
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe vibrational spectrum of 1,4-dioxane in aqueous solution – theory and experiment
Piotr
Borowski
*a,
Wojciech
Gac
a,
Peter
Pulay
b and
Krzysztof
Woliński
a
aFaculty of Chemistry, Maria Curie-Sklodowska University, Maria Curie-Sklodowska Sq. 3, 20-031 Lublin, Poland. E-mail: pibcio@vsop408.umcs.lublin.pl
bDepartment of Chemistry and Biochemistry, University of Arkansas, Fayetteville 72701, USA
First published on 8th July 2016
Abstract
Continuum solvation models become unreliable when the dissolved molecule has specific interactions with the solvent. The present work focuses on comparison between a representative continuum solvation model, the conductor-like screening model (COSMO) and the explicit solvation model (ESM). Other continuum solvation models behave similarly to COSMO. Vibrational spectroscopy is a sensitive test of these models. Their performance for reproducing vibrational frequencies is examined by comparison with our experimental results on 1,4-dioxane dissolved in water. COSMO reproduces the frequency shifts only in the CH stretching vibration region but fails in the lower frequency range dominated by CC and CO stretching and deformations. It often predicts the wrong signs for the frequency shifts and the root-mean-square (RMS) deviation between the calculated and observed shifts is large (nearly 9 cm−1). The explicit solvation model, in which representative dioxane–water clusters are considered, provides accurate solvent frequency shifts, with the correct sign in the overwhelming majority of cases and the RMS deviation is below 2 cm−1 in the low-frequency range. However, it performs slightly worse than COSMO for CH stretching. Surprisingly it was also found that the combined COSMO + ESM model performs worst of all three models considered in this work.
Introduction
Most chemistry and essentially all biochemistry take place in solutions. The solvent, particularly a polar solvent like water, has a significant effect on the solute. Accurate thermodynamic modeling of solvent effects is very time-consuming, and it is often replaced by continuum solvation models or other simplified methods. It is important to evaluate the accuracy of such methods. Vibrational spectroscopy offers an excellent test. Before the advent of modern NMR, vibrational spectroscopy was perhaps the main generally applicable spectroscopic method for molecular characterization. Its disadvantages compared to NMR are twofold. First, most vibrations are strongly delocalized, precluding an easy assignment of a structure based on the spectra, and restricting vibrational spectroscopy to fingerprinting. Second, the resolution of vibrational spectra is limited. Vibrational spectroscopy has a major advantage over NMR: much higher sensitivity. Modern quantum chemical methods can predict vibrational frequencies with remarkable accuracy, and intensities fairly well with modest computational effort. This effectively eliminates the first disadvantage if the identity of the species can be narrowed down to a small number of candidates. Thus we foresee that vibrational spectroscopy, in conjunction with quantum chemical calculations, will become again a fundamental structural tool. It should be mentioned at this point that in the case of complex IR spectra the use of the factor analysis method also turned out to be very successful in molecular identification (see ref. 1 and references therein).Solvent effects are particularly important if there are specific solvation effects like hydrogen bonds between the solute and the solvent. In this paper, we compare a continuum solvation model (COSMO)2–6 for 1,4-dioxane (Dx) with a simple method of simulating its spectrum in aqueous solution: calculating the spectra of stable solute–water hydrogen-bonded complexes. Dioxane and its solutions have been extensively studied using vibrational spectroscopy.7–13 The earliest reports date from the mid-1900s.7–9 Malherbe and Bernstein9 gave a tentative assignment of all bands. Fratiello and Luongo10 were the first to report the IR shifts of dioxane upon dilution with water. The red shifts of the CO stretching vibrations were attributed to hydrogen bond formation. However, the discussion was based on the wrong assignment of the CO stretching modes. No explanation was provided for the blue shift in the CH stretching. Mizuno et al.11 performed NMR and IR measurements for 1,4-dioxane/water mixtures at different concentrations and temperatures. They proposed the formation of a bifunctional hydrogen-bonded complex in which water and dioxane molecules are connected by two hydrogen bonds: OW–H⋯ODx and CDx–H⋯OW (the superscript “W” refers to water) to explain the blue shifts of the CH stretching bands. Chang et al.12 investigated 1,3-dioxane/water mixtures using high-pressure IR and Raman spectroscopy and DFT calculations and concluded that the weak CDx–H⋯OW bonds are formed outside of the first coordination sphere, in contrast to Mizuno's proposal.11 Equatorial CH bonds were found to form stronger bonds than axial ones. The CH stretching shifts toward higher frequencies upon bonding. Recently, Wada and Kato13 determined the proportions of the 2,5- and 1,4-twist-boat conformers in liquid dioxane and in some solutions (including water) using Raman spectroscopy.
The aim of the present study is the theoretical modeling of solvent frequency shifts of dioxane by both dielectric continuum and explicit solvation models. This paper is organized as follows. In the next section experimental and computational details are reported. Then we report the band assignment based on potential energy distribution (PED)14 analysis, and compare the COSMO and ESM frequency shifts upon mixing with water with experiment.
Experimental and computational details
1,4-Dioxane of HPLC purity was purchased from POCh Poland S.A. (now Avantor Performance Materials Poland S.A.) and was used without further purification. The water solvent was distilled after deionization.A Nicolet 8700 (Thermo Scientific) spectrometer operating in the Attenuated Total Refection (ATR Smart Orbit) mode was used. Unless otherwise stated the resolution was 1 cm−1. 256 scans were averaged for each spectrum. The background was recorded prior to each run. Solution spectra were recorded relative to water. A sample droplet was placed on the ATR diamond crystal with an incidence angle of 45°, and an advanced ATR corrections algorithm (OMNIC, Thermo Scientific Nicolet) was applied for intensities. All but one IR active fundamental of dioxane in the range of 400–4000 cm−1 have frequencies well above 650 cm−1. Thus we could use a sensitive mercury cadmium telluride (MCT/A) detector to obtain high quality spectra. The lowest frequency band in the fingerprint region (at 613 cm−1) had to be measured using a less sensitive deuterated triglycine (DTGS) detector, and therefore its frequency is probably less precise.
Raman spectra were recorded using a single scan taken by a Renishaw inVia Raman Microscope also with a resolution of 1 cm−1, with 785 nm excitation light from a semiconductor laser.
To determine accurate experimental frequencies for close-lying vibrational modes (e.g., the modes near 1450 cm−1 in the IR spectrum) a deconvolution procedure using two Voigt profiles was applied.
The solvent (water) effect on the vibrational frequencies of dioxane was investigated using three solvation models: the conductor-like screening model (COSMO),2–6 the explicit solvation model (ESM) and the combined COSMO + ESM model.
ESM calculations were carried out in the following way. Equilibrium geometries and harmonic vibrational frequencies of dioxane and its hydrogen bonded complexes with water were determined at the B3LYP15 level using the 6-311++G** basis set.16,17 All optimized structures were local minima (all frequencies are real). Calculations were performed using the PQS quantum chemistry package.18,19 Since the effects that we are investigating are very subtle we decided to do all calculations with higher accuracy. We tightened the integral prescreening thresholds (by two orders of magnitude) and used a high quality integration grid in the SCF procedure. This step was followed by potential energy distribution (PED) analysis14 and ESFF frequency scaling20 (for the dioxane molecule only) to confirm the band assignments. Scaling parameters were taken from ref. 21, and the 9-parameter redundant primitive internal coordinate based scaling formalism22 was used. Assignment to valence coordinates was based on the PED coefficients and visualization of the vibrational modes.
We have also determined the interaction energies of the dioxane–water clusters. In addition to B3LYP/6-311++G**, we also performed full MP2/6-311++G** calculations (i.e., MP2 geometry optimization and frequency calculations). This enabled us to verify the correctness of the relative DFT energetics. The interaction energies were corrected for the basis set superposition error using the counterpoise correction (CP) scheme.23 Note that the discussion devoted to frequency shifts will be based on B3LYP vibrational frequencies since MP2 is known to poorly predict molecular force fields.21
For the COSMO solvation model (DFT calculations only), we used ε = 78.39 for the dielectric constant of the water solvent in all calculations. We followed exactly the same procedure, i.e., we performed geometry optimization and frequency calculations of the dioxane molecule (COSMO model) and dioxane–water complexes (the COSMO + ESM model).
Results and discussion
Dioxane – band assignment
As reported recently13 liquid 1,4-dioxane contains – in addition to the dominant chair conformer – also small amounts of the 2,5- and 1,4-twisted-boat conformers at room temperature. Their bands can be detected only in the magnified spectrum. Therefore we neglected these conformers in our model.The IR and Raman spectra of neat liquid 1,4-dioxane are given in Fig. 1 and 2. Table 1 provides the detailed assignment of bands based on the calculations for the C2h chair conformer. In the column denoted “Description” contributions from 6 types of internal coordinates (HX, XX, XXX, HXX, HXH, and torsions) are reported. The following notation is used: X denotes a general second row atom, two and three letter symbols refer to bond lengths and valence angles, respectively. The description was based on PED coefficient analysis; the percentage contributions refer to the sums of the diagonal PEDs for all internal coordinates belonging to a given type. Only values greater than 20% are reported. Note that when using the redundant primitive internal coordinates some of the CH2 bending vibrations acquire character of torsions24 (HCCO or HCOC) due to the definition of the primitive coordinates. In the next column, a detailed assignment based on the visualization of the vibrational modes is provided.
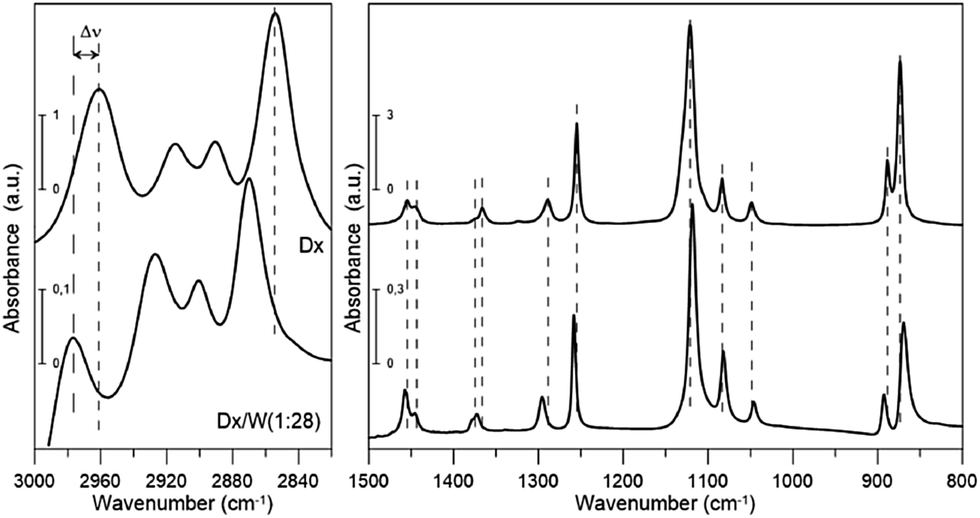 | ||
Fig. 1 FT-IR spectra of dioxane and dioxane–water mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :28). :28). | ||
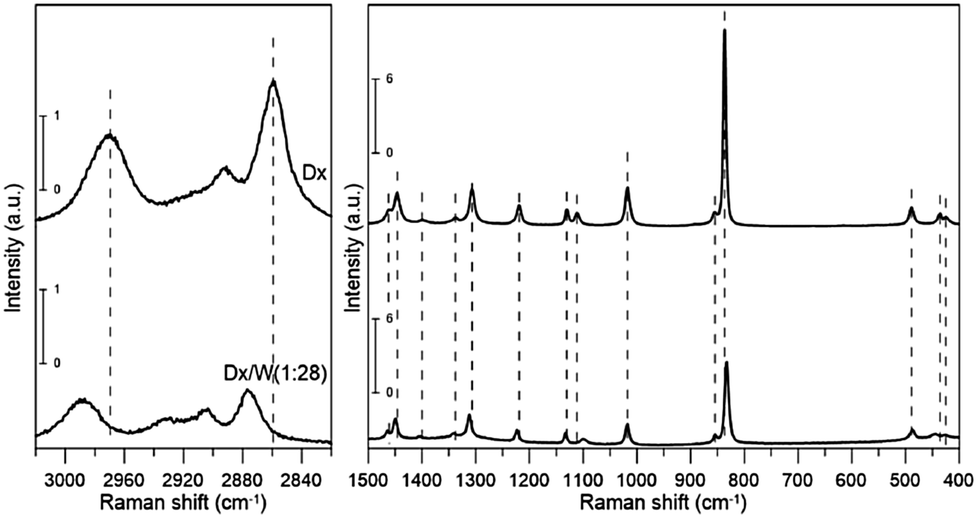 | ||
| Fig. 2 Raman spectra of dioxane and dioxane–water mixture (1:28). | ||
| B3LYP/6-311++G** | Expt. | ν–ν(scal) | Description | Assignment | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| No. | ν (harm) | ν (scal) | Symm. | I IR | I RAM | ν | From | |||
| a Shoulder band. b Numerical noise. c Calculated as an average for two overlapping modes, i.e., 2854.2 − (2860.8 + 2876.4)/2. d Calculated as 2859.6 − (2867.2 + 2872.0)/2. e Calculated as 2961.0 − (2971.6 + 2972.8)/2. f Calculated as 2969.4 − (2971.8 + 2973.4)/2. | ||||||||||
| Fingerprint region | ||||||||||
| 1 | 254.1 | 249.4 | A u | 1.0 | 0.0 | — | — | — | 95% torsions | Ring torsion |
| 2 | 267.9 | 263.1 | B u | 22.0 | 0.0 | — | — | — | 91% torsions | Ring torsion |
| 3 | 416.5 | 409.0 | A g | 0.0 | 0.6 | 425.1 | Ram | 16.1 | 92% torsions | Ring torsion |
| 4 | 440.3 | 435.0 | A g | 0.0 | 0.9 | 435.3 | Ram | 0.3 | 44% XXX + 41% torsions | COC deformation |
| 5 | 490.8 | 484.0 | B g | 0.0 | 3.0 | 488.7 | Ram | 4.7 | 30% XXX + 47% torsions | Ring deformation |
| 6 | 616.8 | 608.1 | B u | 14.8 | 0.0 | 613.3 | IR | 5.2 | 26% XXX + 43% HXX + 30% torsions | CH2 rocking + ring deformation |
| 7 | 840.2 | 824.7 | A g | 0.0 | 16.0 | 837.0 | Ram | 12.3 | 82% XX | COC sym. stretch |
| 8 | 860.9 | 845.2 | B g | 0.0 | 2.4 | 854.2 | Ram | 9.0 | 27% XX + 34% HXX + 38% torsions | CH2 rocking |
| 9 | 881.9 | 866.1 | B u | 75.6 | 0.0 | 873.6 | IR | 7.5 | 87% XX | COC sym. stretch |
| 10 | 891.8 | 875.8 | A u | 21.7 | 0.0 | 888.4 | IR | 12.6 | 79% XX | CC stretch |
| 11 | 1016.2 | 997.8 | A g | 0.0 | 11.2 | 1017.4 | Ram | 19.6 | 89% XX | CC stretch |
| 12 | 1060.9 | 1044.6 | B u | 9.7 | 0.0 | 1048.7 | IR | 4.1 | 36% HXX + 31% torsions | Ring trigonal deformation |
| 13 | 1102.2 | 1082.2 | A u | 13.8 | 0.0 | 1083.4 | IR | 1.2 | 28% HXX + 59% torsions | CH2 rocking |
| 14 | 1126.9 | 1106.7 | B g | 0.0 | 2.3 | 1111.1 | Ram | 4.4 | 83% XX | COC asym. stretch |
| 15 | 1133.0 | 1111.9 | A u | 177.9 | 0.0 | 1121.2 | IR | 9.3 | 95% XX | COC asym. stretch |
| 16 | 1143.2 | 1123.2 | A g | 0.0 | 2.7 | 1130.2 | Ram | 7.0 | 40% HXX + 40% torsions | CH2 rocking |
| 17 | 1238.1 | 1216.2 | B g | 0.0 | 11.1 | 1219.3 | Ram | 3.1 | 82% HXX | CH2 twisting |
| 18 | 1279.4 | 1256.6 | A u | 39.7 | 0.0 | 1254.8 | IR | −1.8 | 60% HXX + 21% torsions | CH2 twisting |
| 19 | 1316.0 | 1293.0 | B u | 9.1 | 0.0 | 1289.1 | IR | −3.9 | 70% HXX + 22% torsions | CH2 twisting |
| 20 | 1326.0 | 1302.4 | A g | 0.0 | 19.1 | 1307.1 | Ram | 4.7 | 55% HXX + 29% torsions | CH2 twisting |
| 21 | 1358.1 | 1334.1 | B g | 0.0 | 3.7 | 1337.8 | Ram | 3.7 | 78% HXX + 21% torsions | CH2 wagging |
| 22 | 1387.2 | 1362.4 | A u | 15.1 | 0.0 | 1366.3 | IR | 3.9 | 21% XX + 62% HXX | CH2 wagging + CC stretch |
| 23 | 1405.1 | 1380.1 | B u | 3.7 | 0.0 | 1375.0 | IRa | −5.1 | 68% HXX | CH2 wagging |
| 24 | 1416.5 | 1391.2 | A g | 0.0 | 4.5 | 1398.4 | Ram | 7.2 | 30% XX + 53% HXX | CH2 wagging + CC stretch |
| 25 | 1481.1 | 1450.4 | B g | 0.0 | 24.1 | 1446.4 | Ram | −4.0 | 35% HXH + 50% torsions | CH2 sym. deformation |
| 26 | 1482.2 | 1451.5 | A u | 1.5 | 0.0 | 1445.0 | IR | −6.5 | 34% HXH + 47% torsions | CH2 sym. deformation |
| 27 | 1489.5 | 1459.0 | B u | 15.2 | 0.0 | 1454.9 | IR | −4.1 | 32% HXH + 50% torsions | CH2 sym. deformation |
| 28 | 1494.8 | 1464.1 | A g | 0.0 | 4.7 | 1460.5 | Ram | −3.6 | 32% HXH + 48% torsions | CH2 sym. deformation |
| CH region | ||||||||||
| 29 | 2971.1 | 2860.8 | A u | 51.6 | 0.0 | 2854.2 | IR | −14.4c | 100% HX | CH stretch (axial) |
| 30 | 2977.7 | 2867.2 | A g | 0.0 | 432.9 | 2859.6 | Ram | −10.0d | 100% HX | CH stretch (axial) |
| 31 | 2982.9 | 2872.0 | B g | 0.0 | 35.5 | 2859.6 | Ram | −10.0d | 100% HX | CH stretch (axial) |
| 32 | 2987.4 | 2876.4 | B u | 170.2 | 0.0 | 2854.2 | IR | −14.4c | 100% HX | CH stretch (axial) |
| 2893.4 | Ram | Overtone | ||||||||
| 2890.6 | IR | Overtone | ||||||||
| 2914.7 | IR | Overtone | ||||||||
| 33 | 3086.3 | 2971.6 | A u | 92.1 | 0.1b | 2961.0 | IR | −11.2e | 100% HX | CH stretch (equatorial) |
| 34 | 3086.5 | 2971.8 | B g | 0.0 | 172.7 | 2969.4 | Ram | −3.2f | 100% HX | CH stretch (equatorial) |
| 35 | 3087.5 | 2972.8 | B u | 44.1 | 0.0 | 2961.0 | IR | −11.2e | 100% HX | CH stretch (equatorial) |
| 36 | 3088.1 | 2973.4 | A g | 0.0 | 316.7 | 2969.4 | Ram | −3.2f | 100% HX | CH stretch (equatorial) |
| RMS (fingerprint region) | 7.7 | |||||||||
| RMS (CH region) | 10.5 | |||||||||
| RMS (all frequencies) | 8.5 | |||||||||
All but the two lowest (<400 cm−1) vibrational modes were identified either from IR or Raman spectra. Our experimental frequencies agree with previous results.9,11 Three bands: two in IR (2890.6 and 2914.7 cm−1) and one in the Raman spectrum (2893.4 cm−1) were assigned to overtones or combination bands since no fundamentals are expected in this range. We will not discuss their frequency shifts. The bands at 2854.2 and 2961.0 cm−1 (IR) as well as 2859.6 and 2969.4 cm−1 (Raman) are attributed to two close lying CH stretching modes (the procedure for calculating deviations between theoretical and experimental wavenumbers is specified in the footnote of Table 1). They correspond primarily to stretching vibrations of axial (lower frequency) and equatorial (higher frequency) CH bonds, respectively, rather than CH2 symmetric and asymmetric stretching vibrations. This is due to the fact that those bonds differ in length by 0.008 Å (Table 2), which results in a difference in force constant values high enough to prevent local vibrations mixing. There is a well-known correlation25–27 between isolated CH vibrational frequencies and bond lengths, giving a rule of thumb that a 0.001 Å contraction in the CH bond length increases the isolated CH frequency by 10 cm−1. The value we obtain is slightly larger, 13.5 cm−1/(−0.001 Å).
| Dx | COSMO | 2a | 2b | |
|---|---|---|---|---|
| B3LYP | ||||
| CH (axial) | 1.100 | 1.098 | 1.098 | 1.097 |
| CH (equatorial) | 1.092 | 1.091 | 1.091 | 1.091 |
| CO | 1.426 | 1.434 | 1.433 | 1.435 |
| CC | 1.523 | 1.520 | 1.520 | 1.522 |
| MP2 | ||||
| CH (axial) | 1.101 | — | 1.099 | 1.098 |
| CH (equatorial) | 1.093 | — | 1.092 | 1.093 |
| CO | 1.423 | — | 1.429 | 1.431 |
| CC | 1.520 | — | 1.517 | 1.520 |
The scaled theoretical spectrum reproduces the experimental one very well, with a root-mean-square (RMS) deviation between the ESFF-scaled and observed wavenumbers of 7.7 cm−1 for the 26 non-CH stretching fundamentals, and 8.5 cm−1 for all 34 identified modes. The calculated IR and Raman intensities, which are usually much less accurate than the frequencies, reproduce the observed pattern in most cases.
Dioxane–water IR and Raman spectra
Shifts in the vibrational frequencies of dioxane upon mixing it with water, defined here as Δν = νsolution − νdioxane, depend on the concentration. It is expected that the shifts reach a saturation plateau as the water to dioxane molar ratio is increased. Our goal is to determine the limiting frequency shifts, corresponding to infinite dilution. As the intensities of the dioxane bands diminish with increasing dilution, we measured the spectra of dioxane–water mixtures with molar ratios of 1:1, 1:4, 1:8, 1:16, 1:28, and 1:56, and recorded their differential IR spectra relative to water. An MCT/A detector was used with a resolution of 4 cm−1. We could track the shifts for 15 IR active modes (13 bands).
The results are reported in Fig. 3. The mode labels refer to the experimental frequencies reported in Table 1. It is clear that saturation for all modes is achieved at a molar ratio of 1:28. Slight changes in the CH stretching mode shifts are observed upon further dilution but they are probably caused by errors in the determination of the band positions due to too low a dioxane concentration. It was assumed that for this molar ratio we would achieve a plateau for the shifts for all bands observed in the Raman spectrum (as well as for the IR active mode at ca. 613 cm−1). From now on under the term “experimental frequency shift” the difference between the band position for 1:28 solution and neat dioxane is understood.
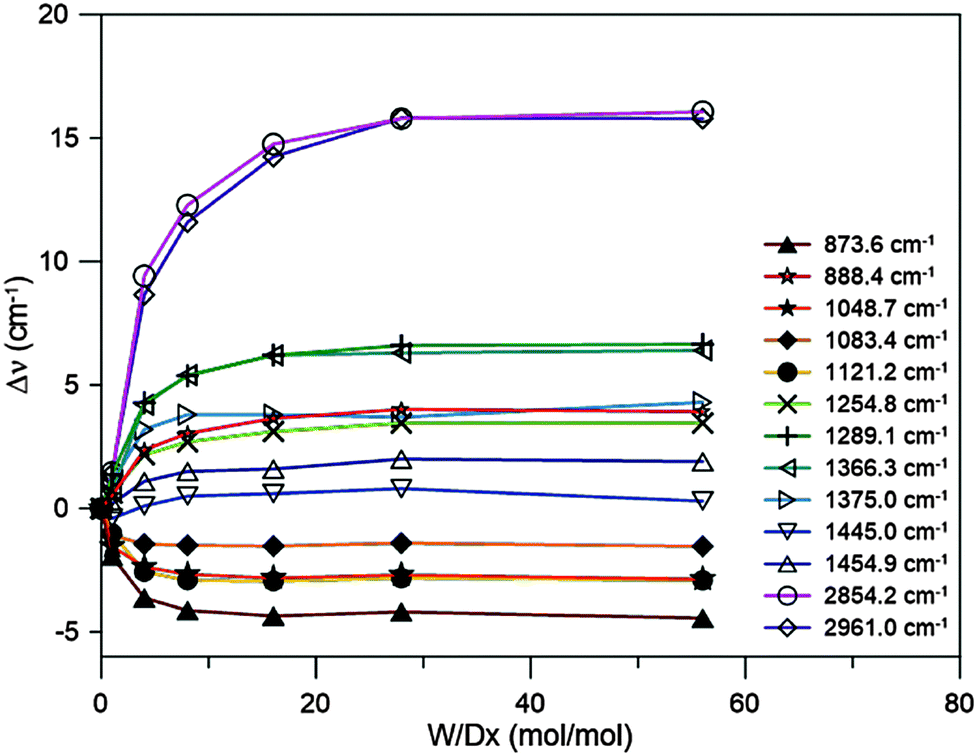 | ||
| Fig. 3 Shifts in the vibrational frequencies upon dilution of dioxane with water. | ||
Dioxane–water clusters
A search for stable dioxane–water clusters, in which water molecules are directly attached to the dioxane molecule via OW–H⋯ODx hydrogen bonds, was performed. We tried a number of initial geometries with up to 4 water molecules attached to dioxane, two per one dioxane oxygen atom. Only the five stable clusters shown in Fig. 4 were found. The 1:1 clusters 1a (0.24/0.88) and 1b (0.00/0.00) are of Cs symmetry, and the 1:2 clusters 2a (0.93/2.39), 2b (0.00/0.00), and 2c (0.74/1.52) are of C2h, C2h, and Cs symmetries, respectively. The values in parentheses next to the cluster symbols refer to B3LYP/MP2 relative energies, respectively (in kcal mol−1). The water molecules directly attached to dioxane form the first coordination sphere. No other stable clusters with more than two water molecules hydrogen bonded directly to dioxane were found. In particular, we did not obtain the bifunctional hydrogen bonded clusters proposed in ref. 11.
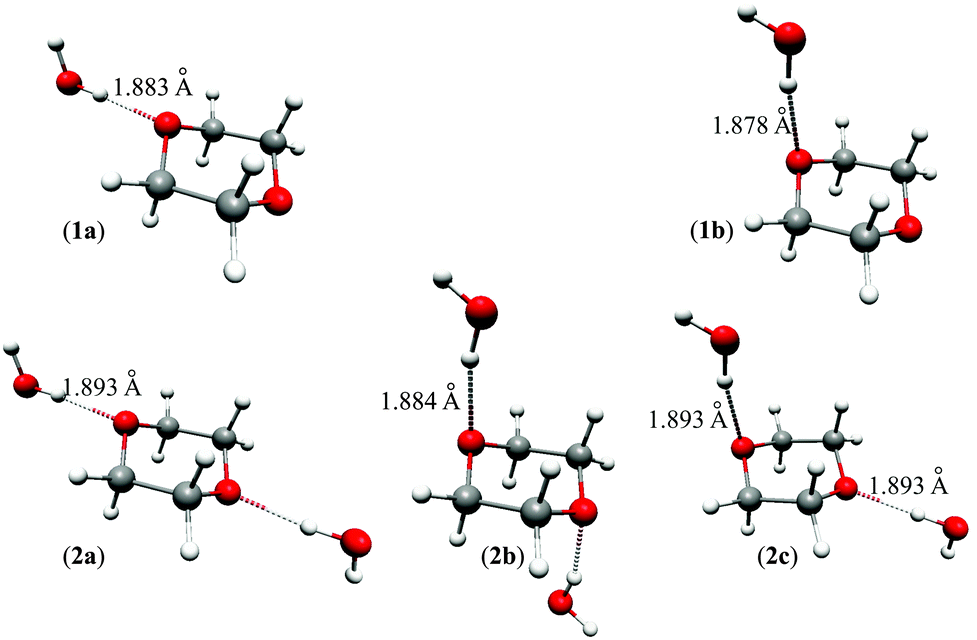 | ||
| Fig. 4 Dioxane–water clusters considered in this work. | ||
The B3LYP and MP2 interaction energies are given in Table 3. MP2 interaction energies calculated using B3LYP-optimized geometries are nearly identical to the latter ones. They are all negative indicating attractive interactions between dioxane and water molecule(s). The interaction energies per hydrogen bond are typically slightly above 5 kcal mol−1 and differ to some extent for various clusters. However, interaction energies (which are obviously approximate) are not the only factors responsible for the content of the particular cluster in the bulk phase (e.g., entropic effects are also important). In principle, one could calculate the percentage contributions of individual complexes using the so-called Quantum Cluster Equilibrium (QCE) theory of liquids (see, e.g., ref. 28–33 and references therein) but our experience indicates that the results are not accurate enough to be applicable in this study. It was therefore assumed that the considered clusters are roughly equally probable to exist in the bulk phase.
| Complex | B3LYP | MP2 | ||||
|---|---|---|---|---|---|---|
| ΔE | BSSE | ΔE + BSSE | ΔE | BSSE | ΔE + BSSE | |
| 1a | −5.84 | 0.67 | −5.17 | −6.77 | 1.84 | −4.93 |
| 1b | −6.14 | 0.80 | −5.34 | −7.69 | 2.33 | −5.36 |
| 2a | −11.27 | 1.30 | −9.97 | −13.38 | 3.74 | −9.64 |
| 2b | −12.34 | 1.64 | −10.70 | −15.88 | 4.92 | −10.96 |
| 2c | −11.56 | 1.43 | −10.13 | −14.21 | 4.15 | −10.06 |
In passing we note that the CP-corrected MP2 interaction energies are nearly identical to those calculated at the B3LYP level. A close agreement like this generally indicates that the energies are close to the converged values, which would be obtained from high level calculations using large basis sets.
Vibrational frequency shifts upon dilution
The solvent effect on molecular properties is nowadays often included by a dielectric continuum solvation model such as COSMO.2–6 However, continuum solvation models are unable to account for the specific interactions, like hydrogen bonding. An alternative is the explicit solvation model (ESM), in which solvation is represented by a supramolecular complex of the solute and a few solvent molecules. Calculated shifts of the harmonic frequencies using both approaches were compared with the experimental frequency shifts. Although harmonic frequencies typically overestimate fundamentals, the error in the shifts is probably much less significant.
Table 4 contains the calculated harmonic frequencies of 1,4-dioxane (the calculated IR and Raman intensities are reported in Table 1), as well as the experimental frequencies for neat liquid dioxane (column “Dx”), its water solution in a 1:28 molar ratio (column “Dx + 28H2O”) and the experimental and calculated frequency shifts. The experimental shifts in the CH stretching region are all positive (blue shift) and range between 16 and 21 cm−1. The solvation shifts of the remaining fundamentals (below 1500 cm−1) vary in the range from −11 to +9 cm−1. CO stretching vibrations (modes 7, 9, 14, and 15) are red shifted, while most of the modes corresponding to the CH2 deformations exhibit blue shifts. The calculated shifts using the COSMO solvation model (column “COSMO”), and their deviation from experiment (column “ΔCOSMO”) are also shown in Table 4. The COSMO shifts agree remarkably well with experiment in the CH stretching region, both in sign and magnitude. This observation is consistent with ref. 34, according to which the CH stretching shift upon solvation can be explained by the electrostatic interaction with the dielectric medium, and does not have to assume C–H⋯O hydrogen bond formation. However, low frequencies (CC and CO stretching and various types of bending vibrations) are poorly described using the COSMO model. In more than half of cases (16 out of 26) the sign of the shift is incorrect and deviations between the calculated and observed shifts are frequently larger than 10 cm−1, up to 16 cm−1 in two cases. The RMS difference between the experimental and calculated shifts is close to 9 cm−1 for the 26 identified modes in this range. COSMO predicts the correct sign for the CO stretching vibrations but overestimates the magnitudes.
| B3LYP | Expt. | Shifts | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| COSMO | Δ COSMO | Explicit solvation model | |||||||||||
| No. | ν | Dx | Dx + 28H2O | Shift | 1a | 1b | 2a | 2b | 2c | Mean | Δ ESM | ||
| a Calculated as an average for two overlapping modes, i.e., (19.9 + 13.4)/2 − 15.8; the same applies to other modes assigned to one band (cf. footnotes c, d, e, and f of Table 1) in both ΔCOSMO and ΔESM columns. | |||||||||||||
| Fingerprint region | |||||||||||||
| 1 | 254.1 | — | — | — | 2.9 | — | 2.5 | −1.5 | 2.7 | 8.8 | 4.2 | 3.3 | — |
| 2 | 267.9 | — | — | — | −4.0 | — | −4.1 | 13.0 | −9.1 | 30.4 | 2.0 | 6.4 | — |
| 3 | 416.5 | 425.1 | 426.0 | 0.9 | 2.8 | 1.9 | 2.4 | 3.6 | 3.5 | 14.5 | 6.7 | 6.1 | 5.2 |
| 4 | 440.3 | 435.3 | 443.9 | 8.6 | −1.8 | −10.4 | 11.4 | 1.4 | 22.6 | 4.2 | 11.9 | 10.3 | 1.7 |
| 5 | 490.8 | 488.7 | 487.2 | −1.5 | −2.8 | −1.3 | −0.7 | −0.9 | −2.1 | −0.6 | −1.1 | −1.1 | 0.4 |
| 6 | 616.8 | 613.3 | 614.3 | 1.0 | −1.3 | −2.3 | 3.9 | −1.4 | 6.4 | 2.6 | 4.0 | 3.1 | 2.1 |
| 7 | 840.2 | 837.0 | 833.0 | −4.0 | −10.9 | −6.9 | −2.4 | −4.3 | −4.4 | −6.3 | −5.6 | −4.6 | −0.6 |
| 8 | 860.9 | 854.2 | 854.6 | 0.4 | −4.7 | −5.1 | −1.2 | −1.5 | −3.3 | 2.0 | −0.7 | −0.9 | −1.3 |
| 9 | 881.9 | 873.6 | 869.4 | −4.2 | −17.5 | −13.3 | −1.8 | −3.1 | −4.5 | −8.3 | −5.2 | −4.6 | −0.4 |
| 10 | 891.8 | 888.4 | 892.4 | 4.0 | −1.5 | −5.5 | 4.9 | 1.5 | 8.8 | 2.7 | 5.2 | 4.6 | 0.6 |
| 11 | 1016.2 | 1017.4 | 1017.7 | 0.3 | −7.3 | −7.6 | 0.4 | −1.4 | 1.6 | −3.3 | −1.1 | −0.8 | −1.1 |
| 12 | 1060.9 | 1048.7 | 1046.1 | −2.6 | −7.8 | −5.2 | −1.1 | −2.7 | −3.2 | −2.7 | −3.1 | −2.6 | 0.0 |
| 13 | 1102.2 | 1083.4 | 1082.0 | −1.4 | −11.7 | −10.3 | −1.1 | −1.0 | −2.3 | 0.3 | −0.8 | −1.0 | 0.4 |
| 14 | 1126.9 | 1111.1 | 1099.8 | −11.3 | −20.5 | −9.2 | −9.7 | −10.7 | −11.7 | −16.9 | −13.8 | −12.6 | −1.3 |
| 15 | 1133.0 | 1121.2 | 1118.4 | −2.8 | −18.9 | −16.1 | 0.6 | −1.2 | −7.1 | −8.4 | −7.7 | −4.8 | −2.0 |
| 16 | 1143.2 | 1130.2 | 1133.0 | 2.8 | −5.4 | −8.2 | −0.3 | 2.2 | −1.6 | 8.8 | 3.6 | 2.5 | −0.3 |
| 17 | 1238.1 | 1219.3 | 1223.2 | 3.9 | −0.6 | −4.5 | 1.7 | 2.4 | 2.7 | 6.2 | 3.6 | 3.3 | −0.6 |
| 18 | 1279.4 | 1254.8 | 1258.3 | 3.5 | −4.3 | −7.8 | 1.0 | 1.0 | 1.5 | 2.7 | 2.0 | 1.6 | −1.9 |
| 19 | 1316.0 | 1289.1 | 1295.7 | 6.6 | −2.6 | −9.2 | 1.6 | 3.4 | 2.5 | 8.7 | 5.0 | 4.2 | −2.4 |
| 20 | 1326.0 | 1307.1 | 1311.8 | 4.7 | −1.5 | −6.2 | 2.5 | 2.7 | 4.7 | 6.0 | 5.2 | 4.2 | −0.5 |
| 21 | 1358.1 | 1337.8 | 1342.1 | 4.3 | 0.7 | −3.6 | 3.1 | 2.1 | 5.0 | 4.8 | 4.3 | 3.9 | −0.4 |
| 22 | 1387.2 | 1366.3 | 1372.6 | 6.3 | 2.9 | −3.4 | 4.7 | 4.3 | 7.7 | 9.3 | 7.5 | 6.7 | 0.4 |
| 23 | 1405.1 | 1375.0 | 1378.7 | 3.7 | −3.6 | −7.3 | 1.6 | −0.9 | 2.6 | −0.2 | 0.6 | 0.7 | −3.0 |
| 24 | 1416.5 | 1398.4 | 1403.4 | 5.0 | −1.2 | −6.2 | 4.0 | 2.8 | 7.0 | 6.2 | 6.1 | 5.2 | 0.2 |
| 25 | 1481.1 | 1446.4 | 1449.4 | 3.0 | −9.2 | −12.2 | 1.4 | 0.5 | 2.9 | 7.0 | 2.7 | 2.9 | −0.1 |
| 26 | 1482.2 | 1445.0 | 1445.8 | 0.8 | −9.8 | −10.6 | 2.3 | 1.5 | 3.0 | 7.7 | 5.6 | 4.0 | 3.2 |
| 27 | 1489.5 | 1454.9 | 1456.9 | 2.0 | −11.3 | −13.3 | 1.4 | 0.7 | 2.7 | 8.1 | 3.5 | 3.3 | 1.3 |
| 28 | 1494.8 | 1460.5 | 1464.6 | 4.1 | −12.4 | −16.5 | 1.5 | 0.8 | 2.4 | 7.9 | 4.6 | 3.4 | −0.7 |
| CH region | |||||||||||||
| 29 | 2971.1 | 2854.2 | 2870.0 | 15.8 | 19.9 | 0.8a | 7.7 | 17.9 | 27.2 | 39.7 | 28.6 | 24.2 | 9.1 |
| 30 | 2977.7 | 2859.6 | 2876.9 | 17.3 | 18.2 | 0.7 | 6.7 | 16.3 | 25.4 | 37.1 | 26.7 | 22.4 | 7.4 |
| 31 | 2982.9 | 2859.6 | 2876.9 | 17.3 | 17.8 | 0.7 | 19.6 | 19.4 | 26.1 | 37.0 | 32.7 | 27.0 | 7.4 |
| 32 | 2987.4 | 2854.2 | 2870.0 | 15.8 | 13.4 | 0.8a | 18.7 | 18.5 | 24.8 | 35.0 | 31.0 | 25.6 | 9.1 |
| 33 | 3086.3 | 2961.0 | 2976.8 | 15.8 | 12.1 | −3.4 | 4.9 | 4.2 | 14.4 | 12.2 | 13.0 | 9.7 | −4.8 |
| 34 | 3086.5 | 2969.4 | 2990.3 | 20.9 | 13.4 | −8.0 | 6.1 | 5.3 | 15.5 | 13.3 | 14.0 | 10.8 | −9.2 |
| 35 | 3087.5 | 2961.0 | 2976.8 | 15.8 | 12.8 | −3.4 | 10.2 | 8.8 | 14.8 | 13.6 | 14.0 | 12.3 | −4.8 |
| 36 | 3088.1 | 2969.4 | 2990.3 | 20.9 | 12.5 | −8.0 | 10.6 | 9.1 | 15.0 | 13.9 | 14.3 | 12.6 | −9.2 |
| RMS (fingerprint region) | 8.8 | 1.7 | |||||||||||
| RMS (CH region) | 4.3 | 7.8 | |||||||||||
| RMS (all frequencies) | 8.0 | 4.1 | |||||||||||
The five dioxane–water clusters discussed above were used as representative supermolecules in the ESM model. The significant interaction energy (about 5 kcal mol−1 per hydrogen bond, compared to kT ≈ 0.6 kcal mol−1 at room temperature) indicates that such clusters exist in the diluted aqueous dioxane solution long enough to be mostly responsible for its vibrational spectrum, and the average effect is observed due to thermal equilibrium in the bulk phase. Indeed, the average lifetime τ of a cluster can be estimated from the relation τ−1 = f0 × exp(−ΔE/kT) with f0 = kT/h. For ΔE = 5 kcal mol−1 and T = 293 K we obtain τ ≈ 10−9 s. This value refers to the gas phase; in liquids τ is expected to be longer. A period of typical molecular vibration is of the order of 10−14–10−13 s so the complexes oscillate at least 104–105 times before they disintegrate.
Cluster spectra contain, in addition to the vibrations of dioxane, water intramolecular vibrations and low frequency intermolecular modes. In order to calculate the frequency shifts we have to identify dioxane modes. This is straightforward in our case since the frequencies of water normal vibrations (two OH stretches, ν(harm) > 3600 cm−1, and one bending, ν(harm) > 1600 cm−1) differ much from those of dioxane and do not mix strongly with the latter. We have developed a numerical indicator showing the percentage of dioxane vibrations in a normal vibration:
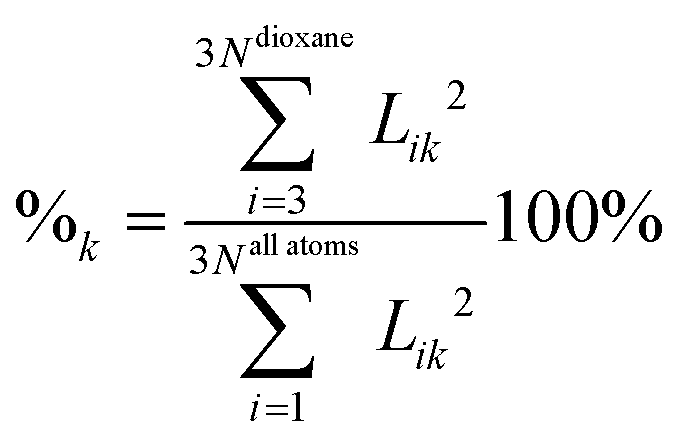
Table 4 contains harmonic frequency shifts for each of the clusters relative to dioxane (columns “1a”,…,“2c”), as well as their mean values (column “Mean”) and the differences between the mean and experimental shifts (column “ΔESM”). As can be seen in all but two cases the sign of the shift is correctly predicted when the specific interactions are explicitly incorporated in the calculations. In addition, the RMS value between the experimental and calculated frequency shifts for the 26 mid-range modes is as low as 1.7 cm−1 compared to 9 cm−1 obtained using COSMO. Apparently ESM is much more accurate in describing the solvent effect in the fingerprint region when hydrogen bonding occurs. In particular, it is much superior for the shifts in the CO stretching vibrations, giving almost perfect agreement with experiment. However, COSMO outperforms ESM in the CH stretching range. The RMS deviation of the shifts using COSMO model for the 8 CH modes (4.3 cm−1) is nearly two times lower than the deviation of ESM (7.8 cm−1). Part of the electrostatic effect34 properly reproduced by COSMO is also reproduced by the ESM model, though.
We also extended our model by considering the additional complex obtained by adding water molecules outside the first coordination sphere. However, changes in the calculated frequencies of dioxane turned out to be insignificant.
In addition, we tried to add the COSMO model to the dioxane–water hydrogen-bonded complexes, in the hope that it may improve the agreement in the CH stretching region. The effects turned out to be nearly additive in the case of the equatorial CH stretching modes – the frequency shifts are 19.4, 20.6, 22.0, and 21.6 cm−1 for modes 33, 34, 35, and 36, respectively. This is to be compared with the values of 21.8, 24.2, 25.1, and 25.1 obtained by direct summation of values taken from Table 4 (columns “COSMO” and “Mean”). In the case of axial CH stretching modes (modes 29, 30, 31, and 32) the effects are less additive – the frequency shifts are 34.6, 30.2, 33.4, and 30.4 cm−1, respectively, compared to 44.1, 40.6, 44.8, and 39.0 cm−1 obtained by direct summation. This results in a large RMS value for the CH stretching of 11.3 cm−1. In addition, the COSMO model deteriorated the ESM frequencies in the fingerprint region yielding an RMS value as large as 8.5 cm−1, and 9.2 cm−1 in the entire spectral range. Thus the joint COSMO + ESM model performs worst out of all three models investigated. This observation is a bit surprising taking into account recent findings.35
The observed trends in the CH and CO stretching vibration frequency shifts can be correlated with the changes in the dioxane geometrical parameters upon solvation. For CH stretchings, the increase in the frequency is mainly due to a decrease in the length of the CH bonds by about 0.001 Å (Table 2). As a consequence the force constants increase, which leads to a blue shift of bands by ca. 15 cm−1, a value reported before. The effect of the HOH⋯ODx hydrogen bond formation on the CO bonds of dioxane is opposite. This lowers the CO force constant, which results in a red shift. Both COSMO and ESM provide similar geometrical changes (cf.Table 2). However, the significant overestimation of the CO stretching shifts by COSMO indicates that the model is not capable of reproducing properly changes in the dioxane force field. The good performance of ESM confirms the correctness of our model, which assumes that most of those changes are caused by the solvent molecules forming quite well defined molecular complexes.
Conclusions
In this work, the solvent effect on the vibrational frequency shifts of dioxane was investigated by means of the dielectric continuum (COSMO) and explicit solvation (ESM) models. The solvent (water) was chosen since it interacts with dioxane with the aid of specific forces (hydrogen bonds). It is shown that the COSMO model is not capable of reproducing the experimental results properly for low-frequency vibrations. In more than half of cases the sign of the frequency shift is incorrectly predicted and the RMS value between the calculated and observed frequency shifts is high. On the other hand, the explicit solvation model properly describes the changes in the dioxane force field and vibrational frequencies upon dilution with water. The observed frequency shifts are correct both in sign and magnitude by considering only water molecules forming a few well-defined complexes with dioxane through hydrogen bonding. The COSMO solvation model seems to be somewhat superior to ESM in the CH region. ESM using only one or two water molecules coordinated to the dioxane oxygen atom(s) reproduces the signs of the C–H solvent shifts but has a larger error than COSMO in this region (8 vs. 4 cm−1). The good performance of the continuum solvation model (COSMO in our case) indicates that the dominant effect for the C–H vibrations is electrostatic interaction with a dielectric environment and it is not necessary to postulate weak C–H⋯O hydrogen bonds, in agreement with ref. 34. Finally it should be emphasized that the combined COSMO + ESM model fails to properly predict the frequency shifts of dioxane upon dilution.Notes and references
- D. C. Lee, P. I. Haris, D. Chapman and R. C. Mitchell, Biochemistry, 1990, 29, 9185–9193 CrossRef CAS PubMed.
- A. Klamt and G. Schüürmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- J. Andzelm, C. Kölmel and A. Klamt, J. Chem. Phys., 1995, 103, 9312–9320 CrossRef CAS.
- A. Klamt, J. Phys. Chem., 1995, 99, 2224–2235 CrossRef CAS.
- A. Klamt and V. Jonas, J. Chem. Phys., 1996, 105, 9972–9981 CrossRef CAS.
- K. Baldridge and A. Klamt, J. Chem. Phys., 1997, 106, 6622–6633 CrossRef.
- D. A. Ramsay, Proc. R. Soc. London, 1947, 190, 562–574 CrossRef CAS.
- S. C. Burket and R. M. Badger, J. Am. Chem. Soc., 1950, 72, 4397–4405 CrossRef CAS.
- F. E. Malherbe and H. J. Bernstein, J. Am. Chem. Soc., 1952, 74, 4408–4410 CrossRef CAS.
- A. Fratiello and J. P. Luongo, J. Am. Chem. Soc., 1963, 85, 3072–3075 CrossRef CAS.
- K. Mizuno, S. Imafuji, T. Fujiwara, T. Ohta and Y. Tamiya, J. Chem. Phys., 2003, 107, 3972–3978 CrossRef CAS.
- H. C. Chang, J. C. Jiang, C. W. Chuang, J. S. Lin, W. W. Lai, Y. C. Yang and S. H. Lin, Chem. Phys. Lett., 2005, 410, 42–48 CrossRef CAS.
- R. Wada and M. Kato, Chem. Phys. Lett., 2015, 641, 74–79 CrossRef CAS.
- Y. Morino and K. Kuchitsu, A Note on the Classification of Normal Vibrations of Molecules, J. Chem. Phys., 1952, 20, 1809–1810 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- M. J. Frisch, J. A. Pople and J. S. Binkley, J. Chem. Phys., 1984, 80, 3265–3269 CrossRef CAS.
- PQS version 4.0, Parallel Quantum Solutions, 2013 Green Acres Road, Fayetteville, Arkansas 72703.
- J. Baker, K. Wolinski, M. Malagoli, D. Kinghorn, P. Wolinski, G. Magyarfalvi, S. Saebo, T. Janowski and P. Pulay, J. Comput. Chem., 2009, 30, 317–335 CrossRef CAS PubMed.
- P. Borowski, M. Fernández-Gómez, M. P. Fernández-Liencres and T. Peña Ruiz, Chem. Phys. Lett., 2007, 446, 191–198 CrossRef CAS.
- P. Borowski, J. Phys. Chem. A, 1012, 116, 3866–3880 CrossRef PubMed.
- P. Borowski, J. Mol. Spectrosc., 2010, 264, 66–74 CrossRef CAS.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- P. Borowski, S. Pasieczna-Patkowska, M. Barczak and K. Pilorz, J. Phys. Chem. A, 2012, 116, 7424–7435 CrossRef CAS PubMed.
- D. C. McKean, J. Phys. Chem. A, 2000, 104, 8995–9008 CrossRef CAS.
- K. Siam, J. D. Ewbank, L. Schafer and C. van Alsenoy, THEOCHEM, 1988, 41, 83–92 CrossRef CAS.
- J. S. Wong, R. A. MacPhail, C. B. Moore and H. L. Strauss, J. Phys. Chem., 1982, 86, 1478–1484 CrossRef CAS.
- F. Weinhold, J. Chem. Phys., 1998, 109, 367–372 CrossRef CAS.
- F. Weinhold, J. Chem. Phys., 1998, 109, 373–384 CrossRef CAS.
- R. Ludwig, F. Weinhold and T. C. Farrar, Mol. Phys., 1999, 97, 479–486 CrossRef CAS.
- P. Borowski, J. Jaroniec, T. Janowski and K. Woliński, Mol. Phys., 2003, 101, 1413–1421 CrossRef CAS.
- M. Brüssel, E. Perlt, S. B. C. Lehmann, M. von Domaros and B. Kirchner, J. Chem. Phys., 2011, 135, 194113 CrossRef PubMed.
- G. Matisz, A.-M. Kelterer, W. M. F. Fabian and S. Kunsági-Máté, J. Phys. Chem. B, 2011, 115, 3936–3941 CrossRef CAS PubMed.
- P. J. A. Ribeiro-Claro and P. D. Vaz, Chem. Phys. Lett., 2004, 390, 358–361 CrossRef CAS.
- R. Vianello and J. Mavri, New J. Chem., 2012, 36, 954–962 RSC.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2016 |