
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOctahedral Werner complexes with substituted ethylenediamine ligands: a stereochemical primer for a historic series of compounds now emerging as a modern family of catalysts†
Andreas
Ehnbom
 ,
Subrata K.
Ghosh
,
Kyle G.
Lewis
and
John A.
Gladysz
*
,
Subrata K.
Ghosh
,
Kyle G.
Lewis
and
John A.
Gladysz
*
Department of Chemistry, Texas A&M University, P.O. Box 30012, College Station, Texas 77842-3012, USA. E-mail: gladysz@mail.chem.tamu.edu
First published on 20th October 2016
Abstract
As reported by Alfred Werner in 1911–1912, salts of the formally D3 symmetric [Co(en)3]3+ (en = ethylenediamine) trication were among the first chiral inorganic compounds to be resolved into enantiomers, the absolute configurations of which are denoted Λ (left handed helix) or Δ (right handed helix). After a >100 year dormant period during which few useful reactions of these substitution inert complexes were described, carbon substituted derivatives have recently been found to be potent catalysts for enantioselective organic synthesis. This review systematically outlines the fascinating range of stereoisomers that can arise, such as conformers associated with the five membered chelate rings (λ/δ), alignment modes of the C–C bonds with the C3 symmetry axis (lel/ob), geometric isomers (fac/mer), and configurational diastereomers (R/S) arising from carbon stereocenters. These analyses demonstrate a profound stereochemical diversity that can be applied in catalyst optimization. Efforts are made to bridge the often orthogonal nomenclature systems inorganic and organic chemists employ to describe these phenomena.
 Andreas Ehnbom | Andreas received his MSc from Lund University (LU) in 2013. His awards include the Oxford University Press Award (2009), the Dean's Summer Student Scholarship (2010) from University College London (UCL), a Ludvig Stenberg Scholarship (2014), and the American Institute of Chemists Student Award (2014). In 2012, Andreas participated in an exchange abroad program working in the lab of Prof. Suzanne Blum at UC-Irvine. Prior to that, he worked with Prof. Ebbe Nordlander (2009–2011), collaborated with Prof. Graeme Hogarth at UCL, and researched with Prof. Selwyn Mapolie at Stellenbosch University (2011). In 2016 he coauthored a chapter in Organic Reactions with Prof. Peter Somfai (LU). Since June 2015, he has worked for Profs. John A. Gladysz and Michael B. Hall at Texas A&M University, with the goal of developing novel Werner catalysts and studying them using computational tools. |
 Subrata K. Ghosh | Subrata received his MSc from Texas A&M University-Commerce in 2010 where he worked with Profs. Allan D. Headley and Bukuo Ni. He then began work on his Doctoral degree with Prof. John A. Gladysz at Texas A&M University, where his research involves the development of novel catalysts family based on Werner complexes and resulted in several papers (see citations) and a patent. His honors include a Pathways to the Doctorate Fellowship (2010–2012), an A. E. Martell Graduate Student Prize (2014), an Eastman Chemical travel grant (2015), and the Derek and Christiane Barton Graduate Award for Outstanding Research (2016). On the personal side, he is married to Swagata Ghosh who is a homemaker, and he plays cricket in his spare time. |
 Kyle G. Lewis | Kyle received his BS in Chemistry at Baylor University in 2008 where he worked for Prof. C. M. Garner and graduated Phi Beta Kappa. He earned his PhD at Texas A&M University in 2013 with Prof. J. A. Gladysz where he developed Werner-type cobalt complexes for enantioselective hydrogen bond mediated catalysis that resulted in several papers (see citations) and a patent. He has given invited presentations at the BASF-CaRLa Winter School (Heidelberg) and the ACS Division of Organic Chemistry Graduate Research Symposium (Boulder); the latter was assisted by an Eastman Chemical travel grant. He is currently a Senior Research Chemist at ExxonMobil Chemical Company in Houston, Texas. |
 John A. Gladysz | John is a native of the Kalamazoo Michigan area, and since receiving his doctoral degree (Stanford, 1974) has held academic appointments at UCLA, the University of Utah, the Universität Erlangen-Nürnberg, and Texas A&M University. He has received awards in organometallic chemistry from the ACS (1994) and the RSC (2013), and is a Fellow of the ACS (2009) and RSC (2013). From June 1984 through July 2010, he served as the Associate Editor of Chemical Reviews. He then succeeded Dietmar Seyferth as the Editor in Chief of Organometallics, a position he held until January 2015. On the personal side, he is married to Janet Blümel, a Professor of Chemistry at Texas A&M. They live on the Crow's Nest Ranch, which consists of 137 acres (56 hectares) a few miles east of College Station, Texas. |
Key learning points(1) Representations, symmetry, absolute configuration of the chiral octahedral trication [Co(en)3]3+.(2) Conformations of the en ligands in [Co(en)3]3+, and lel/ob orientations. (3) Stereoisomers of analogs with 1,2-propylenediamine ligands. (4) Stereoisomers of analogs with vicinally disubstituted ethylenediamine ligands. (5) Diastereomer stabilities and catalysis. |
1. Introduction
Werner complexes with chelating 1,2-diamines have played prominent roles in the development of inorganic and coordination chemistry, particularly with regard to structure and stereochemistry.1,2 For example, octahedral cobalt(III) adducts – the most widespread family of Werner diamine complexes – were the first chiral inorganic compounds to be isolated in enantiomerically pure form.2 However, until recently they have had few, if any, applications in synthetic organic chemistry, either as reagents or catalysts. One reason is that cobalt(III) complexes are, by virtue of their low spin d6 electronic configurations, substitution inert,3 thereby preventing the coordination and activation of organic substrates at reasonable temperatures.Recently, there have been conceptual and practical breakthroughs that have enabled chiral Werner complexes to serve as highly effective catalysts for enantioselective organic reactions.4–6 One has been the realization that the NH bonds associated with coordinated amines are capable of functioning as hydrogen bond donors to Lewis basic organic substrates.4,7 Over the last 20 years, a number of chiral organic hydrogen bond donors have been found to be effective catalysts for a multitude of enantioselective transformations.8 Not surprisingly, coordination compounds can function similarly, and frequently offer architectures and binding site arrays that have no counterparts in organic systems. Another has been the development of lipophilic4 and/or fluorophilic9 Werner complexes, such that catalysis can be conducted in the absence of water, which would otherwise saturate (or compete with substrate access to) the hydrogen bond donor sites.
Given this new interest in chiral Werner complexes, the authors thought it would be helpful to review the many hierarchal levels of stereochemistry that can be embodied in octahedral cobalt(III) complexes of chelating 1,2-diamines, for which ethylenediamine (en) is the archetype. This has already seen extensive analysis,10 but the literature is fragmented, and many studies present crystal structures or CD spectra with little accompanying analysis. A potential entry level stumbling block is that organic and inorganic chemists often favor different vocabularies in analyzing the same phenomena. Hence, efforts are made to employ both syntaxes side by side throughout this review.
Several conspicuous omissions deserve note at the outset. First, some cobalt N,N,N′,N′-tetramethyl ethylenediamine (TMEDA) complexes have been reported.11 However, there are questions regarding the existence of the cobalt(III) tris(chelate).12 Disecondary diamine chelates, which feature NHRR′ donor groups, are easily accessed,13 but introduce additional stereocenters. The “asymmetric nitrogen atoms” rapidly invert in the free ligands, but become fixed upon coordination. This leads to a further level of stereoisomerism, which can be subject to either kinetic or thermodynamic control. Outside of efforts by Searle,13b–d there have been no systematic studies of such coordination environments, and it is left to motivated readers to derive the extensive families of stereoisomers that can result.
2. The chiral [Co(en)3]3+ trication: cobalt stereocenter
All octahedral tris(chelate) complexes are chiral, and the enantiomers of the [Co(en)3]3+ trication can be rendered in the “Star of David” motifs shown in Fig. 1a. These non-superimposable mirror images differ in whether the connections between the “front triangle” and “rear triangle” run in counter clockwise or clockwise directions. This constitutes an example of helical chirality, with the first motif being left handed and the second right. As detailed in many inorganic textbooks, the former is designated Λ and the latter Δ.14 In the organic literature, enantiomers that can be viewed as left or right handed helices are often designated M and P. Other common representations of the enantiomers of [Co(en)3]3+ are given in Fig. 1b and c. Such compounds are sometimes referred to – together with nonplanar complexes of formula M(A)(B)(C)(D) – as “chiral at metal”. Note also that the two NH2 protons, and the two CH2 protons, are diastereotopic and can give different 1H NMR signals.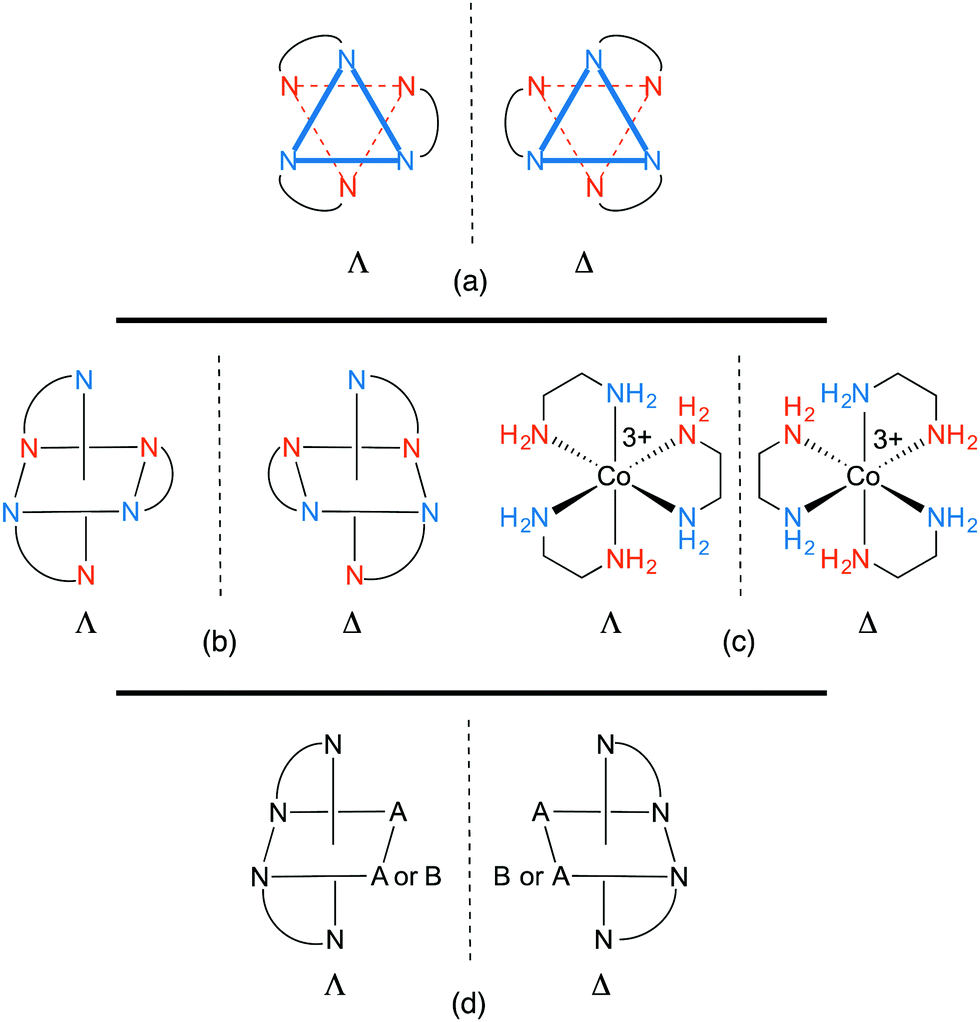 | ||
| Fig. 1 Representations of the enantiomeric Λ and Δ-[Co(en)3]3+ trications (a–c) and the related bis(chelates) [Co(en)2(A)2]3+ and [Co(en)2(A)(B)]n+ (d). | ||
In 1912, Werner reported that the enantiomers of [Co(en)3]3+ can be separated by fractional crystallization of the diastereomeric tartrate salts.2e Chloride anion exchange then afforded the resolved enantiomers, Λ- and Δ-[Co(en)3]3+ 3Cl−. In the preceding year, Werner described analogous resolutions of cations of the formulae [Co(en)2(A)(B)]n+ and [Co(en)2(A)2]n+. As is evident from Fig. 1d, two chelating ligands are sufficient to render an octahedral complex chiral. Tricationic tris(ethylenediamine) adducts of other metals ([M(en)3]3+, M = Cr,15,16 Rh,16,17 Ir18) and the tetracation19 [Pt(en)3]4+ were similarly resolved via salts of various chiral anions.
Despite being chiral, the [Co(en)3]3+ trication possesses several symmetry elements. However, for reasons that become obvious in Section 3, this initial analysis approximates the chelate backbones as planar, or having identical conformations. With this proviso, there is a principal C3 axis that runs perpendicular to the plane of the paper in Fig. 1a and exchanges each blue nitrogen atom (and each vermillion nitrogen atom). There are furthermore three C2 axes in a perpendicular plane (the plane of the paper) that exchange blue and vermillion nitrogen atoms. This corresponds to the chiral point group D3. However, the authors are not aware of any crystal structures where the trication exhibits this idealized symmetry,7 as the counter anions (and/or solvate molecules) hydrogen bond to the NH groups in motifs that lower the symmetry.
The enantiomers of [Co(en)3]3+ are extremely stable with respect to ligand dissociation or racemization under ambient conditions. The half-lives for the hydrolysis and racemization of [Co(en)3]3+ 3Cl− in 0.10 M aqueous NaOH at 25 °C have been estimated as 3.2 years (38 kcal mol−1) and >3.2 years, respectively.3b Other reports confirm that no racemization of [Co(en)3]3+ 3Cl− occurs in aqueous solution during (a) 3 months at room temperature, (b) 75 minutes at 85 °C in the presence of 100 equiv. of NaNO2, or (c) 15 hours at reflux in the presence of HCl.20 However, when activated charcoal is added, racemization takes place within two minutes at 90 °C.21 The charcoal is believed to function as a redox catalyst, allowing the generation of small amounts of substitution labile cobalt(II).
Naturally, the enantiomers give mirror image CD spectra with opposite signs for Δε or [θ].22 When additional ligand based stereocenters are introduced, such that diastereomers result, the shapes of the CD spectra remain largely a function of the cobalt configuration.5b,23 Hence, cobalt configurations can be reliably assigned from the sign of the Cotton effect.
3. The chiral [Co(en)3]3+ trication: ligand conformations
In the previous section, the conformations of the ethylenediamine chelate rings were not considered. With respect to the CH2CH2 backbone, a gauche orientation of the two NH2 groups (torsion angle ca. 60°) would be expected to be much more stable than an eclipsed orientation (torsion angle ca. 0°). Thus, as illustrated by A and B in Fig. 2, nonplanar rings would be anticipated. For each Co(en) fragment, two nonsuperimposable mirror images are possible. These are commonly designated with the lower case Greek letters λ and δ and correspond to left handed and right handed helices, respectively (the counterclockwise and clockwise senses are illustrated with green arrows in Fig. 2). Each NH2 and CH2 group features one hydrogen atom that is pseudoaxial, and one that is pseudoequatorial.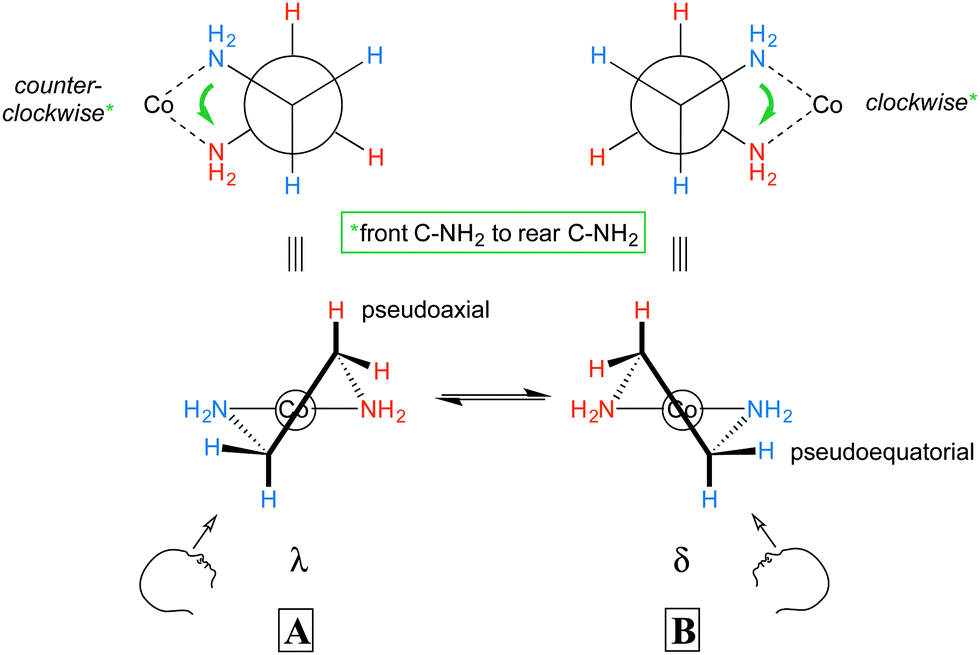 | ||
| Fig. 2 Chelate conformations of ethylenediamine. | ||
The axial and equatorial hydrogen atoms of cyclohexane rapidly exchange via a “ring flip”, and the same holds for the hydrogen atoms of the ethylenediamine chelate rings. Thus, the λ and δ conformers will readily equilibrate (see Fig. 2, bottom). However, in the absence of disorder, each chelate will crystallize in one conformation or the other.
Importantly, the conformation of the chelate affects the relative orientations of the CH2–CH2 bonds and the symmetry of the [Co(en)3]3+ trication. First, consider the “docking” of three λ ethylenediamine units to give a trication with a Δ configuration at cobalt. The initial step is shown in Fig. 3A, and when all three chelates are in place, the structure represented as Δ-λλλ results. Next, consider the docking of three δ ethylenediamine units to give a trication with the same Δ configuration at cobalt (i.e., an identical spatial orientation of chelating cobalt–nitrogen bonds). The initial step is shown in Fig. 3B. In order for the cobalt–nitrogen bond locations to “match up”, the δ ethylenediamine must first be rotated by 90°. When all three chelates have been similarly put in place, the structure represented as Δ-δδδ results.
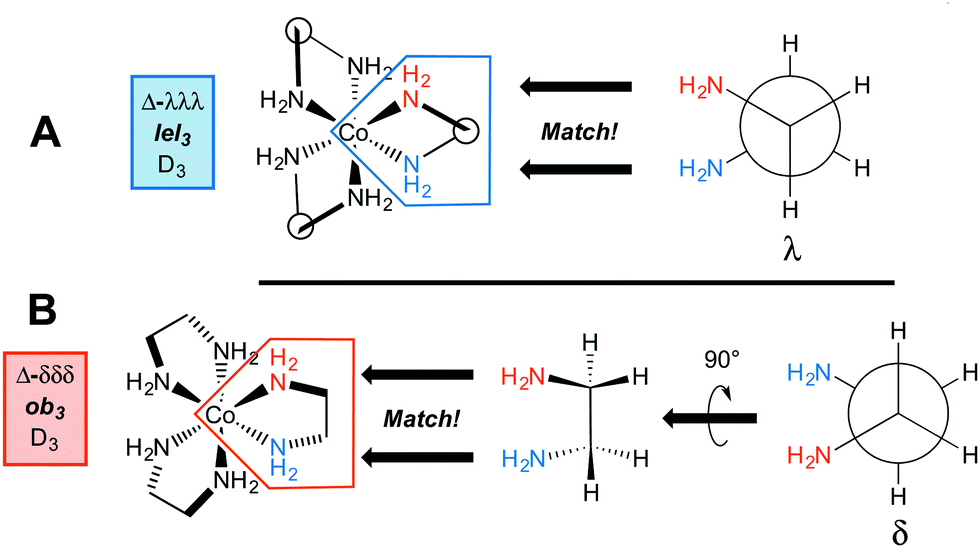 | ||
| Fig. 3 Accommodation of ethylenediamine ligands with three λ or three δ conformations in the coordination sphere of an octahedral cobalt atom with a Δ configuration. | ||
In the first structure (Δ-λλλ; Fig. 3A), the λ chelates are said to adopt lel orientations, so named because the three CH2–CH2 bonds are parallel to the C3 symmetry axis.24 These linkages are often incorporated into Newman type projections, as indicated by hollow circles. In the second structure (Δ-δδδ; Fig. 3B), the δ chelates are said to adopt ob orientations, so named because the three CH2–CH2 bonds are oblique to the C3 axis. The designations Δ-λλλ and Δ-lel3 (Fig. 3A) are both found in the literature, and can be used interchangeably, but with one important caveat that soon follows below. The same holds for Δ-δδδ and Δ-ob3.
When the [Co(en)3]3+ trication has a Λ configuration at cobalt, these relationships are reversed. A λ chelate leads to an ob orientation while a δ chelate results in a lel orientation. Furthermore, each of the three ethylenediamine ligands can independently adopt either an ob or lel orientation. Therefore, for a complex with a given cobalt configuration, four diastereomers exist.
All possibilities are depicted in Fig. 4. Note that the complexes analyzed in Fig. 3, and their mirror images, have D3 symmetry. However, the new diastereomers introduced in Fig. 4, with mixed lel and ob orientations, have C2 symmetry.
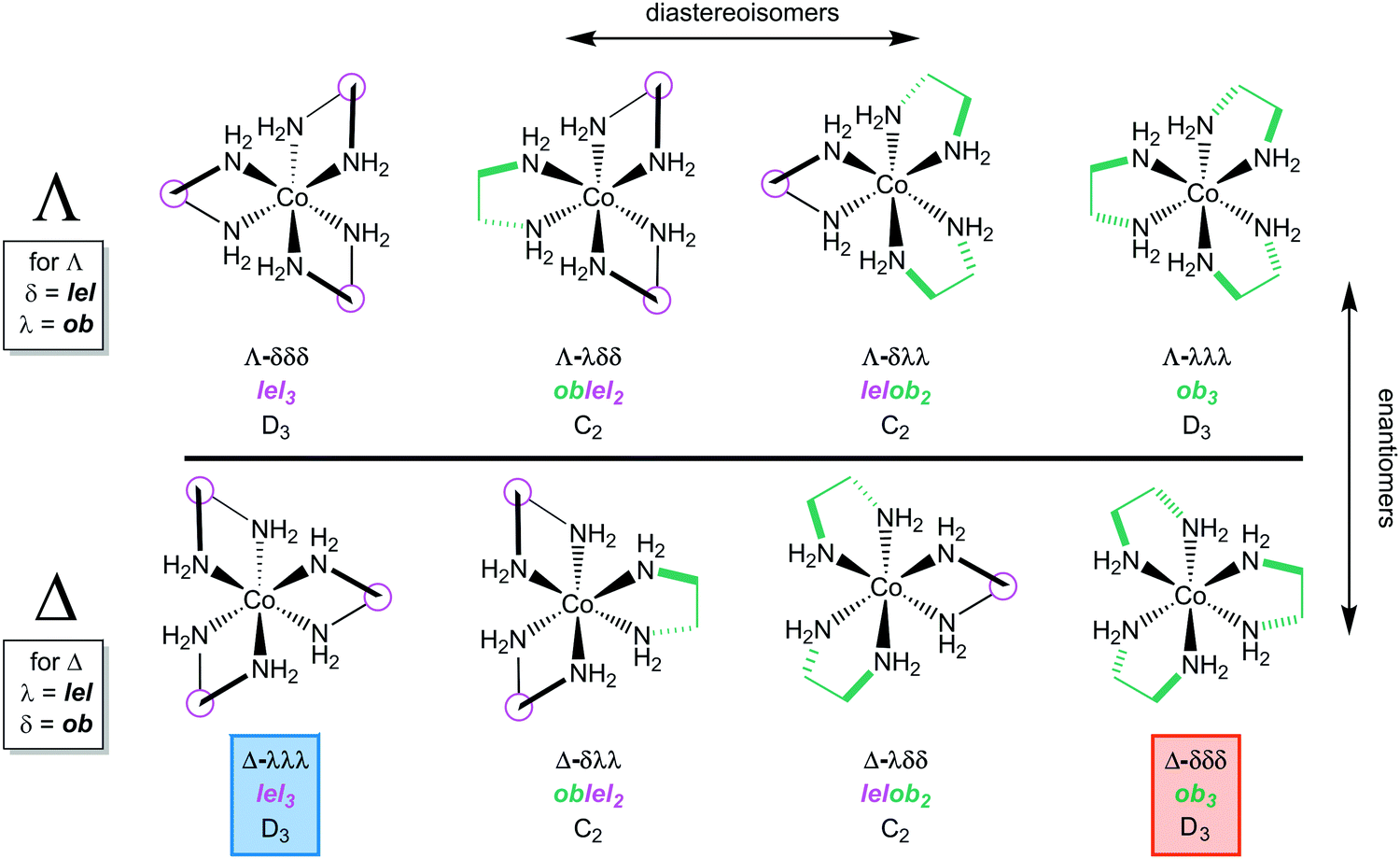 | ||
| Fig. 4 All possible stereoisomers of the [Co(en)3]3+ trication. | ||
Fig. 4 also illustrates a “trap” or potential error in identifying enantiomers. Just like the descriptors R/S always identify enantiomers of compounds with a single tetrahedral carbon stereocenter, so will the family of descriptors consisting of upper/lower case delta/lambda identify mirror image components of the [Co(en)3]3+ trication. In other words, the stereoisomer Λ-λλλ can automatically be regarded as the enantiomer of Δ-δδδ.
However, this is not the case with lel/ob. Instead, Λ-lel3 and Δ-ob3 are diastereomers, as is easily seen in the upper left and lower right structures in Fig. 4. Rather, it is Λ-lel3 and Δ-lel3 that are enantiomers. Another way to look at this is as follows: if the C3 axis is perpendicular to the plane of the paper, and one enantiomer is reflected in the plane of the paper to give the other, a “parallel” (lel) orientation of the CH2–CH2 bond with respect to the C3 axis must be preserved. For these reasons, the authors generally refer to lel/ob as “orientations” or “perspectives”.
In practice, the conformations of the ethylenediamine chelate rings rapidly interconvert and do not have to be considered when analyzing the complexes in Fig. 1. In other words, the four diastereomers with Λ configurations in Fig. 4, as well as the four diastereomers with Δ configurations, will not normally be distinguishable in solution. However, all of these motifs can be observed in crystal structures.7 They also become important with certain types of substituted ethylenediamine ligands as described below.
Tris(chelate) complexes with larger rings, such as derived from 1,3-diaminopropane25 and 1,4-diaminobutane26 ligands, have also been reported. These similarly yield Λ and Δ enantiomers, but the additional CH2 units give rise to larger numbers of chelate conformations. These have not yet been analyzed in comparable detail. However, parallels between chair, boat, and twist-boat cyclohexane and 1,3-diaminopropane chelate conformations have been noted.25
4. Octahedral Werner complexes with monosubstituted ethylenediamine ligands
The simplest monoalkylated diamine ligand is 1,2-propylenediamine (pn).27 Whereas ethylenediamine is limited to two conformational enantiomers (see Fig. 2), this ligand allows for two configurational enantiomers: (R)-1,2-propylenediamine ((R)-pn) and (S)-1,2-propylenediamine ((S)-pn). In the following analysis, cobalt(III) tris(chelate) complexes with homochiral ligands (i.e., all R or all S) are treated first.4.1 Stereoisomerism at the cobalt center
As with ethylenediamine (Fig. 1), three pn ligands can chelate to give either a Λ or Δ cobalt configuration. However, the methyl group lowers the chelate symmetry from an “A–A” to an “A–B” motif. This in turn leads to facial (fac) and meridional (mer) stereoisomers, which can be viewed as counterparts to geometric isomers in organic compounds. Fig. 5 illustrates the four possible adducts of cobalt(III) and (R)-pn that can result (ignoring the chelate conformations). Four additional adducts can be generated with (S)-pn.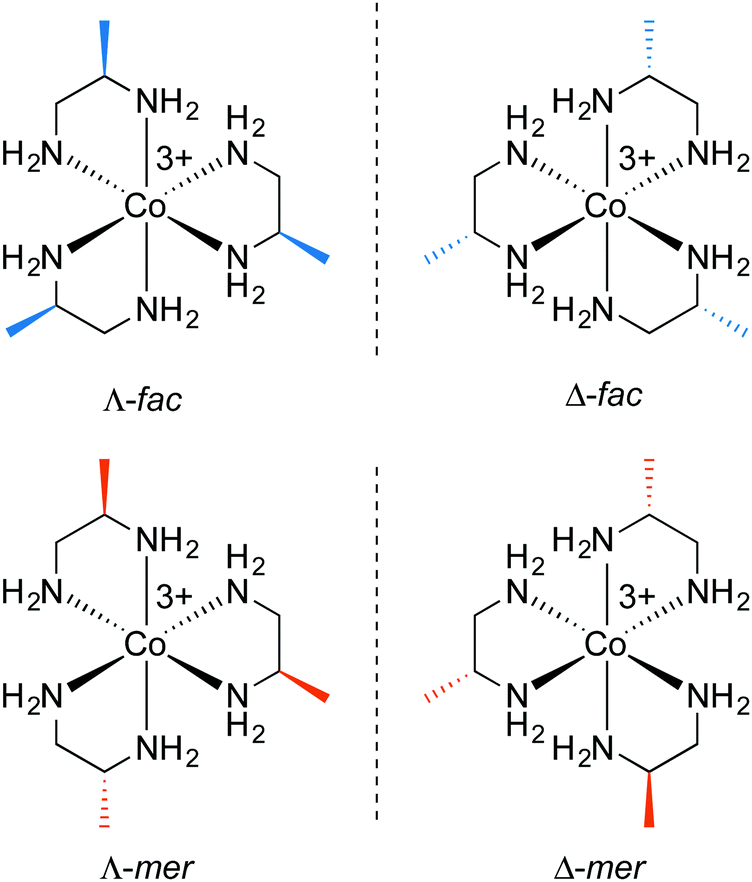 | ||
| Fig. 5 The fac/mer isomers of the [Co((R)-pn)3]3+ trication. | ||
4.2 Chelate conformation
The conformation of the five membered chelate ring plays a more involved role with substituted ethylenediamine complexes than with the parent ligand. For 1,2-propylenediamine, the chelate will logically prefer conformations that place the methyl group in pseudoequatorial positions over pseudoaxial positions. Fig. 6 illustrates the pseudoequatorial and pseudoaxial versions of both (R)-pn and (S)-pn ligands.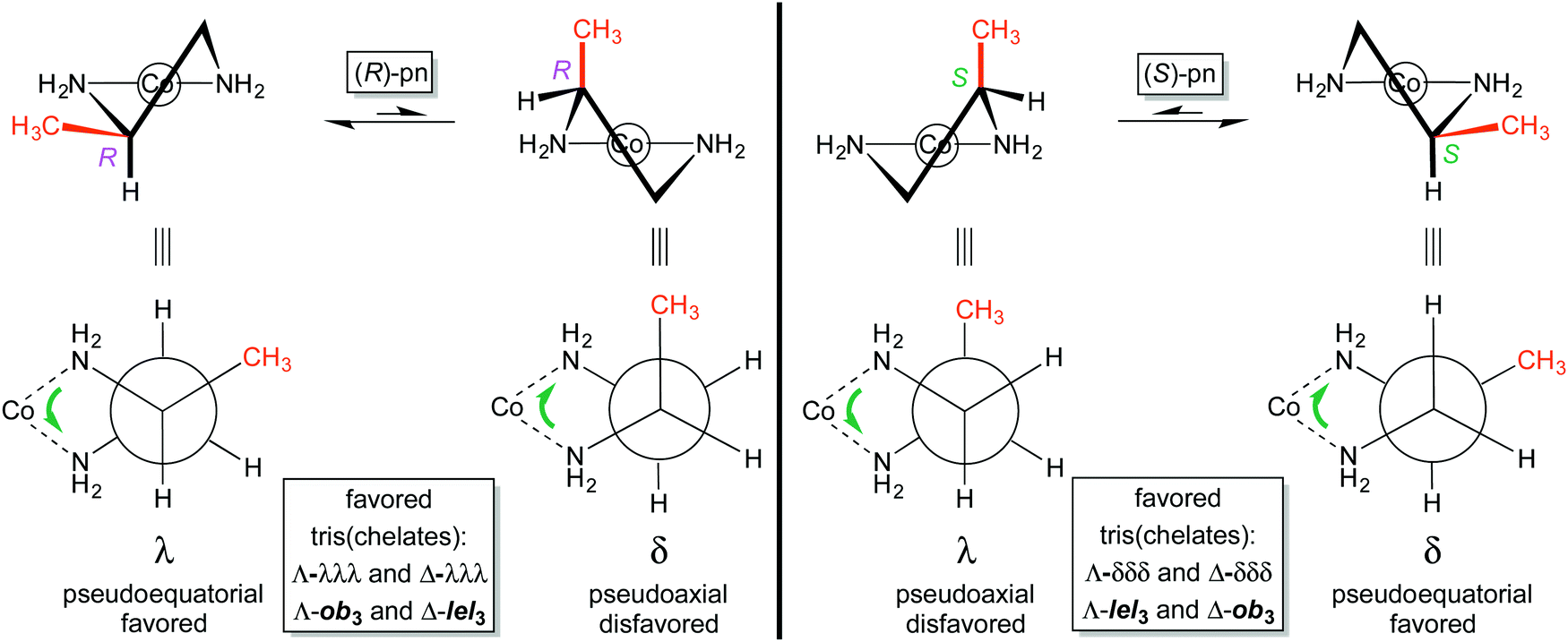 | ||
| Fig. 6 Chelate conformations of (R)-pn and (S)-pn. | ||
For (R)-pn, the λ conformation of the chelate directs the methyl group into a pseudoequatorial position. A “ring flip” to the δ conformation is unfavorable because the methyl group must occupy a pseudoaxial position. It then follows that the tris(chelate) [Co((R)-pn)3]3+ should, irrespective of mer/fac geometry, preferentially exist as either Λ-λλλ or Δ-λλλ stereoisomers. With reference to the analyses of ob/lel orientations of unsubstituted ethylenediamine (Fig. 3 and 4), this means that in the Λ complex, the chelate CH2–CHCH3 linkages will all be oblique to the C3 axis (Λ-ob3). Similarly, in the Δ complex the chelate CH2–CHCH3 linkages will align parallel to the C3 axis (Δ-lel3).
In contrast, the (S)-pn ligand will be more stable in the δ conformation (Fig. 6, right). Thus, [Co((S)-pn)3]3+ should preferentially exist as either Λ-δδδ or Δ-δδδ stereoisomers, which correspond to Λ-lel3 and Δ-ob3. In any event, Fig. 7 summarizes the four preferred stereoisomers of [Co((R)-pn)3]3+, considering all possible combinations of metal configurations, chelate conformations, and fac/mer geometries. As a side comment on nomenclature, one could ask whether the first example, labeled Λ-fac-λλλ, might equally well be represented as Λ-fac-RRR. The authors would discourage this practice, as the absolute configurations of the carbon stereocenters are already specified in the formula [Co((R)-pn)3]3+, and the absence of the λλλ designation leaves open the possibility that one of the chelate rings might display an alternative δ conformation as, for example, a consequence of crystallization.
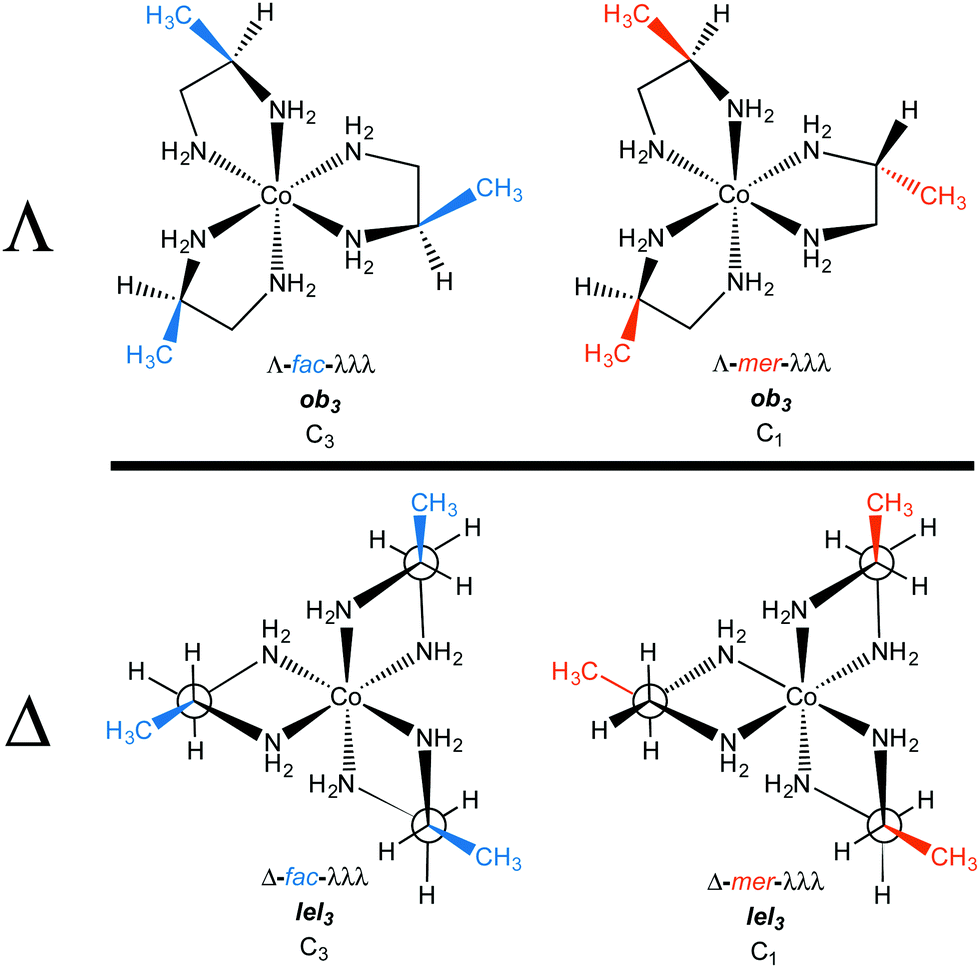 | ||
| Fig. 7 Principal stereoisomers of the [Co((R)-pn)3]3+ trication. | ||
Alert readers will note that upon going from Fig. 1a–c to Fig. 4 to Fig. 7, structures of progressively lower symmetries are encountered (D3, C3, C2, C1). Thus, it is essential to define a common reference point. Accordingly, this is taken as the “formal” or “pseudo” C3 axis that is perpendicular to the two “Star of David” triangles (Fig. 1a), each comprised of one NH2 group from each of the three chelate rings. This corresponds to a true C3 axis for structures with D3 or C3 symmetry, and is always depicted perpendicular to the plane of the paper.
4.3 Stereoisomers allowing all possible combinations of ligand configurations
Cobalt(III) complexes of racemic pn are also chiral. This leads to a series of stereoisomers consisting of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 or 1:2 (heterochiral) mixtures of (R)-pn and (S)-pn ligands, in addition to those enumerated above. Table 1 summarizes these various isomers, which were originally enumerated by Harnung.27 However, drawing the new structures is left as an exercise for the reader – one that would be very appropriate for a graduate student take-home exam. The answers are supplied in Fig. S2 of the ESI.†
:1 or 1:2 (heterochiral) mixtures of (R)-pn and (S)-pn ligands, in addition to those enumerated above. Table 1 summarizes these various isomers, which were originally enumerated by Harnung.27 However, drawing the new structures is left as an exercise for the reader – one that would be very appropriate for a graduate student take-home exam. The answers are supplied in Fig. S2 of the ESI.†
| Cobalt configuration | Ligand configuration | Preferred chelate conformation | Perspective down the C3 axisa | Geometric type isomers | Number of stereoisomers |
|---|---|---|---|---|---|
| a Or an equivalent axis as defined in the text. b This increases if chelate conformations that have pseudoaxial methyl groups are allowed (see Fig. 6). | |||||
| Λ | SSS | δδδ | lel 3 | fac (1), mer (1) | 2 |
| Δ | RRR | λλλ | fac (1), mer (1) | 2 | |
| Λ | RSS | λδδ | oblel 2 | fac (1), mer (3) | 4 |
| Δ | SRR | δλλ | fac (1), mer (3) | 4 | |
| Λ | SRR | δλλ | lelob 2 | fac (1), mer (3) | 4 |
| Δ | RSS | λδδ | fac (1), mer (3) | 4 | |
| Λ | RRR | λλλ | ob 3 | fac (1), mer (1) | 2 |
| Δ | SSS | δδδ | fac (1), mer (1) | 2 | |
| Number of stereoisomers | 24b | ||||
5. Octahedral Werner complexes with symmetrically disubstituted ethylenediamine ligands
Many Werner complexes have been prepared from ethylenediamine ligands with two identical vicinal substituents. Examples include 2,3-butanediamine(1,2-dimethylethylenediamine),28 1,2-cyclohexanediamine (chxn),29,30 and 1,2-diphenylethylenediamine (dpen).23,31,32 In all cases, three ligand stereoisomers are possible, as depicted for chxn in Fig. 8. The first two are chiral and constitute the familiar “rac” pair, whereas the last is the achiral meso diastereomer.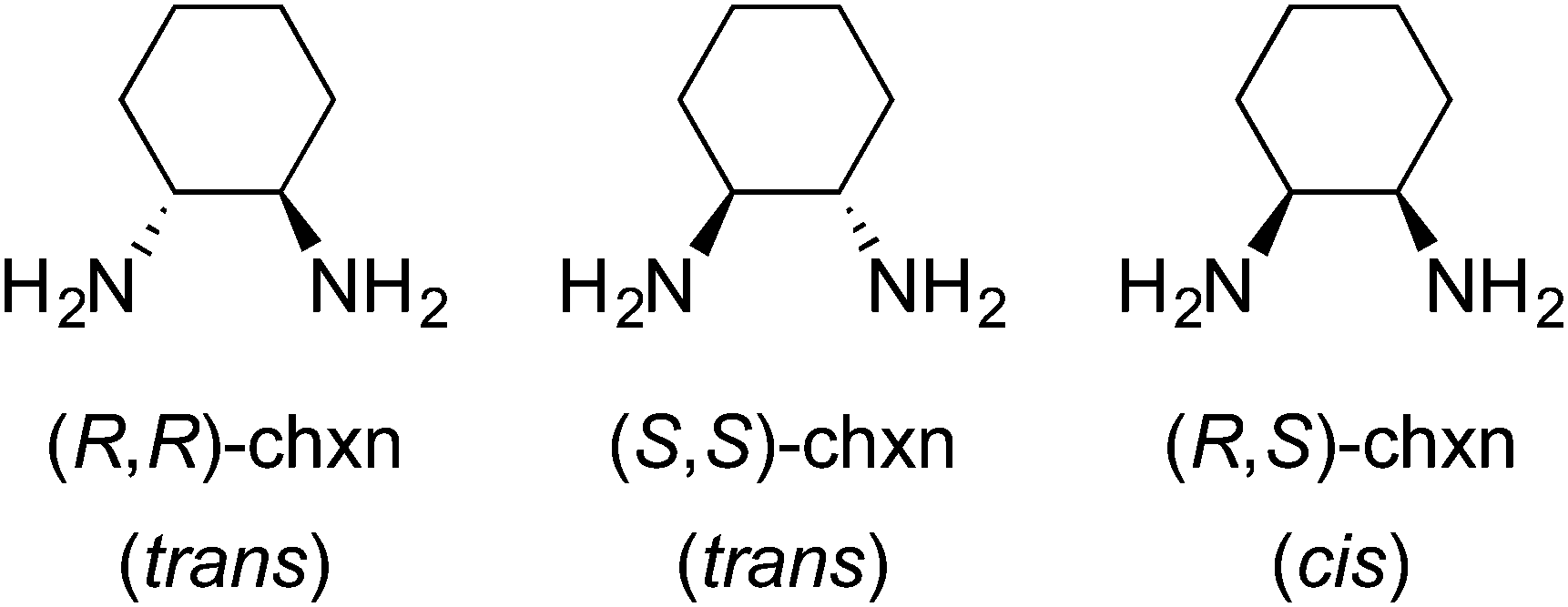 | ||
| Fig. 8 Configurational stereoisomers of 1,2-cyclohexanediamine. | ||
Due to the metal centered chirality, meso ligands can yield chiral adducts, and enantiopure complexes have in fact been isolated.28a,30a Since chxn complexes have received the most attention in the literature, these are treated first.
5.1 Stereoisomerism at the cobalt center
Consider first the chelation of three identical ligands of any of the preceding types to cobalt. As with the other cases above, either Λ or Δ configurations can result. As a general rule, disubstituted ethylenediamine ligands that have C2 symmetry, such as trans-chxn or (S,S)- or (R, R)-dpen, do not lead to fac/mer isomers. For cis or meso (R,S)-chxn or (R,S)-dpen, which are not C2 symmetric, fac/mer isomers result. In a fac isomer, the three Co–NH2 bonds that connect to carbon atoms with R configurations would all have bond angles of ca. 90°. In a mer isomer, one of these angles would be ca. 180°.5.2 Ligand conformation: trans-1,2-cyclohexanediamine (trans-chxn)29
As shown in Fig. 9, the chelation of trans-chxn requires that both NH2 groups occupy equatorial positions (a single atom cannot span axial substituents that have 1,2 relationships). With (R,R)-chxn (top left), this in turn requires a λ chelate conformation. Thus, for the tris(chelate) [Co((R,R)-chxn)3]3+, only Λ-λλλ (Λ-ob3) and Δ-λλλ (Δ-lel3) stereoisomers are possible (bottom left). In contrast, (S,S)-chxn requires a δ chelate conformation (Fig. 9, top right). For the tris(chelate) [Co((S,S)-chxn)3]3+, only Λ-δδδ (Λ-lel3) and Δ-δδδ (Δ-ob3) stereoisomers are possible (bottom right), each enantiomeric with one of the isomers of [Co((R,R)-chxn)3]3+. There are no other stereoisomers for tris(chelates) in which the trans-chxn ligands are homochiral.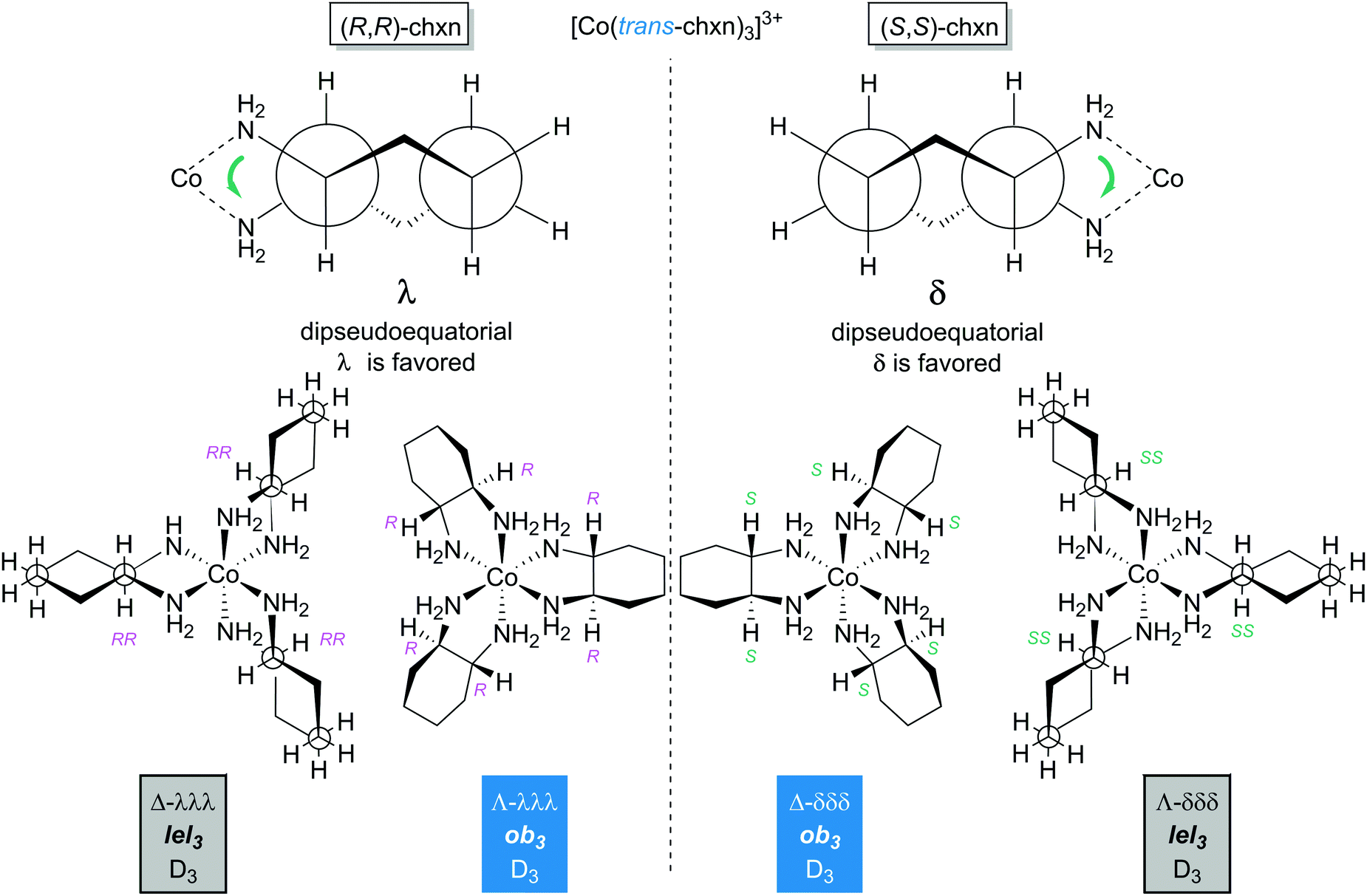 | ||
| Fig. 9 Diamine chelate conformations and principal stereoisomers of the [Co(trans-chxn)3]3+ trication with homochiral (all S,S or all R,R) ligands. | ||
Next, consider the complexes possible when the trans-chxn ligands are no longer restricted to be homochiral (as with syntheses carried out with racemic diamine).29 This is reminiscent of the scenario entertained for pn in Table 1, but more tractable due to the higher ligand symmetry and constraints imposed by the cyclohexane ring. The eight possible stereoisomers are depicted in Fig. 10, four with a Λ cobalt configuration (top), and four with a Δ configuration (bottom). Each set of four contains a complex derived from (i) three (R,R)-chxn ligands (depicted in fuller form in Fig. 9), (ii) two (R,R)-chxn ligands and one (S,S)-chxn ligand, (iii) one (R,R)-chxn ligand and two (S,S)-chxn ligands, and (iv) three (S,S)-chxn ligands (depicted in Fig. 9).
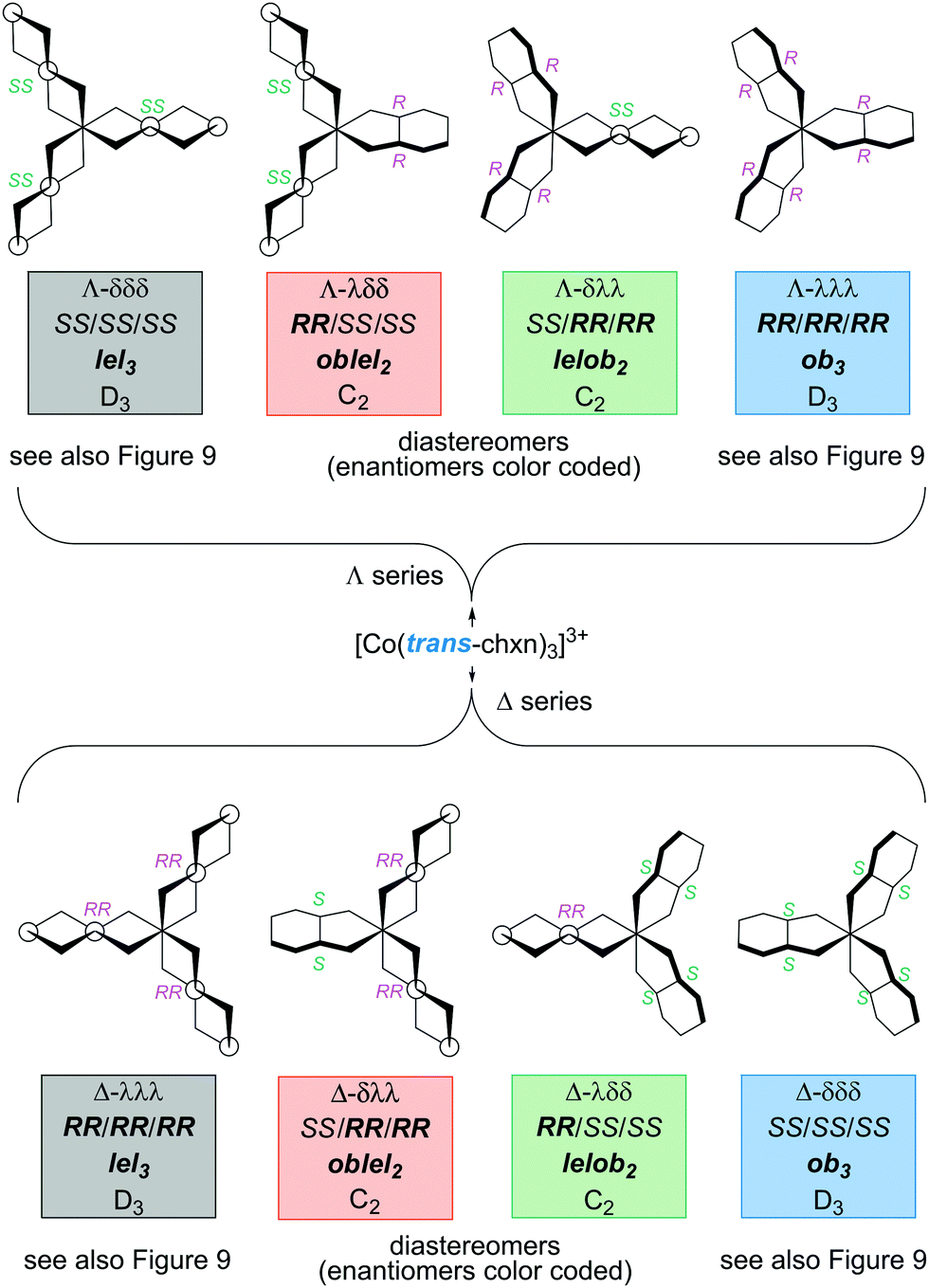 | ||
| Fig. 10 Stereoisomers of the [Co(trans-chxn)3]3+ trication with all possible combinations of (R,R)-chxn and (S,S)-chxn ligands. | ||
Enantiomeric relationships are color coded in Fig. 10. Here, designations such as Λ-λλλ and Λ-RR/RR/RR would be fully equivalent, given the inability of trans-chxn to chelate when the amino groups occupy axial positions. The latter expression may be more intuitive for organic chemists.
5.3 Ligand conformation: cis-1,2-cyclohexanediamine (cis-chxn)30
As illustrated in Fig. 11, the chelation of the meso ligand, cis- or (R,S)-chxn, requires that one amino group occupy an equatorial position on the cyclohexane ring, and the other an axial position. With respect to the five membered chelate ring, one C![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif)
![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif)
![[2 with combining low line]](https://www.rsc.org/images/entities/char_0032_0332.gif) substituent must occupy a pseudoequatorial position, and the other a pseudoaxial position. The partial structures in Fig. 11 (top) can be interconverted by a cyclohexane “ring flip”, analogous to those of related bicyclic molecules such as cis-perhydroindane and cis-decalin.
substituent must occupy a pseudoequatorial position, and the other a pseudoaxial position. The partial structures in Fig. 11 (top) can be interconverted by a cyclohexane “ring flip”, analogous to those of related bicyclic molecules such as cis-perhydroindane and cis-decalin.
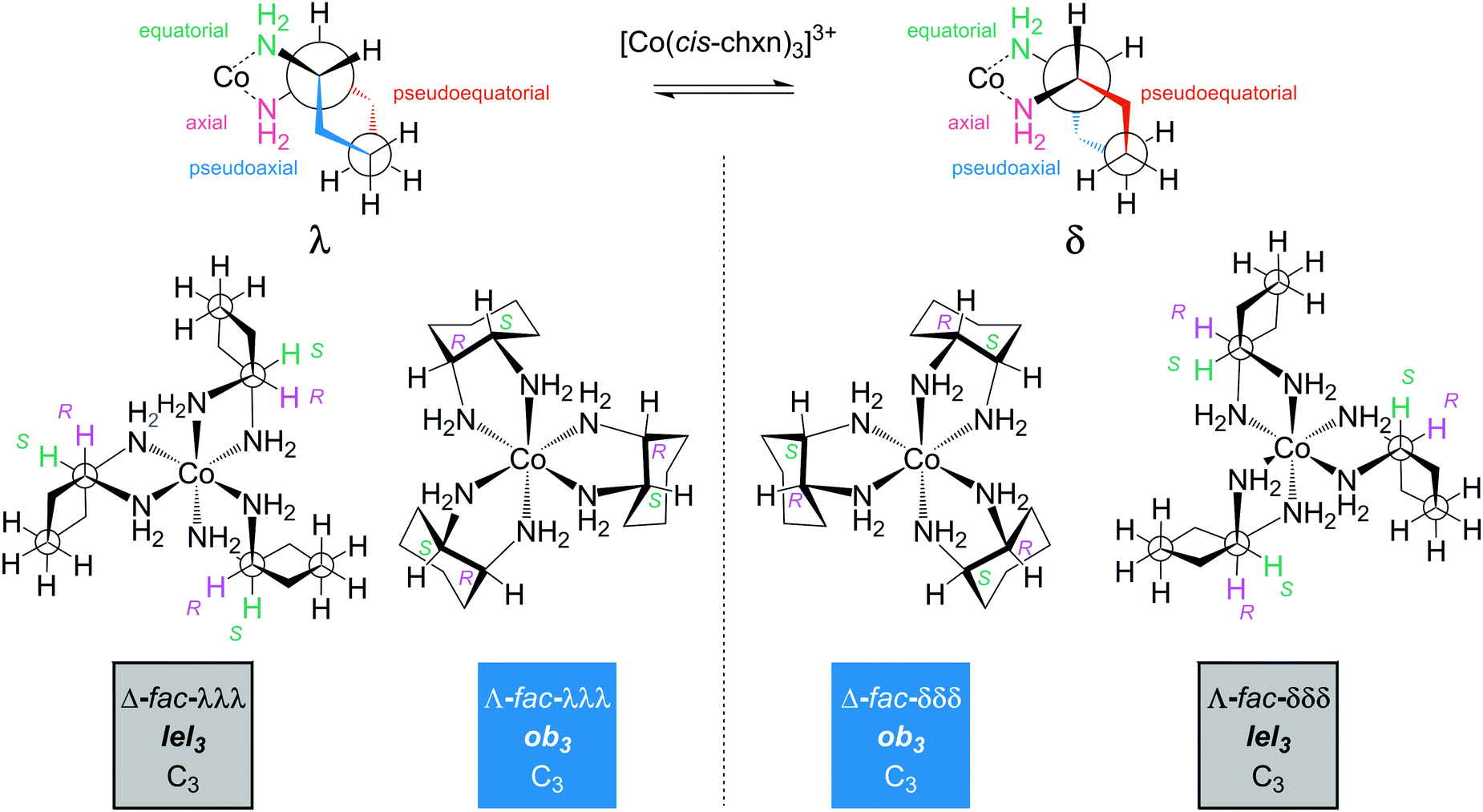 | ||
| Fig. 11 Diamine chelate conformations and representative stereoisomers of the [Co(cis-chxn)3]3+ trication; axial and equatorial are used to designate C–N bond positions on the cyclohexane ring, and pseudoaxial and pseudoequatorial are used to designate C– bond positions on the chelate ring. | ||
Four representative stereoisomeric cobalt tris(chelates) are shown in Fig. 11 (bottom). These have cobalt and chelate configurations as well as lel/ob orientations and enantiomeric relationships analogous to those of [Co(trans-chxn)3]3+ in Fig. 9. However, additional fac/mer descriptors are required; all of those illustrated are fac isomers (as is easily derived from the R/S labels).
As one generates all possible permutations of cobalt and chelate configurations and fac/mer arrays, a plethora of stereoisomers proves possible. Only by simultaneously building molecular models of each (to ensure no duplicates and optimally test for mirror images) were the authors able to convince themselves of the existence of 24 stereoisomers (12 pairs of enantiomers). Given this somewhat overwhelming set of data, the structures are depicted in the ESI† (Fig. S3) and summarized in Table S1. However, many of them can be interconverted by a cyclohexane “ring flip”, and when this is applied to all three chelates only four distinct families of isomers remain. These consist of Λ-fac and Δ-fac groupings (enantiomeric, four members each), and Λ-mer and Δ-mer groupings (enantiomeric, eight members each).
Interestingly, although all four types of stereoisomers have been isolated in enantiopure form, they have not been extensively analyzed.30 Given the low barriers for most cyclohexane “ring flips”, these likely exist as mixtures of conformers in solution. In any case, Fig. S3 (ESI†) testifies as to the incredible stereochemical diversity associated with the title compounds.
5.4 Ligand conformation: (S,S)- and (R,R)-1,2-diphenylethylenediamine (dpen)
As illustrated in Fig. 12, the chelation of either enantiomer of dpen requires that both phenyl groups occupy either pseudoequatorial or pseudoaxial positions. Naturally the former will be greatly favored. This results in a λ chelate conformation for (R,R)-dpen and a δ conformation for (S,S)-dpen. It then follows that for the tris(chelate) [Co((R,R)-dpen)3]3+, the stereoisomers Λ-λλλ (Λ-ob3) and Δ-λλλ (Δ-lel3) should be the more stable (Fig. 12, bottom left). Similarly, for [Co((S,S)-dpen)3]3+, the stereoisomers Λ-δδδ (Λ-lel3) or Δ-δδδ (Δ-ob3) will be the more stable (Fig. 12, bottom right; mirror images of the bottom left structures). All of these are analogous to the isomers given for trans- or (S,S)- and (R,R)-chxn in Fig. 9 (in which the C substituents on the chelate ring are constrained to occupy pseudoequatorial positions).
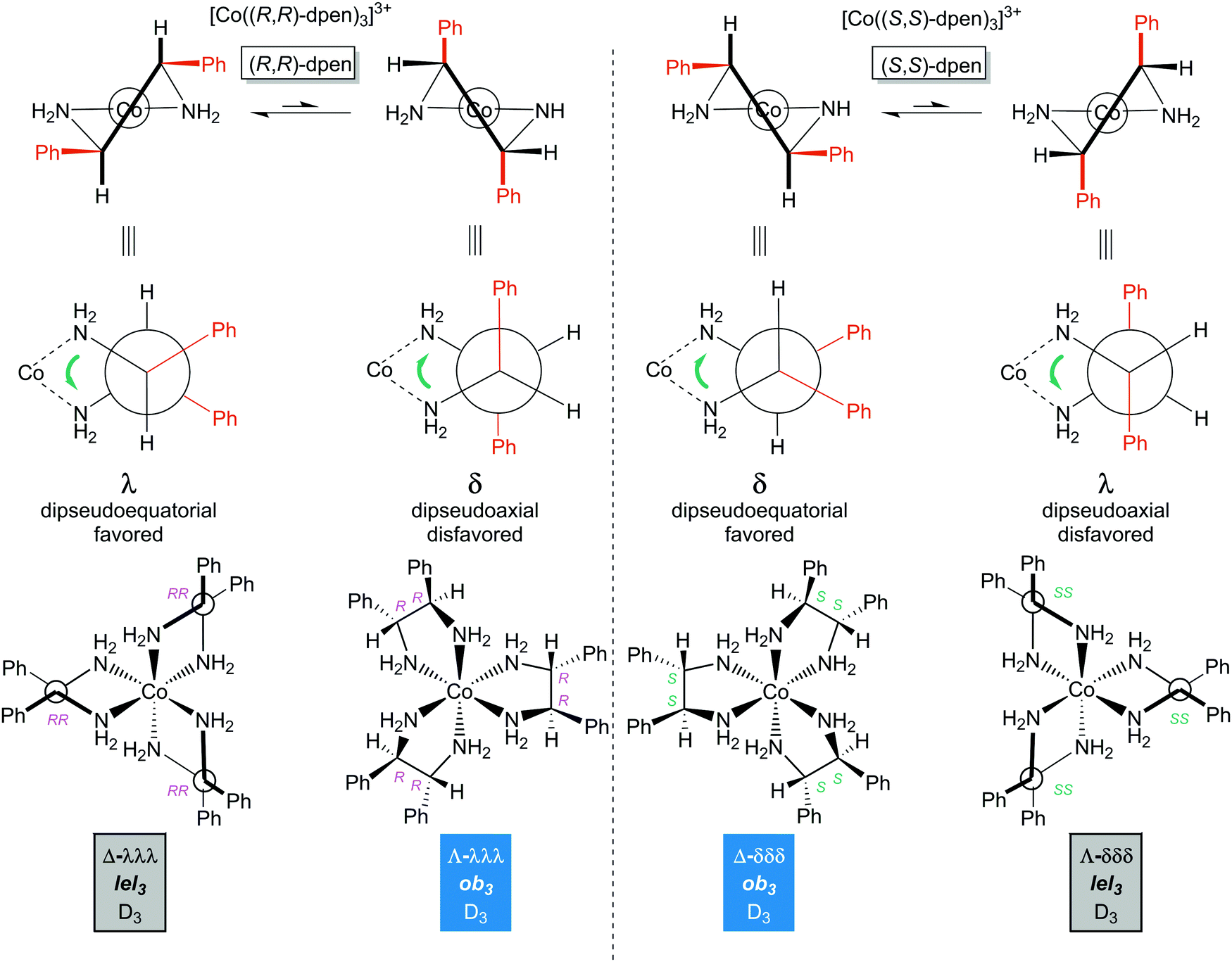 | ||
| Fig. 12 Diamine chelate conformations and principal configurational stereoisomers of the [Co(dpen)3]3+ trication with homochiral (all S,S or all R,R) ligands. | ||
As was analyzed for other chiral chelate ligands above, tris(chelate) complexes of dpen can be generated from a racemate. As shown in Fig. 13, eight stereoisomers are possible, four with a Λ configuration at cobalt and four with a Δ configuration. Half of these feature combinations of heterochiral (R,R)-chxn and (S,S)-chxn ligands. The situation is closely related to that for [Co(trans-chxn)3]3+ in Fig. 10.
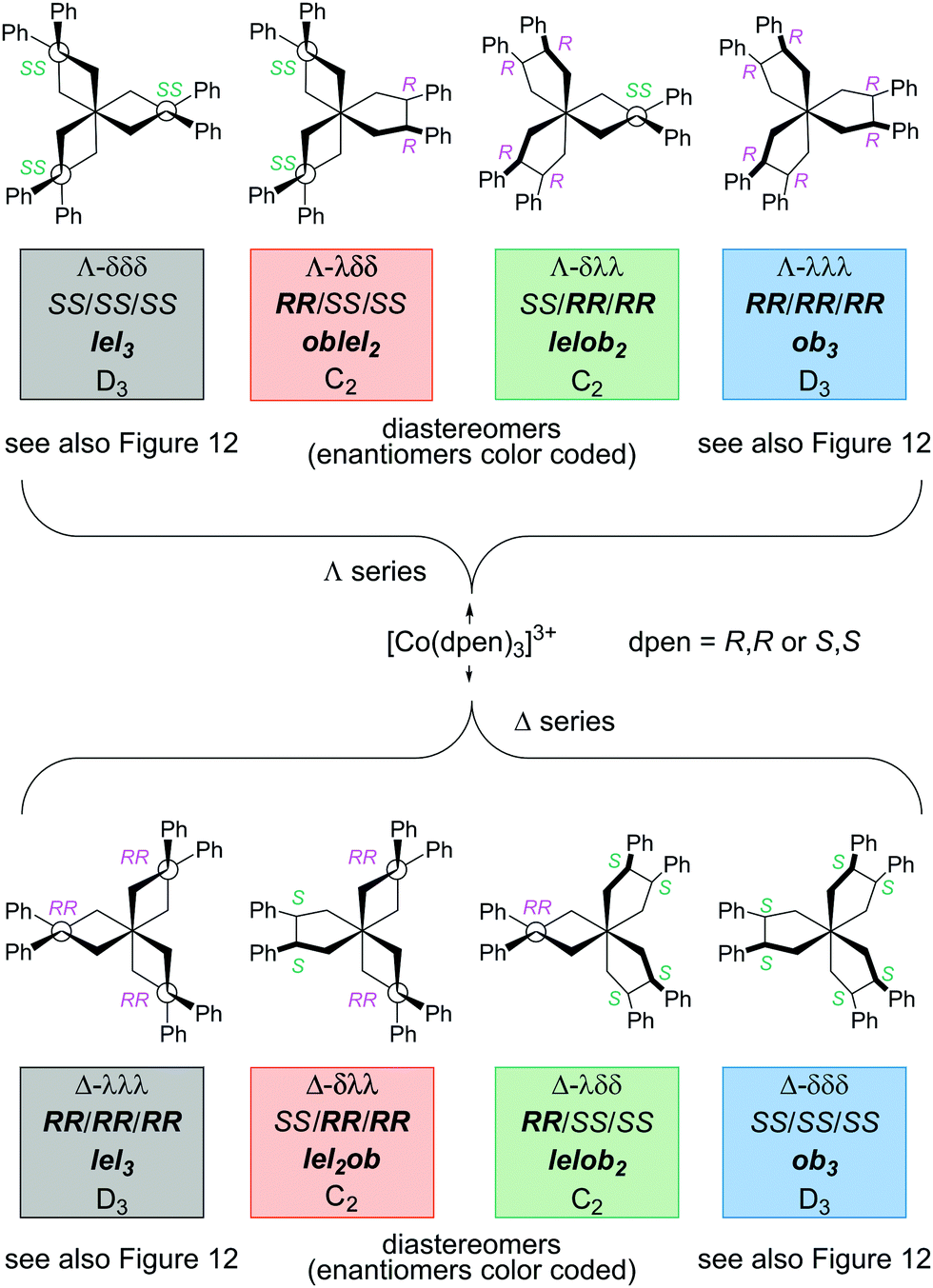 | ||
| Fig. 13 Stereoisomers of the [Co(dpen)3]3+ trication with all possible combinations of (R,R)-dpen and (S,S)-dpen ligands. | ||
A similar analysis for the meso ligand (R,S)-dpen leads to a set of isomers analogous to those of [Co(cis-chxn)3]3+ in Fig. 11 and Fig. S3 (ESI†). However, reports to date indicate that such adducts are labile, presumably due to steric interactions resulting from the phenyl group that must occupy a pseudoaxial position on the chelate ring.32 Several isomeric tris(chelate) cobalt(III) complexes of the meso ligand (R,S)-2,3-butanediamine have been isolated.28a,b
6. Diastereomer stabilities
A detailed discussion or analysis of the relative stabilities of the preceding types of diastereomers would be beyond the scope of this review. Indeed, there is strong evidence that interactions with anions can affect stability orders in solution,5a,26b and this issue is currently being probed in the authors' laboratory. Nonetheless, available literature is briefly summarized.In their seminal study of the [Co(en)3]3+ trication, Corey and Bailar estimated Δ-λλλ-[Co(en)3]3+ (which has a lel3 orientation per Fig. 3) to be more stable than Δ-δδδ-[Co(en)3]3+ (which has an ob3 orientation) by 1.8 kcal mol−1.24 This was based upon intramolecular carbon–hydrogen and hydrogen–hydrogen interactions, a treatment the authors admitted was a rough approximation. Isomers with oblel2 and lelob2 orientations were thought to have intermediate stabilities. More recent DFT results suggest that Δ-λλλ-[Co(en)3]3+ is 1.5 kcal mol−1 more stable than Δ-δδδ-[Co(en)3]3+ in water.22a
In another study, Harnung was able to equilibrate diastereomers of the trichloride salt [Co(trans-chxn)3]3+ 3Cl−.29 As shown in Fig. 14, an aqueous solution of the racemate, both enantiomers of which have lel3 orientations (Fig. 10), was refluxed over charcoal21 in the presence of added racemic trans-chxn. All of the stereoisomers depicted in Fig. 10 were generated, and the ratios (determined after chromatographic separation) showed a distinct trend. Namely, the original stereoisomers with lel3 orientations dominated, and quantities monotonically decreased as the proportion of ob orientations increased.
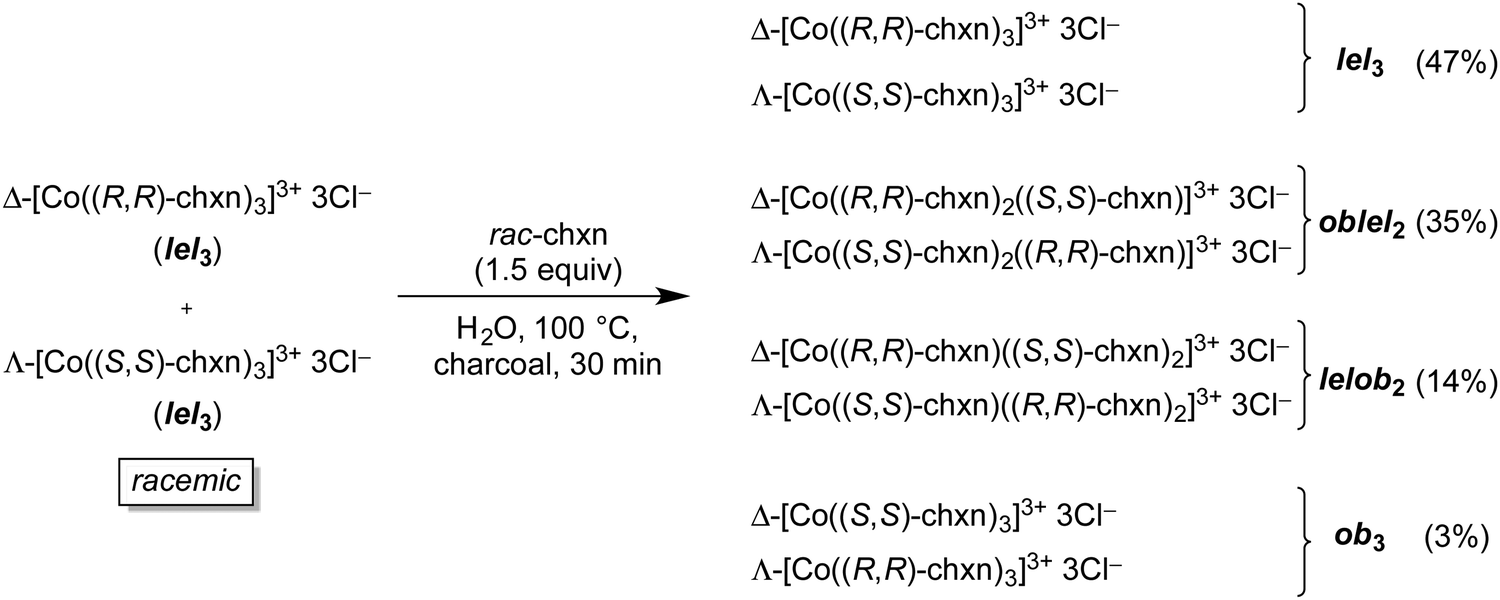 | ||
| Fig. 14 Thermal equilibration of racemic [Co(trans-chxn)3]3+ 3Cl− in the presence of excess ligand. | ||
In a follow up experiment shown in Fig. 15,27 an aqueous solution of enantiopure Δ-[Co((R,R)-chxn)3]3+ 3Cl− (which has an lel3 orientation) was refluxed over charcoal in the presence of enantiopure (R,R)-chxn. After the same time, only 7% of a new stereoisomer had been generated, Λ-[Co((R,R)-chxn)3]3+ 3Cl− (which per Fig. 10 has an ob3 orientation).
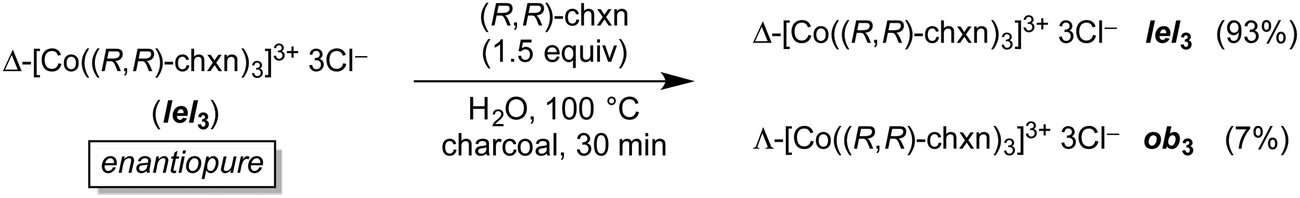 | ||
| Fig. 15 Thermal equilibration of enantiopure Δ-[Co((R,R)-dpen)3]3+ 3Cl− in the presence of excess ligand. | ||
An experiment analogous to that in Fig. 14 was conducted with the salt [Co(pn)3]3+ 3Cl− that had been generated from racemic pn and an achiral cobalt(II) precursor.27 The many possible isomers of this trication were analyzed in Table 1. Chromatography gave separate fractions for the lel3, oblel2, lelob2, and ob3 structures (each fraction a mixture of stereoisomers) in a 35.0:41.1:18.0:4.0 ratio with excellent mass balance. Additional background regarding the often surprisingly efficient chromatographic separation of isomeric (even enantiomeric) tris(diamine) cobalt(III) complexes is provided elsewhere.33 A version of this experiment with non racemic [Co(pn)3]3+ 3X− has also been reported.34 An equilibration similar to that in Fig. 14 but with the meso or cis chxn adduct [Co((R,S)-chxn)3]3+ 3X− has been mentioned, but the isomer ratios were not given.30a
Thus, a clear bias for isomers with lel rich orientations is observed in the preceding experiments. However, it should be noted that in Fig. 14 and 15, reactants already possessing lel3 orientations were employed. Also, the authors did not definitively establish that equilibrium had been attained. Perhaps longer reaction times would have resulted in greater proportions of products with ob-rich orientations. Furthermore, the counter anion, chloride, is a strong hydrogen bond acceptor and may influence equilibrium ratios. Nonetheless, the aggregate data strongly suggest a trend, which is furthermore consistent with preliminary DFT computational results obtained by the authors.
7. Catalysis
A detailed treatment of the catalytic properties of the preceding types of complexes will be deferred until additional results from the authors' laboratory are published. However, applications reported to date are summarized in Fig. 16.4–6 These include Michael additions of malonate esters to α,β-unsaturated nitro compounds (top) and ketones, additions of 1,3-dicarbonyl compounds to di(t-butyl) azodicarboxylate, yielding amino acid precursors (middle), and ring opening polymerizations of lactide (bottom).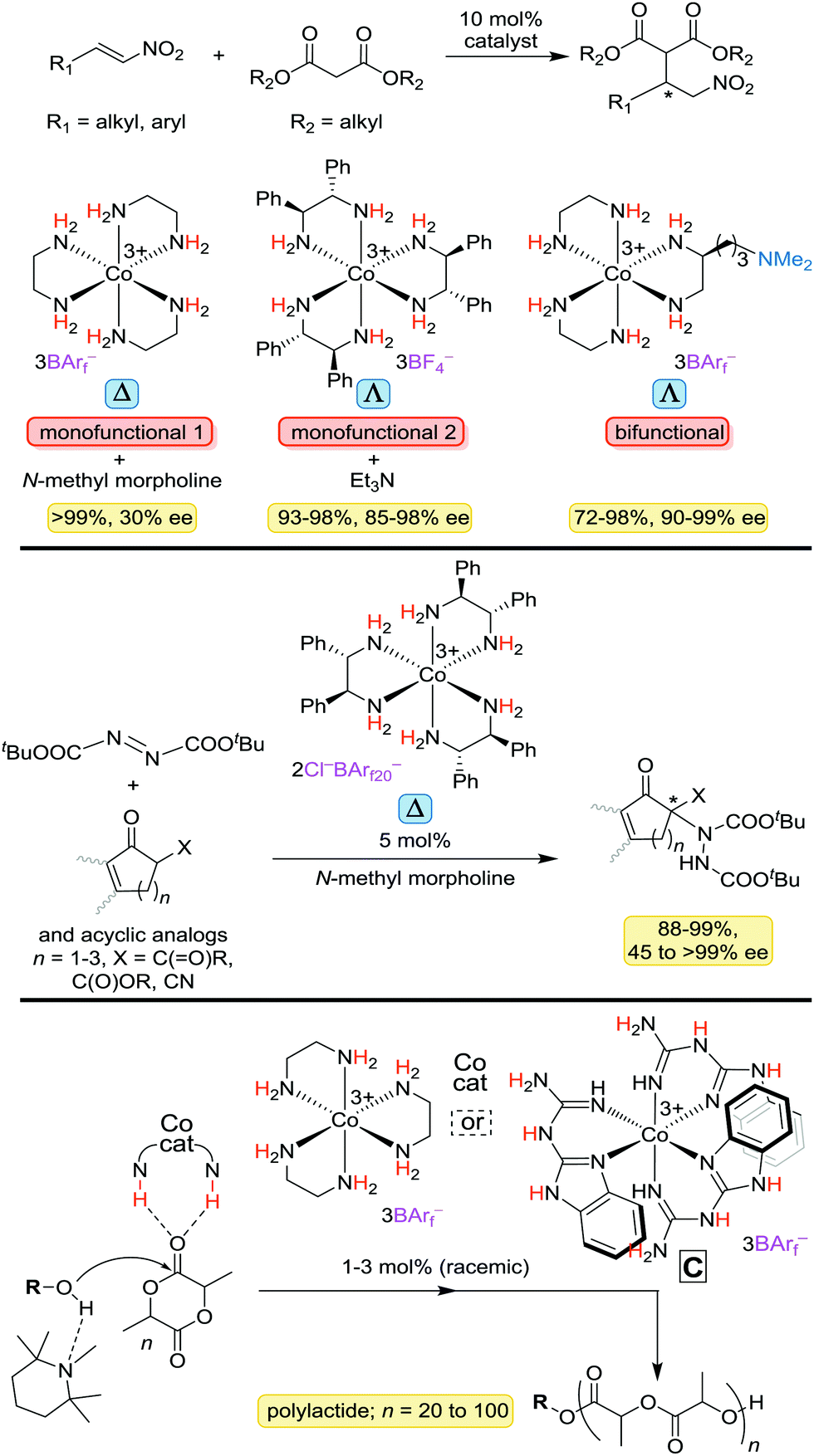 | ||
| Fig. 16 Organic transformations that can be catalyzed by cobalt(III) complexes with ethylenediamine or substituted ethylenediamine ligands (BArf− = B(3,5-C6H3(CF3)2)4−; BArf20− = B(C6F5)4−). | ||
With the first reaction, a bifunctional catalyst that incorporates a tertiary amine proves somewhat more effective than a monofunctional catalyst with dpen ligands that is used in conjunction with Et3N. With the last reaction, another type of chelating nitrogen donor ligand, in which the activating NH groups are found in the chelate backbone, proves to be especially effective (see C). Although the polymerizations have yet to be conducted with an enantiopure catalyst, the other reactions have and under optimum conditions deliver products of >90% ee. Many mechanistic questions surrounding these processes remain extant.
8. Summary and recommendations
Although all of the examples cited in this survey involve cobalt, the principles and conclusions can clearly be extrapolated to all octahedral tris(chelate) metal complexes of ethylenediamines. Also, most types of carbon substituted ethylenediamine ligands should be closely modeled by one of the examples analyzed. For instance, pn can be viewed as representative of all monosubstituted diamines. For geminally disubstituted analogs such as H2NCRR′CH2NH2, the larger R/R′ group would prefer a pseudoequatorial position in the chelate ring, leading to stereoisomers analogous to those of pn. The chxn and dpen complexes define the stereoisomer space for ethylenediamines with identical vicinal substituents. In cases where the substituents are rendered unequal (e.g., H2NCHRCHR′NH2), what were formerly rac type ligands will now have an added layer of fac/mer isomerism. However, such cobalt tris(chelate) complexes remain, to the authors' knowledge, unknown.Underneath the many structures given above can be found a variety of types of “names”. In the authors' view, the most appropriate one will be situational. If one wishes to specify a reactant or product, representations of the type in Fig. 14 and 15 are sufficient. If one wishes to denote a specific three dimensional structure, all of the descriptors Λ/Δ, R/S, λ/δ, and (for diamines without C2 symmetry) fac/mer will generally be necessary. With certain conformationally restricted diamines (e.g., chxn), the R/S and λ/δ designations may be formally redundant, but both are recommended nonetheless. The authors consider it a disservice to the reader to replace the configuration of a ligand, such as (R,R)-chxn in Fig. 14 and 15, with the sign of the optical rotation, such as (−)-chxn. However, the sign of the optical rotation of the complex may be added at the front of the name if desired.
In the authors' opinion, the descriptors lel and ob do not represent primary stereochemical phenomena but rather chelate orientations or perspectives that are enforced by fundamental stereochemical variables. Hence, these designations are given separately. However, they are clearly valuable for purposes of geometric classification. Furthermore, as noted in the previous section, this feature appears to be a major determinant with respect to chromatographic retention times and thermodynamic stabilities. Efforts to drill down deeper on this relationship are underway.
These are exciting times for Werner complexes. They have always been a classroom favorite, but most often for illustrating tried and true physical principles established long ago. Now they have contemporary applications in enantioselective catalysis,4–6,35,36 and the literature and new analyses summarized above should help to better understand the underlying phenomena and more methodically mine this emerging field.
Acknowledgements
The authors thank the Welch Foundation (Grant A-1656) for support.Notes and references
- General treatments of inorganic stereochemistry that include introductions to Werner complexes of 1,2-diamines: (a) A. von Zelewsky, Stereochemistry of Coordination Compounds, John Wiley & Sons, Chichester, 1996 Search PubMed; (b) H. Amouri and M. Gruselle, Chirality in Transition Metal Chemistry, John Wiley & Sons, Chichester, 2008 Search PubMed; (c) C. J. Hawkins, Absolute Configuration of Metal Complexes, Wiley-Interscience, New York, 1971 Search PubMed; (d) See also G. B. Kauffman, Coord. Chem. Rev., 1974, 12, 105–149 CrossRef CAS.
- (a) A. Werner, Chem. Ber., 1911, 44, 1887–1898 CrossRef CAS . V. L. King is listed as an author for the Experimental section; (b) A. Werner, Chem. Ber., 1911, 44, 2445–2455 CrossRef; (c) A. Werner, Chem. Ber., 1911, 44, 3272–3278 CrossRef; (d) A. Werner, Chem. Ber., 1911, 44, 3279–3284 CrossRef; (e) A. Werner, Chem. Ber., 1912, 45, 121–130 CrossRef CAS.
- (a) H. Taube, Chem. Rev., 1952, 50, 69–126 CrossRef CAS; (b) Data for [Co(en)3]3+: J. A. Friend and E. K. Nunn, J. Chem. Soc., 1958, 1567–1571 RSC; (c) For an entry level rationale, see W. U. Malik, G. D. Tuli and R. D. Madan, Selected Topics in Inorganic Chemistry, S. Chand & Company, New Delhi, 2002, ch. 14 Search PubMed.
- C. Ganzmann and J. A. Gladysz, Chem. – Eur. J., 2008, 14, 5397–5400 CrossRef CAS PubMed.
- (a) K. G. Lewis, S. K. Ghosh, N. Bhuvanesh and J. A. Gladysz, ACS Cent. Sci., 2015, 1, 50–56 CrossRef CAS PubMed; (b) S. K. Ghosh, C. Ganzmann, N. Bhuvanesh and J. A. Gladysz, Angew. Chem., Int. Ed., 2016, 55, 4356–4360 ( Angew. Chem. , 2016 , 128 , 4429–4433 ) CrossRef CAS PubMed; (c) A. Kumar, S. K. Ghosh and J. A. Gladysz, Org. Lett., 2016, 18, 760–763 CrossRef CAS PubMed.
- See also C. Thomas and J. A. Gladysz, ACS Catal., 2014, 4, 1134–1138 CrossRef CAS.
- S. K. Ghosh, A. Ehnbom, K. G. Lewis and J. A. Gladysz, review in preparation.
- (a) M. S. Taylor and E. N. Jacobsen, Angew. Chem., Int. Ed., 2006, 45, 1520–1543 ( Angew. Chem. , 2006 , 118 , 1550–1573 ) CrossRef CAS PubMed; (b) A. G. Doyle and E. N. Jacobsen, Chem. Rev., 2007, 107, 5713–5743 CrossRef CAS PubMed; (c) X. Yu and W. Wang, Chem. – Asian J., 2008, 3, 516–532 CrossRef CAS PubMed.
- S. K. Ghosh, A. S. Ojeda, J. Guerrero-Leal, N. Bhuvanesh and J. A. Gladysz, Inorg. Chem., 2013, 52, 9369–9378 CrossRef CAS PubMed.
- (a) A recent review that is complementary to this one:, Y. Liu, Y. Liu and M. G. B. Drew, Coord. Chem. Rev., 2014, 260, 37–64 CrossRef; (b) Y. Saito, in Topics in Stereochemistry, ed. E. L. Eliel and N. L. Alinger, 1978, vol. 10, pp. 95–174 Search PubMed.
- (a) C. Comuzzi, A. Melchior, P. Polese, R. Portanova and M. Tolazzi, Eur. J. Inorg. Chem., 2002, 2194–2201 CrossRef CAS; (b) A. Johansson, E. Wingstrand and M. Håkansson, Inorg. Chim. Acta, 2005, 358, 3293–3302 CrossRef CAS.
- (a) P. Hendry and A. Ludi, J. Chem. Soc., Chem. Commun., 1987, 891–892 RSC; (b) When a Sci-Finder search was conducted for the tris(TMEDA) trication [Co(Me2NCH2CH2NMe2)3]3+, the previous reference was obtained. However, there are no structural or line formulae given in this article, and a key phrase “producing 2,3-methyl-2,3-diaminobutane or tetramethylethylenediamine (tmen)” can be construed to mean two separate compounds or the same compound (the title and abstract of the article are ambiguous). As can be inferred from subsequent papers in this series involving other metals, the authors must intend tmen to be 2,3-methyl-2,3-diaminobutane, meaning that instead salts of [Co(NH2CMe2CMe2NH2)3]3+ have been synthesized: S. Brönnimann, A. Zilian, H. U. Güdel and A. Ludi, Inorg. Chim. Acta, 1990, 173, 159–162 CrossRef; (c) A Sci-Finder search for [Co(NH2CMe2CMe2NH2)3]3+ does not afford ref. 12a. However, it does yield a corresponding full paper: P. Hendry and A. Ludi, Helv. Chim. Acta, 1988, 71, 1966–1970 CrossRef CAS.
- (a) J. A. Hearson, S. F. Mason and R. H. Seal, J. Chem. Soc., Dalton Trans., 1977, 1026–1034 RSC; (b) G. H. Searle and F. R. Keene, Inorg. Chim. Acta, 1989, 155, 125–138 CrossRef CAS; (c) G. H. Searle and E. R. T. Tiekink, Inorg. Chim. Acta, 1989, 156, 57–63 CrossRef CAS; (d) I. M. Atkinson, F. R. Keene and G. H. Searle, J. Chem. Soc., Dalton Trans., 1991, 45–51 RSC.
- T. S. Piper, J. Am. Chem. Soc., 1961, 83, 3908–3909 CrossRef CAS.
- A. Werner, Ber. Dtsch. Chem. Ges., 1912, 45, 865–869 CrossRef CAS.
- F. Galsbøl in Inorganic Syntheses, ed. R. W. Parry, McGraw-Hill, New York, 1970, pp. 269–280 Search PubMed.
- A. Werner, Chem. Ber., 1912, 45, 1228–1236 CrossRef CAS.
- (a) A. Werner and A. P. Smirnoff, Helv. Chim. Acta, 1920, 3, 472–486 CrossRef CAS; (b) F. Galsbøl and B. S. Rasmussen, Acta Chem. Scand., 1982, A36, 83–87 CrossRef.
- A. Werner, Vierteljahresschr. Naturforsch. Ges. Zürich, 1917, 62, 553–564 Search PubMed.
- W. G. Gehman and W. C. Fernelius, J. Inorg. Nucl. Chem., 1957, 9, 71–81 CrossRef.
- (a) B. D. Douglas, J. Am. Chem. Soc., 1954, 76, 1020–1021 CrossRef CAS; (b) For interesting observations regarding differences between types of charcoals and their catalyst lifetimes, see the final paragraph of the Experimental section of ref. 27.
- (a) F. E. Jorge, J. Autschbach and T. Ziegler, J. Am. Chem. Soc., 2005, 127, 975–985 CrossRef CAS PubMed; (b) C. F. Bell, Syntheses and Physical Studies of Inorganic Compounds, Pergamon, New York, 1972, ch. 22 Search PubMed.
- B. Bosnich and J. MacB. Harrowfield, J. Am. Chem. Soc., 1972, 94, 3425–3437 CrossRef CAS.
- E. J. Corey and J. C. Bailar Jr., J. Am. Chem. Soc., 1959, 81, 2620–2628 CrossRef CAS.
- (a) T. Nomura, F. Marumo and Y. Saito, Bull. Chem. Soc. Jpn., 1969, 42, 1016–1020 CrossRef CAS; (b) Y. Sunatsuki, S. Miyahara, T. Suzuki, M. Kojima, T. Nakashima, N. Matsumoto and F. Galsbøl, New J. Chem., 2010, 34, 2777–2784 RSC.
- (a) J. Fujita and H. Ogino, Chem. Lett., 1974, 57–58 CrossRef CAS; (b) S. Sato, Y. Saito, J. Fujita and H. Ogino, Inorg. Nucl. Chem., 1974, 10, 669–673 CrossRef CAS.
- S. E. Harnung, S. Kallesøe, A. M. Sargeson and C. E. Schäffer, Acta Chem. Scand., 1974, A28, 385–398 CrossRef.
- (a) M. Kojima, H. Funaki, Y. Yoshikawa and K. Yamasaki, Bull. Chem. Soc. Jpn., 1975, 48, 2801–2804 CrossRef CAS; (b) C. J. Hilleary, T. F. Them and R. E. Tapscott, Inorg. Chem., 1980, 19, 102–107 CrossRef CAS; (c) M. F. Gargallo, L. Lechuga, M. C. Puerta, F. González-Vílchez and R. Vilaplana, J. Chem. Educ., 1988, 65, 1018–1019 CrossRef.
- S. E. Harnung, B. S. Sørensen, I. Creaser, H. Maegaard, U. Pfenninger and C. E. Schäffer, Inorg. Chem., 1976, 15, 2123–2126 CrossRef CAS.
- (a) H. Toftlund and T. Laier, Acta Chem. Scand., 1977, 31, 651–656 CrossRef CAS; (b) T. Mizuta, K. Toshitani and K. Miyoshi, Bull. Chem. Soc. Jpn., 1991, 64, 1183–1191 CrossRef CAS.
- R. Kuroda and S. F. Mason, J. Chem. Soc., Dalton Trans., 1977, 1016–1020 RSC.
- For the isolation of racemic [Co((R,S)-dpen)3]3+ 3Cl−, which slowly decomposes in solution, see M. Kojima, M. Ishiguro and J. Fujita, Bull. Chem. Soc. Jpn., 1978, 51, 3651–3652 CrossRef CAS.
- Y. Yoshikawa and K. Yamasaki, Coord. Chem. Rev., 1979, 28, 205–229 CrossRef CAS . See in particular sections F-v and F-vi.
- F. P. Dwyer, F. L. Garvan and A. Shulman, J. Am. Chem. Soc., 1959, 81, 290–294 CrossRef CAS.
- See also (a) L. Gong, L.-A. Chen and E. Meggers, Angew. Chem., Int. Ed., 2014, 53, 10868–10874 ( Angew. Chem. , 2014 , 126 , 11046–11053 ) CrossRef CAS PubMed; (b) J. Ma, X. Ding, Y. Hu, Y. Huang, L. Gong and E. Meggers, Nat. Commun., 2014, 5, 4531 CAS; (c) H. Huo, C. Fu, C. Wang, K. Harms and E. Meggers, Chem. Commun., 2014, 50, 10409–10411 RSC; (d) J. Liu, L. Gong and E. Meggers, Tetrahedron Lett., 2015, 56, 4653–4656 CrossRef CAS; (e) Y. Hu, Z. Zhou, L. Gong and E. Meggers, Org. Chem. Front., 2015, 2, 968–972 RSC.
- V. I. Maleev, M. North, V. A. Larionov, I. V. Fedyanin, T. F. Savel'yeva, M. A. Moscalenko, A. F. Smolyakov and Y. N. Belokon, Adv. Synth. Catal., 2014, 356, 1803–1810 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Figures and tables representing all stereoisomers for the complexes described in Fig. 7 and 11. See DOI: 10.1039/c6cs00604c |
| This journal is © The Royal Society of Chemistry 2016 |