reactivity of LA with respect to that of BL in their copolymerization,8 we reasoned that the presence of bulkier substituents in the ortho positions of the phenolate ligand, thus disfavoring the reaction of LA, would probably favor the copolymerization of the two monomers. A similar conceptual approach was successfully pursued by Nomura et al. in the ε-caprolactone/lactide copolymerization in the presence of salen-aluminum based complexes.11 With this in mind, a salan ligand bearing tert-butyl groups at the ortho positions of the phenolate ring was selected and used for the preparation of the aluminum isopropoxide and yttrium isopropoxide derivatives.
With respect to the aluminum derivatives, yttrium complexes stabilized by tetradentate salen or salan-type ligands have been far less explored12 and have never been used in the copolymerization of BL with LA. The synthesis of the salan-yttrium compound (1) was performed as previously reported.7
The synthesis of the salan-aluminum compound (2) was performed by the stoichiometric reaction of the N,N′-dimethyl-N,N′-bis[(3,5-di-t-butyl-2-hydroxyphenyl)methylene]-1,2-diaminoethane-proligand (Scheme 1) with aluminum isopropoxide in toluene at 80 °C for 6 days.
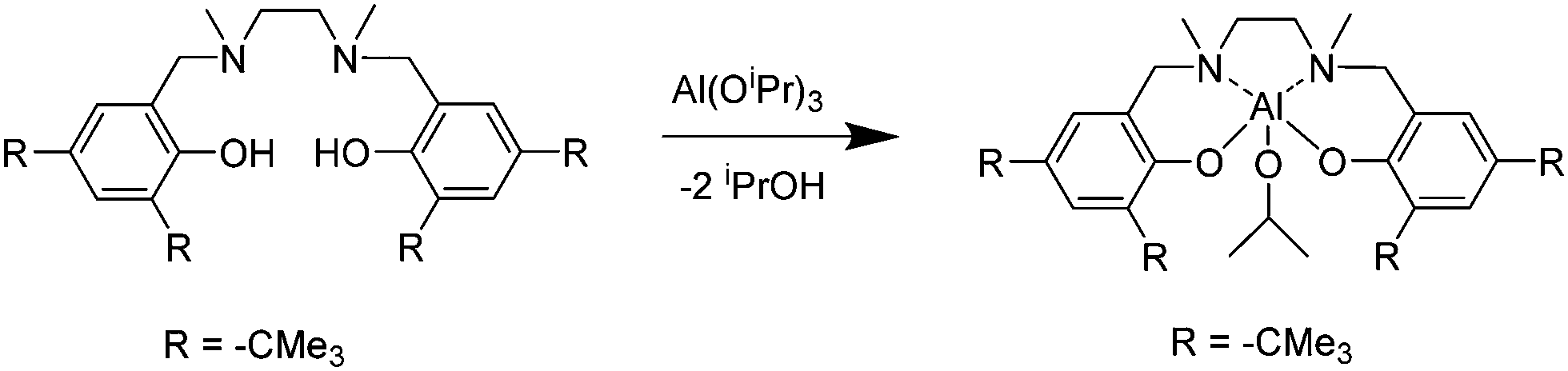 | ||
| Scheme 1 Synthesis of the salan-aluminum compound (2). | ||
The complex was obtained as a white solid in a high yield, and was characterized by 1H, 13C NMR, and bi-dimensional NOESY NMR analyses. The 1H NMR spectrum of complex (2) registered at 298 K in CDCl3 showed the presence of broad signals, probably as a consequence of the flexible nature of the salan ligand and/or the occurrence of fluxional phenomena in solution (Fig. 1).
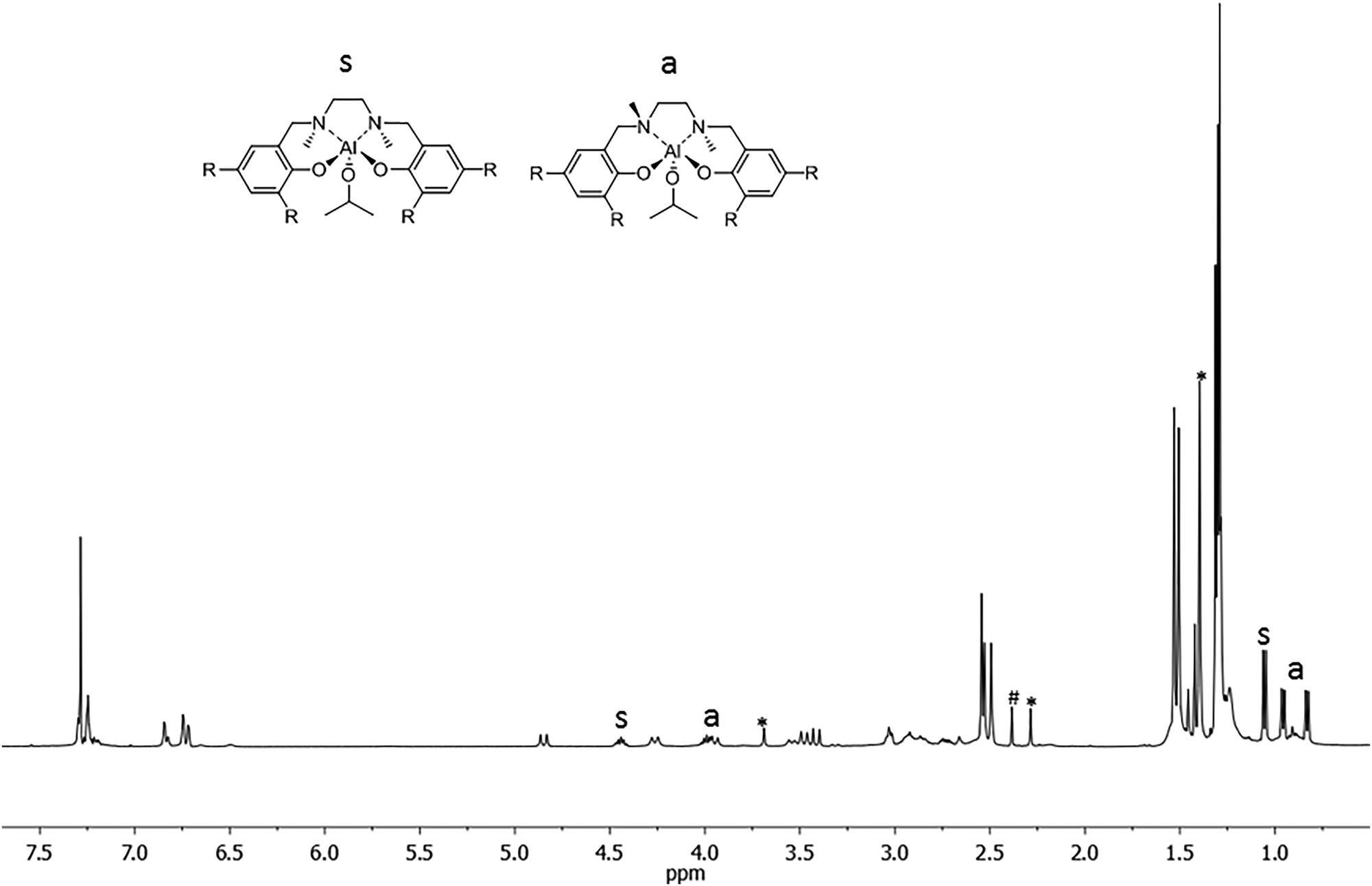 | ||
| Fig. 1 1H NMR spectrum (CDCl3, 25 °C) of the salan-aluminum compound (2). Marked signals are due to the free ligand (*) and solvent (#). | ||
The 1H NMR spectrum showed the presence of at least two species in approximately a 1:1 ratio. Indeed two methine protons for the isopropoxide group (Al–OCHMe2) appeared as septets at δ = 4.44 and 3.98 ppm; the corresponding methyl groups appeared as one doublet at δ = 1.05 ppm and as a double of doublets at δ = 0.95 ppm and 0.83 ppm, respectively. The latter pattern indicated that the two methyl groups are not equivalent and that the ligand could be wrapped around the aluminum in a non-symmetrical way. Of the two species observed, one is symmetric (s), and the other is asymmetric (a). Confirmation of this hypothesis comes from the inspection of the spectrum: indeed one singlet for the methyl groups on the nitrogen (NMe) at δ = 2.54 ppm appeared for the symmetric species s, while two singlets at δ = 2.53 ppm and at δ = 2.49 ppm appeared for the asymmetric species a. Moreover, 4 signals account for the tert-butyl groups in the a species, while just 2 are found in the symmetric species a. Notably the ratio of the integrals between the total OCH(CH3)2 groups and the total NMe groups, indicated a 1:1 ligand:isoproxide molar ratio.
After coordination of the ligand to the aluminum center, the phenoxymethylene protons ArCH2N became diastereomeric and appeared as broad doublets at δ = 4.85, 4.26, 3.95, 3.54, 3.48 and 3.41 ppm. The flexible nature of the salan ligand framework may be the origin of the presence of at least two species, in fluxional equilibrium, in solution.
In the NMR spectrum of the sample the presence of resonances due to the “free ligand” is observed and it accounts for less than 4% in weight of the sample. The formation of the free ligand might be due to adventitious water.
The 13C NMR characterization of compound 2, also supported by DEPT135 NMR analysis, confirmed the presence of two species (see ESI†). Indeed the number of signals observed in the 13C NMR spectrum (i.e. 2 signals for the methine carbon isopropoxide Al–OCHMe2, 3 signals for the methyl groups on the isopropoxide, 3 signals for the NMe carbon, the aromatic carbon pattern) is consistent with the above attribution.
To gain more insight into the nature of these species, a 1H–1H NOESY spectrum was registered at room temperature (Fig. 2). In the NOESY spectrum, the red cross-peaks (in opposite phase with the diagonal) indicated spatial correlation. The positive blue cross-peaks, with the same phase as the diagonal, indicated chemical exchange processes instead.
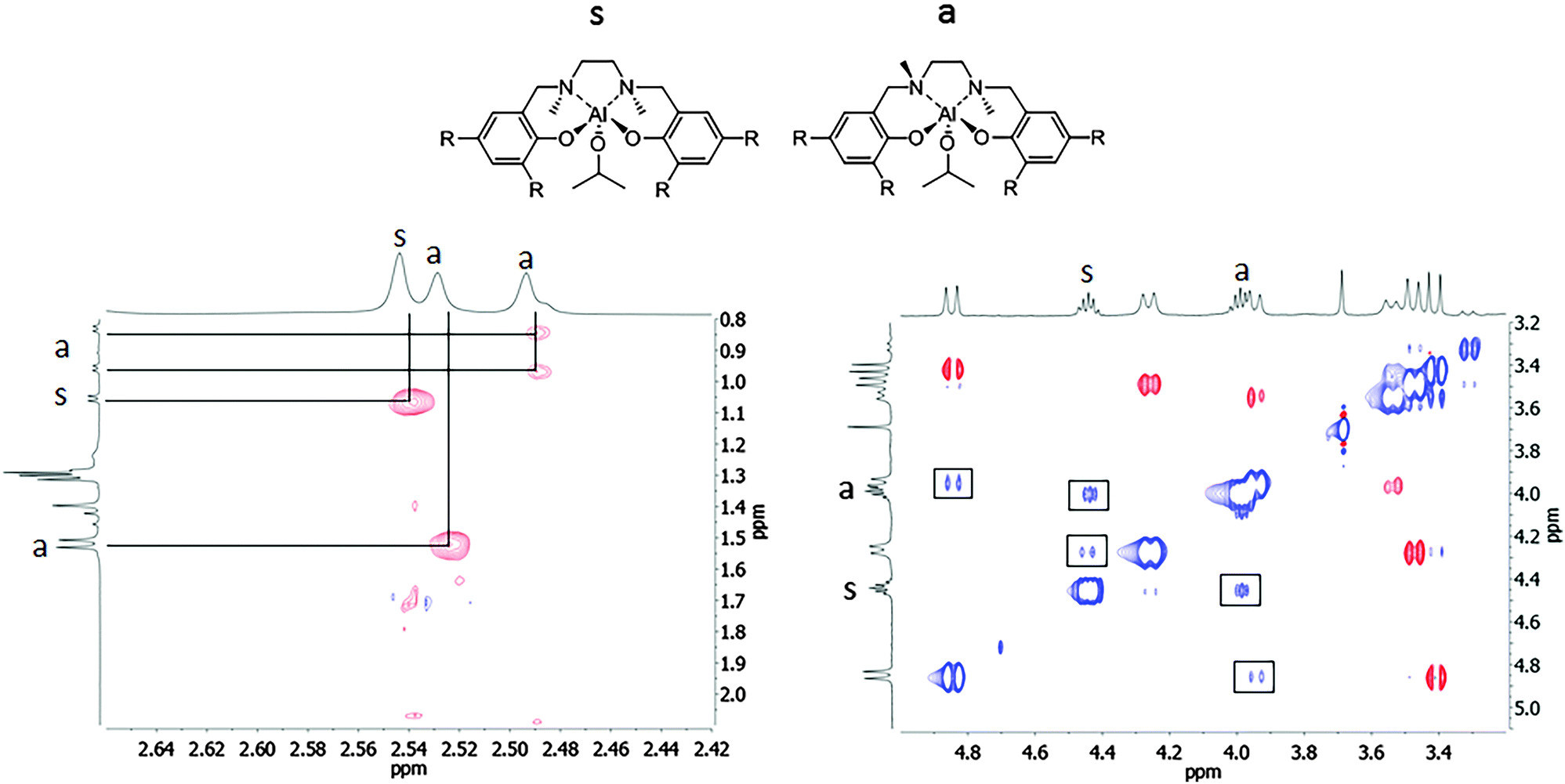 | ||
| Fig. 2 1H–1H NOESY spectrum of the salan-aluminum compound (2) in CDCl3 at 25 °C. The cross-peaks in phase with the diagonal are indicated with a square. | ||
For the symmetric species, a NOE effect was observed between the NMe protons at 2.54 ppm and the singlet Al–OCHMe2 protons at 1.05 ppm (Fig. 2, left), thus indicating that the two equivalent methyl groups on the nitrogen atoms are in a pseudo-cis geometry pointing toward the isoproxide group. In contrast, in the asymmetric species a two NMe2 signals appeared; the NMe protons at 2.49 ppm showed a NOE effect with the two non-equivalent methyl groups in the Al–OCHMe2 (at 0.95 and 0.83 ppm). The other NMe protons at 2.49 ppm showed a NOE effect with a tert-butyl group on the phenoxide ring. This clearly indicated the pseudo-trans positions of the methyl groups on the nitrogen atoms in the species a. Moreover, positive cross-peaks, with the same phase as the diagonal, also arise. In detail, positive cross-peaks are observed between the different ArCH2N protons as well as between the methine protons of the isopropoxide group Al–OCHMe2 (Fig. 2, right). It is reasonable to hypothesize that a chemical exchange, which may involve alkoxy-bridged species, is occurring in solution.
Copolymerization of rac-β-butyrolactone and lactide
Yttrium complexes are among the most efficient catalysts for the ROP of BL with high molecular weight PHB.5a–c,6,7As reported in the literature, the salan-yttrium based catalyst (1) produced syndiotactic enriched PHB (Scheme 2).7
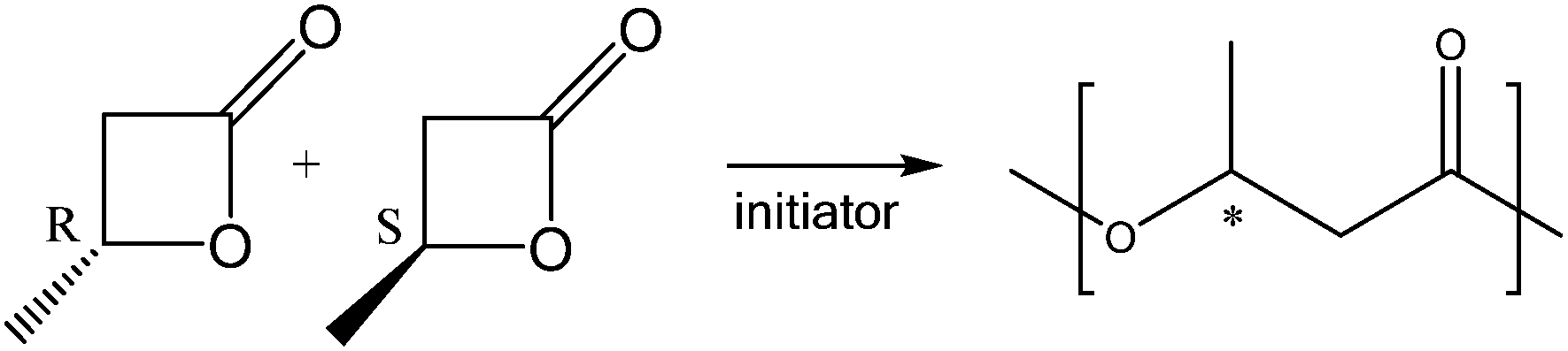 | ||
| Scheme 2 ROP of rac-β-butyrolactone. | ||
The ROP of BL in the presence of the salan-aluminum compound (2) was assessed in a toluene solution under different experimental conditions, by systematically changing the temperature, time, amount of solvent, and monomer/catalyst molar ratio. The products were characterized by NMR, SEC and DSC analysis. The salan-aluminum compound (2) catalyzes the ROP of BL with PHB, with a conversion of 72% in 70 hours at 70 °C. The polymer was atactic, as determined by the 13C NMR analysis in the carbonyl and methylene regions (see ESI†). Both in the methylene and in the carbonyl regions, the four resonances attributed, according to the literature, to the triads rm, mm, rr, and mr showed roughly equally intensities. The probability of racemic linkages between monomer units was Pr = 0.54.13
Copolymerization of BL with LA was performed in the presence of the salan-based-yttrium and aluminum compounds (1 and 2), by varying the polymerization conditions (Scheme 3).
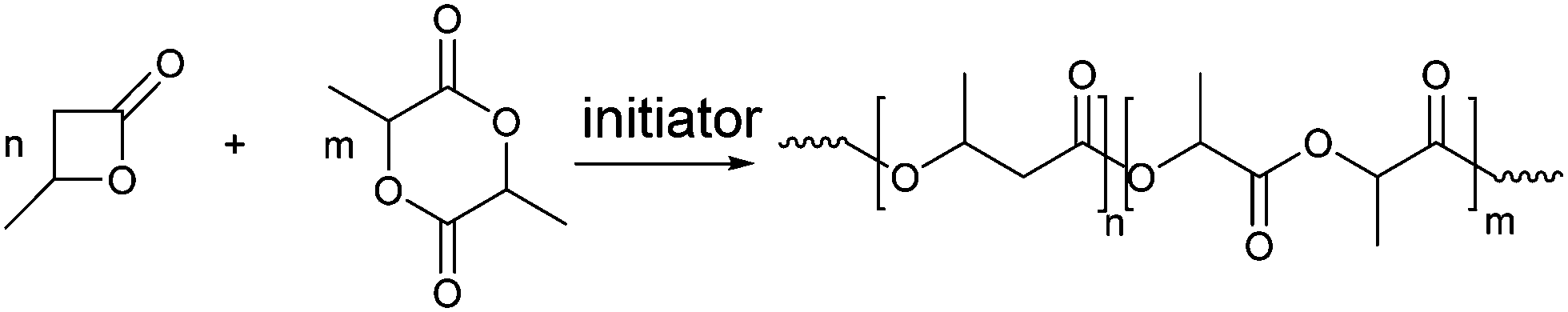 | ||
| Scheme 3 Synthesis of poly(3-hydroxybutyrate)-co-poly(lactide). | ||
The polymers were characterized by 1H and 13C NMR, SEC and DSC. The results of the NMR and SEC characterization are shown in Table 1.
| Entrya | L-LA, D-LA, rac-LA | T [°C] | Cat [×10−2 mol L−1] | Molar feed ratio [mol] | Conversionb [% in mol] | Compositionb [% in mol] | Hetero-sequencesb [% in mol] | Mntheo [g mol−1] | SEC characterization | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cat | BL | LA | BL | LA | BL | LA | Mn | Đ | ||||||
| a Polymerization conditions: catalyst = salan-yttrium (1); solvent = toluene; and time = 2 hours. b The time was 4 hours. The conversion and composition were determined by 1H-NMR analysis, while the heterosequences, i.e. the percent mol ratio of [BL–LA]/[BL–BL + BL–LA], by 1H and 13C NMR analysis. c The time was 144 hours and the catalyst was salan-aluminum (2). | ||||||||||||||
| 1 | L | 70 | 6.3 | 1 | 93 | 91 | 66 | 99 | 45 | 55 | 57 | 18525 |
16668 |
1.50 |
| 2 | L | 20 | 5.9 | 1 | 78 | 80 | 57 | 99 | 48 | 52 | 31 | 15228 |
24160 |
1.26 |
| 3 | L | 50 | 6.0 | 1 | 89 | 92 | 58 | 99 | 34 | 66 | 43 | 17555 |
18826 |
1.65 |
| 4 | L | 50 | 3.0 | 1 | 108 | 111 | 72 | 99 | 47 | 53 | 45 | 22512 |
24212 |
1.27 |
| 5 | L | 50 | 1.5 | 1 | 109 | 104 | 33 | 99 | 20 | 80 | 85 | 17920 |
35338 |
1.08 |
| 6 | L | 20 | 1.5 | 1 | 89 | 93 | 33 | 99 | 39 | 61 | 34 | 15784 |
29329 |
1.12 |
| 7 | L | 50 | 3.1 | 1 | 51 | 134 | 62 | 99 | 28 | 72 | 58 | 21822 |
47528 |
1.11 |
| 8 | L | 50 | 3.1 | 1 | 111 | 41 | 26 | 99 | 44 | 56 | 51 | 8327 | 18653 |
1.09 |
| 9b | L | 50 | 2.7 | 1 | 207 | 239 | 20 | 99 | 17 | 83 | 95 | 37632 |
43073 |
1.09 |
| 10 | rac | 50 | 3.0 | 1 | 88 | 88 | 66 | 99 | 37 | 63 | 35 | 17540 |
28004 |
1.20 |
| 11 | D | 50 | 3.0 | 1 | 74 | 74 | 79 | 99 | 45 | 55 | 40 | 15577 |
20129 |
1.24 |
| 12c | L | 70 | 5.8 | 2 | 82 | 89 | 43 | 51 | 40 | 60 | 56 | 9569 | 9891 | 1.05 |
| 13 | L | 20 | 3.0 | 1 | 81 | 37 | 88 | 99 | 64 | 36 | 20 | 11404 |
16462 |
1.09 |
Polymerization in the presence of salan-yttrium proceeded with a good yield in 2 hours even at 20 °C, with almost complete conversion of LA (99%). Reactions with the salan-aluminum compound, instead, were very slow, and the conversion of both monomers was approximately 50% after six days. The conversion of BL depended on the amount of salan-yttrium catalyst being used; when the concentration was 1.5 mol L−1, the conversion was never higher than 39%, but the conversion increased as the catalyst concentration increased. The average molecular weights of the copolymers, measured by SEC, were also strongly dependent on the catalyst. Monomodal distributions were always observed. When salan-yttrium was used, molecular weights from 16000 to 48000 g mol−1 were obtained with a polydispersity index between 1.09 and 1.65. With salan-aluminum lower molecular weights of approximately 9900 g mol−1 were obtained as well as a very low dispersity index (1.05).
The synthesis was performed at the following three different reaction temperatures: 20, 50 and 70 °C. The highest molecular weight was obtained when the temperature was set to 20 °C but at the same time the number of hetero-sequences (% in mol) was low (20–34%). The highest number of hetero-sequences (57%) was obtained when the highest reaction temperature (70 °C) was used. At high concentrations, the conversion and incorporation of BL increased. The amount of solvent also affected the average molecular weight of the copolymers; at low concentrations the molecular weight increased and a high concentration resulted in a lower molecular weight. This effect is probably related to monomer diffusion.
The molar feed ratio did not correspond to the conversion of BL and LA or the composition of the copolymer. The LA was preferentially incorporated into the copolymer with respect to BL. However, the polymerization runs performed at different feed ratios demonstrated the possibility of modulating the composition and the molecular weight of the polymers to some extent. Very interestingly, the highest conversion of BL was obtained when D-LA was copolymerized with BL suggesting that the stereochemistry of LA could affect the conversion of BL.
Characterization of the copolymer microstructure was performed by 13C and 1H NMR. In the 1H NMR spectrum of the poly(BL-co-LA) sample, the signals attributable to the PHB and PLLA are recognized (Fig. 3). Interestingly, the methylene hydrogens appeared very sensitive to the chemical environment, and two patterns of doublet of doublets, one relative to the BL–BL homosequences and the other to the BL–LA heterosequences, are recognized to be centered at chemical shifts of 2.55 and 2.71 ppm, respectively. The intensities of the signals due to the heterosequences (BL–LA diads) increase by increasing the amount of LA in the copolymer (Fig. 4).
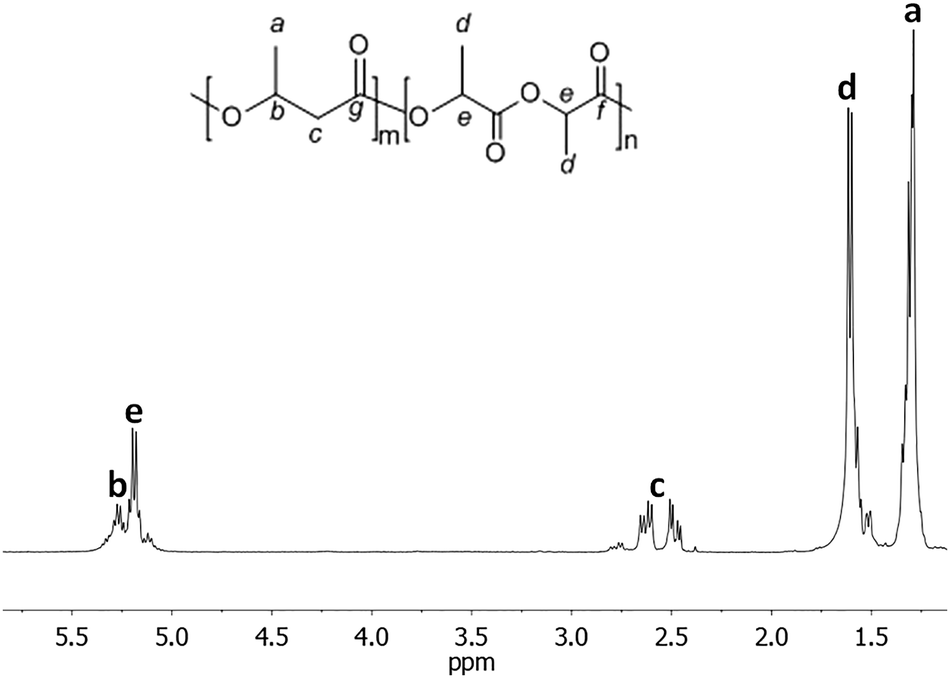 | ||
| Fig. 3 1H NMR spectrum of the poly(3-hydroxybutyrate-co-lactide) sample (Table 1, entry 2) in CDCl3 at 25 °C. | ||
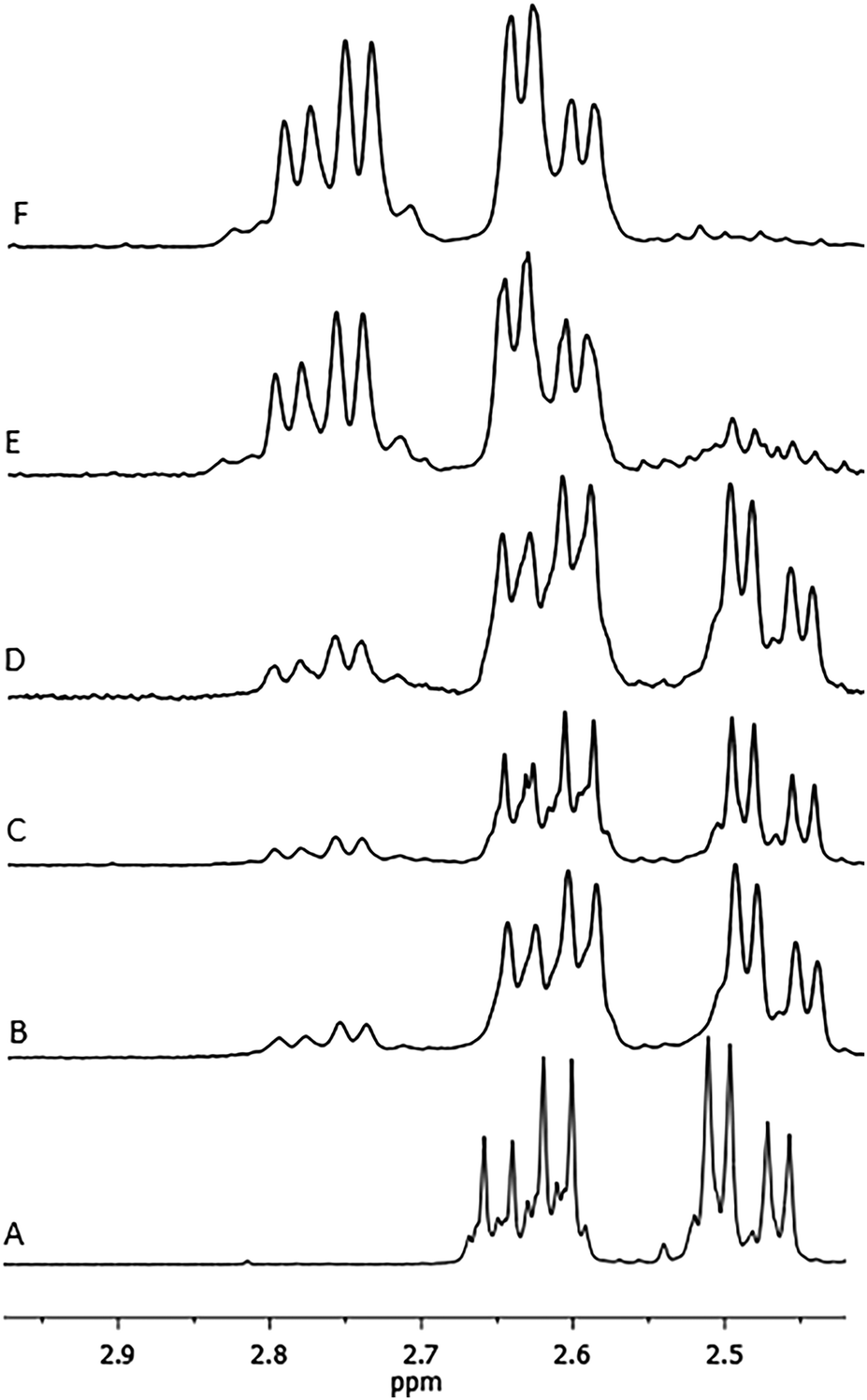 | ||
| Fig. 4 Methylene regions of the 1H NMR spectra of the poly(3-hydroxybutyrate-co-lactide) samples with different monomer compositions: A is a PHB homopolymer, B is entry 2, C is entry 4, D is entry 1, E is entry 5 and F is entry 9 (Table 1). | ||
A low molecular weight sample was prepared: the 1H NMR spectrum (see ESI†) showed the presence of the signals of the hydroxy end groups HOCH(CH3)CO– at δ = 4.36 ppm and of HOCH(CH3)CH2CO– at δ = 4.36 ppm, probably derived from the hydrolysis of a growing chain ending, respectively, with a lactyl unit and a butyrate unit.14a,b The ratio between the two signals was 1:6, thus indicating that the copolymeric chains ended preferentially with a butyrate unit, since the lactide monomer was presumably consumed first.
MALDI-TOF MS analysis of the sample of entry 12 was carried out, and confirmed the copolymeric nature, as well as the presence of hydroxy end groups. Moreover sequences of even numbers of repetitive lactyl units were also observed, thus indicating the occurrence of transesterification reactions. However the MALDI MS spectrum obtained does not depict the entire sample but the less abundant lower molecular weight fraction (see ESI†).
More details on the copolymer microstructure are derived from the 13C NMR spectrum. The attribution has been performed according to the literature.8a Having designated L as a lactyl unit and B a butyrate unit, the signals relative to the LLL and BBB homosequences were recognized. Moreover, signals due to the BL heterosequences are also recognized and attributed to the proper sequence according to the literature8a (Fig. 5).
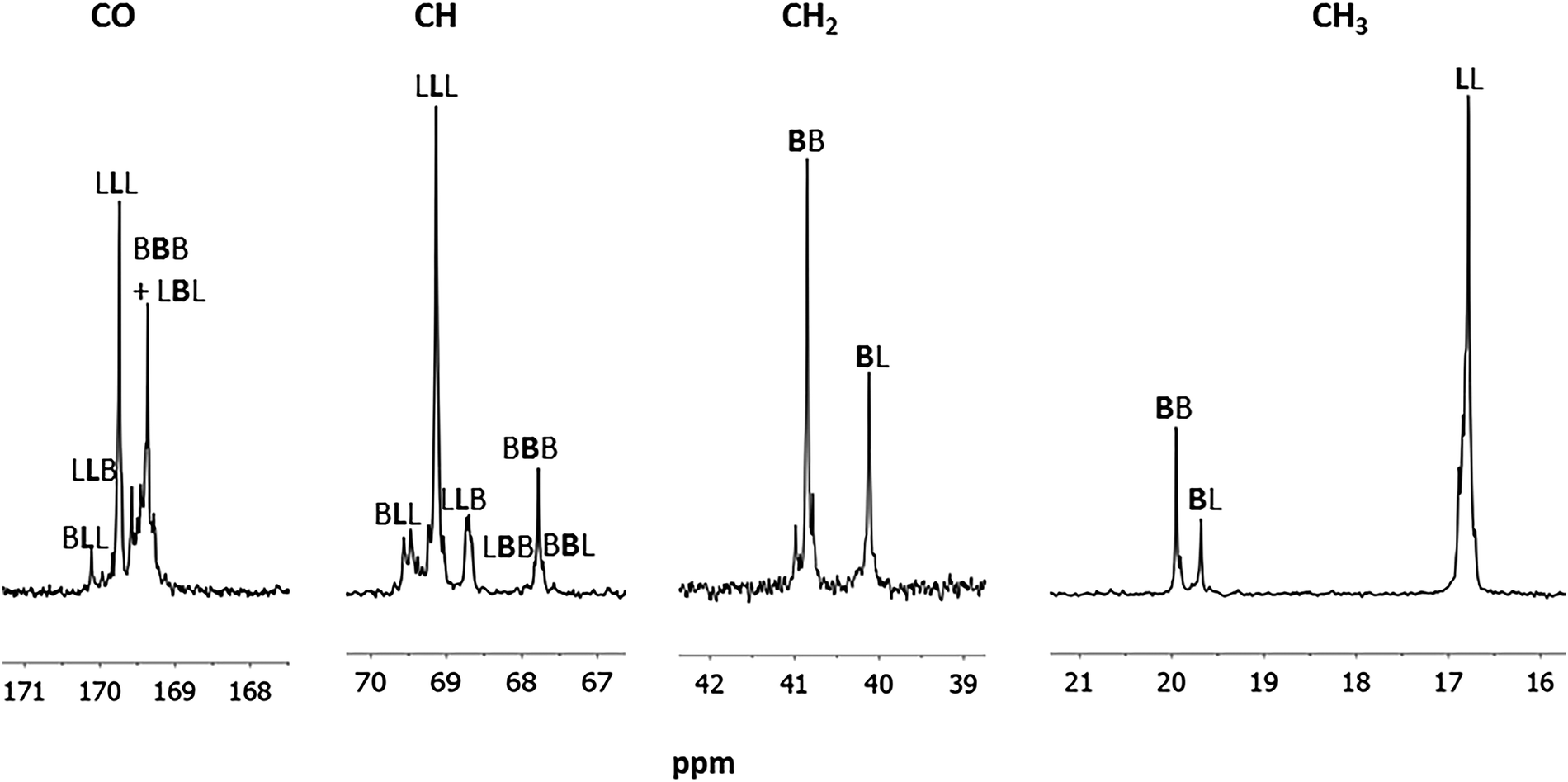 | ||
| Fig. 5 13C NMR (selected regions) spectrum of the poly(hydroxybutyrate-co-lactide) (Table 1, entry 2) in CDCl3 at 25 °C. | ||
Characterization of the microstructure of poly(BL-co-LA) was completed by the 2D HMBC 1H–13C correlation NMR spectrum. As shown in Fig. 6, a correlation cross-peak between the butyrate methylene protons and the methine carbon of the LA relative to the sequence LLB appeared.
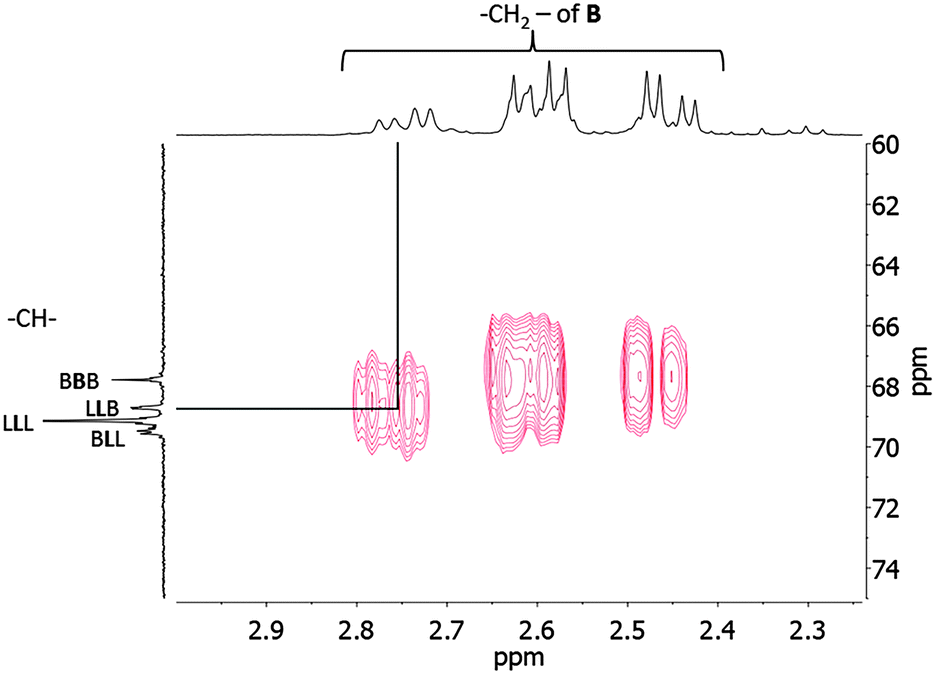 | ||
| Fig. 6 2D HMBC 1H–13C correlation NMR spectrum of the poly(hydroxybutyrate-co-lactide) sample (Table 1, entry 3). | ||
The overall data showed that LA and BL are copolymerized by both of the catalysts, with salan-yttrium being more efficient. The heterosequences are also observed with high frequency, while the copolymers were always more rich in LA.
To gain more insights into the copolymerization, kinetic studies were also performed (Fig. 7). Monitoring of the copolymerization reactions of salan-yttrium with BL and L-LA or D-LA, performed by 1H NMR, disclosed that the rate of LA polymerization was significantly higher than that of BL. Interestingly in the copolymerization with D-LA, the rate of BL polymerization was faster (Fig. 8).
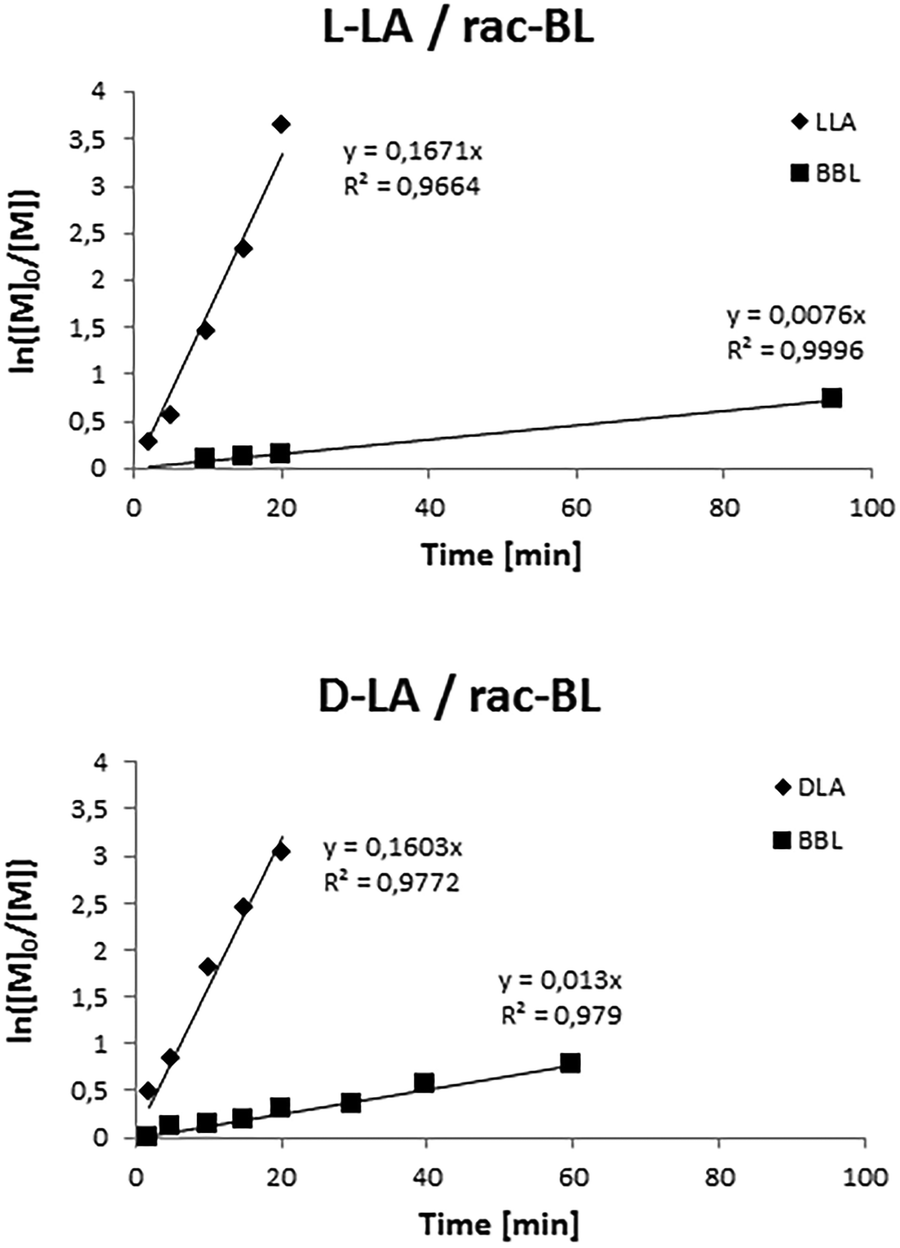 | ||
| Fig. 7 Pseudo-first-order kinetic plots for the ROP of LA and rac-BL promoted by salan-yttrium. Reaction conditions: [Y] = 3 M; [LA]:[BL]:[Y] = 90:90:1; toluene as solvent; and T = 25 °C. | ||
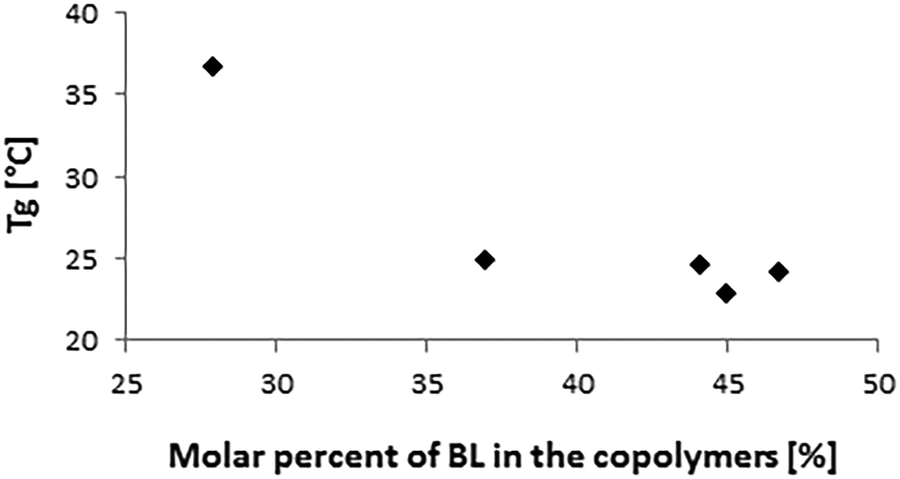 | ||
| Fig. 8 Plot of Tgversus the molar percentage of BL in the copolymers. | ||
The conversion rate of BL in the copolymerization with D-LA was twice as fast as that of the copolymerization of BL and L-LA. This behavior is most likely due to some stereochemical preference in the salan-yttrium catalyst.
Due to the different rates of the copolymerization with the two monomers, the formation of gradient copolymers is proposed, where the composition changes gradually from predominantly sequences of one comonomer to the other as a function of the copolymer chain length. Similar results were obtained by Shaver et al. in the copolymerization of LA and BL in the presence of different salan-aluminum catalysts.8c
The thermal properties of the copolymers were analyzed by DSC (Table 2). The thermograms showed only one glass transition temperature (Tg) for each sample, while no melting temperature was detected. The Tg decreased by increasing the molar percentage of BL in the copolymers (Fig. 8). The polymers were fully amorphous, which suggests that there was no phase separation.
Conclusions
Copolymerization of different monomers opens the way to the synthesis of materials having improved and modulated properties with respect to the parent homo-polymers.We have shown that salan-yttrium is able to promote the copolymerization of rac-β-butyrolactone (BL) and lactide (LA) to high molecular weight copolymers with good control and a monomodal distribution of the molecular weights. The results showed that salan-yttrium was a more effective catalyst compared to salan-aluminum, allowing the preparation of high molecular weight copolymers with a higher monomer conversion. Indeed almost complete conversion of LA was obtained in all reactions when salan-yttrium was used. The NMR characterization showed large amounts of heterosequences and a larger incorporation of LA with respect to BL. The incorporation of BL could be improved by increasing the temperature and adjusting the catalyst concentration. The composition of BL and LA in the copolymer could be modulated to some extent by the molar feed ratio. Interestingly, when D-LA was used instead of L-LA the conversion of BL was 79% and the molar feed ratio corresponded much better to the composition of the copolymer.
The kinetic experiments showed a faster incorporation of LA with respect to BL, while the thermal analysis showed a single glass transition temperature (Tg) for all of the samples. The Tg could be modulated by varying the molar percentage of BL in the copolymer. The above data are consistent with the formation of a gradient copolymer. As recently outlined, such gradient copolymers may be “superior to traditional block polymers for many applications, but few methods are available for synthesizing such polymers.”15 These materials may exhibit promising features for applications in the biomedical field.
Acknowledgements
The authors acknowledge VINNOVA, Mobility for Growth, the Marie Curie Actions FP7-PEOPLE-2011-COFUND (GROWTH 291795) and the Swedish Research Council (Grant ID 621-2013-3764) for financial support of this work. The authors thank Dr Patrizia Iannece (University of Salerno, Italy) for elemental analysis, and Dr Sabrina Carroccio (Consiglio Nazionale delle Ricerche, Catania, Italy) for MALDI-TOF MS analysis.Notes and references
- H. Wölfle, H. Kopacka, K. Wurst, P. Preishuber-Pflügl and B. Bildstein, J. Org. Chem., 2009, 694, 2493 CrossRef
.
- Y. Poirier, D. E. Dennis, K. Klomparens and C. Somerville, Science, 1992, 256, 520 CAS
- M. Zinn, B. Witholt and T. Egli, Adv. Drug Delivery Rev., 2001, 53, 5 CrossRef CAS PubMed
-
(a)
G. Scott and D. Gillead, Degradable Polymers, Chapman & Hall, 1995 Search PubMed
-
(a) C. M. Thomas, Chem. Soc. Rev., 2010, 39, 165 RSC
- D. Pappalardo, M. Bruno, M. Lamberti and C. Pellecchia, Macromol. Chem. Phys., 2013, 1965 CrossRef CAS
- J. Fang, M. J.-L. Tschan, T. Roisnel, X. Trivelli, R. M. Gauvin, C. M. Thomas and L. Maron, Polym. Chem., 2013, 4(2), 360 RSC
-
(a) H. Abe, Y. Doi, Y. Hori and T. Hagiwara, Polymer, 1998, 39, 59 CrossRef CAS
- E. Y. Tshuva, N. Gendeziuk and M. Kol, Tetrahedron Lett., 2001, 42(36), 6405 CrossRef CAS
-
(a) P. Hormnirun, E. L. Marshall, V. C. Gibson, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 2004, 126, 2688 CrossRef CAS PubMed
- N. Nomura, A. Akita, R. Ishii and M. Mizuno, J. Am. Chem. Soc., 2010, 132(6), 1750–1751 CrossRef CAS PubMed
-
(a) J. W. Kramer, D. S. Treitler, E. W. Dunn, P. M. Castro, T. Roisnel, C. M. Thomas and G. W. Coates, J. Am. Chem. Soc., 2009, 131, 16042 CrossRef CAS PubMed
- For comparison purposes, polymerization tests in the presence of the aluminium isoproxide and yttrium isopropoxide precursors were also carried out. Notably, the metal isoproxide precursors produced atactic polymers. The obtained PHB was amorphous, with a glass transition temperature below 0 °C.
-
(a) H. Ma and J. Okuda, Macromolecules, 2005, 38(7), 2665–2673 CrossRef CAS
- R. Zhao and K. J. Shea, ACS Macro Lett., 2015, 4, 58 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of aluminium compounds and of polymers; MALDI mass spectra. See DOI: 10.1039/c6nj00298f |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |