
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOld tricks, new dogs: organocatalytic dienamine activation of α,β-unsaturated aldehydes
Vanesa Marcos
*a and
José Alemán
 *b
*b
aSchool of Chemistry, University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: vanesa.marcosalgaba@manchester.ac.uk
bDepartamento de Química Orgánica (Modulo 1), Facultad de Ciencias, Universidad Autónoma de Madrid, Cantoblanco, 28049, Madrid, Spain. E-mail: jose.aleman@uam.es; Web: http://www.uam.es/jose.aleman
First published on 2nd November 2016
Abstract
Chiral secondary amines are some of the most commonly used kinds of catalysts. They have become a reliable tool for the α- and β-activation of carbonyl compounds, via HOMO, SOMO or LUMO activation pathways. Recently, chemists have turned their attention to the development of novel organocatalytic strategies for remote functionalisation, targeting stereocentres even more distant from the catalyst-activation site, through dienamine, trienamine, and vinylogous iminium ion pathways (γ-, ε- and δ-positions, respectively). Here we outline and discuss the state-of-the-art in dienamine activation, classifying examples according to the different reactive activation pathways followed by the formed dienamine intermediate (1,3-, 1,5-, 2,5- and 4,5-functionalisation) and the reaction type developed, as determined by the structure and the nature of electrophiles and nucleophiles.
 Vanesa Marcos | Vanesa Marcos obtained her PhD from the Universidad Autónoma de Madrid under the Supervision of Prof. García Ruano and Dr Alemán and focussed on sulfinyl carbanions and organocatalytic methodologies. During this period, she spent six months in Prof. Jørgensen's group (Denmark), and four months in Prof. Tiecco's group (Italy). In 2011, she joined Prof. Martín's group as a Postdoctoral Researcher working on the synthesis of chiral fullerenes. Currently, she is the Project Manager in the Leigh group at the University of Manchester (UK) and is focussed on the development of new molecular machines. Her research interests include asymmetric synthesis, catalysis and molecular machines. |
 José Alemán | José Alemán obtained his PhD working on sulfur chemistry, under the supervision of Professor García Ruano in 2005. In 2003, he spent six months in the laboratory of Prof. Padwa at Emory University, Atlanta, USA. He subsequently carried out postdoctoral research (2006–2008) at the Center for Catalysis in Aarhus (Denmark) with Prof. Jørgensen. He returned to Spain in 2009 and is currently a ‘Ramón y Cajal’ researcher at the Universidad Autónoma de Madrid (Spain). His research interests include asymmetric synthesis and catalysis. More recently, he received a Consolidator Grant awarded by the European Research Council (see http://www.uam.es/jose.aleman). |
1. Introduction
In the past decade, organocatalysis has emerged as a powerful and environmentally benign strategy towards numerous organic transformations.1 Since its renaissance it has been proven to be a robust and useful tool, and equally as competent as metal or enzyme catalysis. Organocatalysis can be sub-classified into covalent and non-covalent catalysis. In the covalent catalysis, the carbonyl group has played a central role and has allowed the introduction of several new modes of chemical activations with hundreds of newly developed reactions. Therefore, this bio-mimetic “organocatalytic revolution” has inspired the development of original activation modes, especially those concerning the carbonyl group. Among these, activation accomplished by chiral amines has been shown to be a particularly powerful tool due to the well-established methods of HOMO,2 LUMO,3 and SOMO4 activations widely used in asymmetric synthesis.In 2000, the concepts of LUMO5-lowering (iminium ion, Fig. 1) and HOMO-raising (α-functionalisation via enamine,2a–fFig. 1) organocatalytic strategies were applied to the activation of both, unsaturated and saturated, carbonyl compounds. Since then, the application of asymmetric aminocatalysis to activate a more remote position has been under consideration, but it took several years for it to be accomplished6 (in contrast to the remarkably quick advances in the field of the chiral auxiliaries7). From a historical point of view, the stoichiometric enamine preparation was first made practical by Mannich and Davidsen8 and it was used for the first time, in a catalytic manner, in the well-known Hajos–Parrish–Eder–Sauer–Wiechert reaction (Scheme 1).9
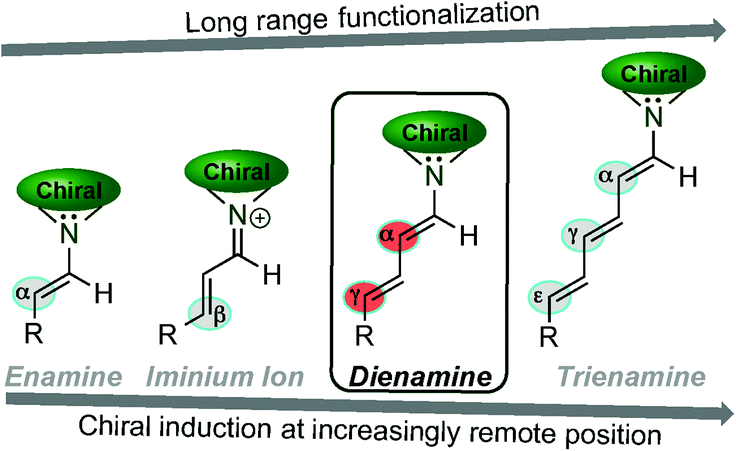 | ||
| Fig. 1 Different long extended functionalisation of aldehydes. | ||
 | ||
| Scheme 1 The first example of asymmetric organocatalysis using HOMO-raising activation. | ||
After this early example from the 70's, in 1993, Serebryakov and colleagues were the first to study the stoichiometric and organocatalytic dienamine activation of α,β-unsaturated aldehydes (Scheme 2).10 In these works, the authors described the synthesis of different polysubstituted cyclohexadienes, using the HOMO-raising activation of the dienamine system. The authors reported that after the formation of the chiral dienamine, a subsequent cycloaddition from its sterically less hindered face to a dienophile (exo or endo approach) can take place to give the final cycloadducts which, after hydrolysis and release of the catalyst, could give the polysubstituted cyclohexadienes. This mechanism was supported experimentally through the isolation of different chiral dienamine intermediates by condensation of an aldehyde with a chiral secondary amine and subsequently by the reaction of these dienamines with the dienophile in a two-step process (see the bottom, Scheme 2).
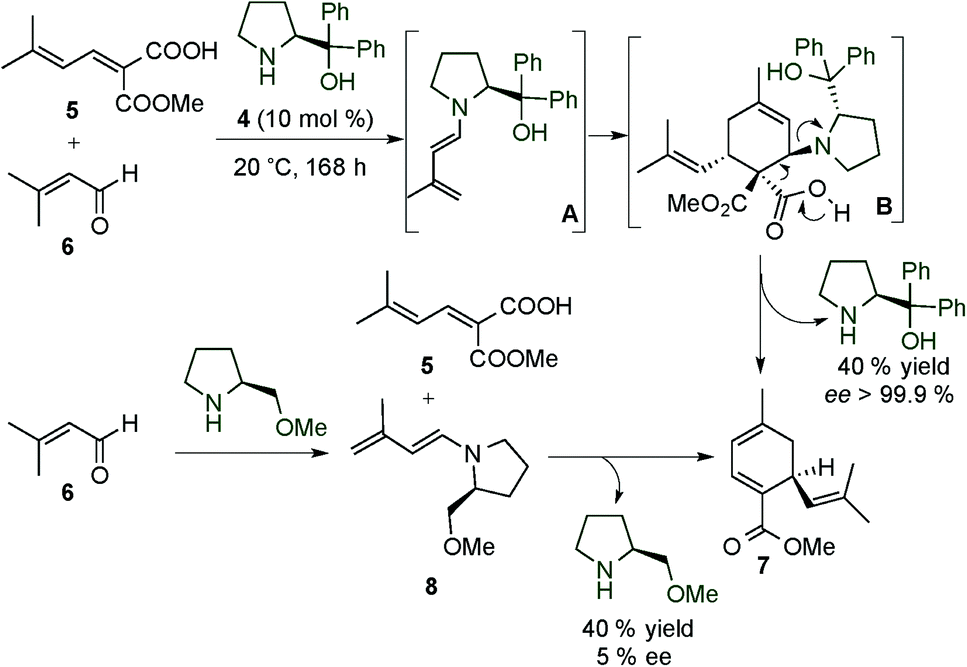 | ||
| Scheme 2 First asymmetric catalytic example using dienamine activation for the synthesis of cyclohexadienes. | ||
In addition, based on computational studies, the authors proposed some stereochemical models for the obtained results (Scheme 3).10c The plane accommodating the five backbone atoms of the delocalized system of a dienamine has sterically non-equivalent faces. Therefore, four favourably oriented complexes can be found; two are related to the endo approaches, whereas the other two correspond to the exo ones. The endo-I and exo-I approaches lead to the same S configuration of the final product (where the intermediate cycloadducts are epimers at the configuration of the C–N bond), resulted from the reaction of the dienophile by the “lower face”. In an analogous manner, when the dienophile reacts through the “upper face”, the endo-II and exo-II approaches allow the synthesis of the R configuration final product (where intermediates are again epimers at the C–N bond).
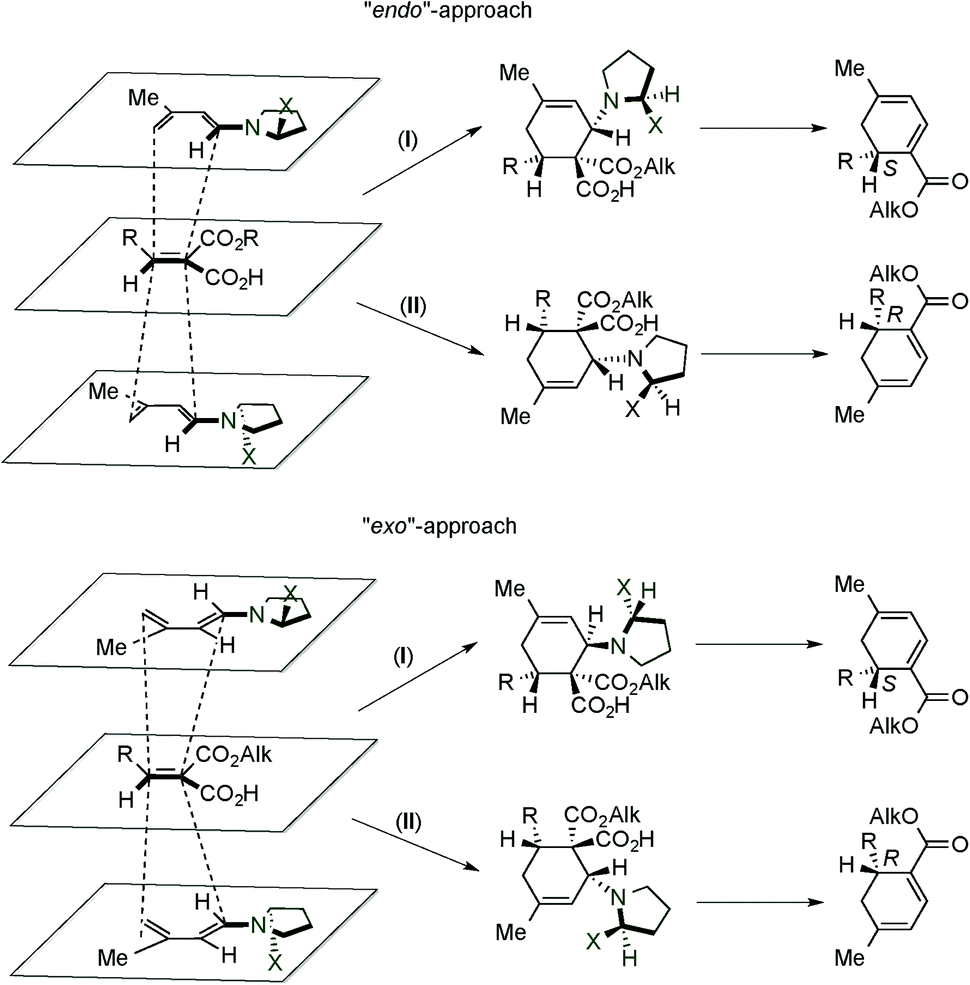 | ||
| Scheme 3 Proposed reaction mechanism and stereochemical model by Serebryakov et al. | ||
Whereas the level of control achieved in this early example was excellent, no representative examples were reported in this field until 2006 when Jørgensen et al. described the aza-1,5-functionalisation of α,β-unsaturated aldehydes.11 In a parallel manner, the dienamine activation of α,β-unsaturated ketones was developed by Barbas III and Ramachary12 and owing to the fact that it has been recently discussed in detail by other revisions,13 only the functionalisation of enals via dienamine intermediates has been discussed in this review.
The dienamine intermediate formed (Fig. 2) presents different activation pathways to react by, and the pathway that the reaction takes is determined by the structure and the nature of electrophiles and nucleophiles (HOMO and LUMO coefficients). The simplest pathway undergoes 1,3-reactivity which allows the α-functionalisation of the aldehyde and leaves an unreacted double bond available for further modification (top-left, Fig. 2). The second reactivity relates to the 1,5-functionalisation, and was the first reported remote functionalisation (bottom-left, Fig. 2).11 Thirdly, by taking into account that the normal Diels–Alder reaction usually proceeds with electron rich-dienes, the formed dienamine has a large tendency to react with dienophiles. Thus, a large number of publications have described using this strategy, in an intra- and inter-molecular manner, for the synthesis of enantio-enriched six membered rings (top-right, Fig. 2). In a final approach, inspired by the pioneer mechanistic studies carried out by Seebach and Blackmond,14 in which a formal organocatalytic [2+2]-cycloaddition between enamine intermediates and nitroolefins was identified as an undesired reaction pathway leading to catalyst inhibition, the asymmetric synthesis of cyclobutanes at the more remote double bond of the dienamine intermediate via [2+2] cycloadditions (bottom-right, Fig. 2) has been developed. Following these seminal examples, other cycloadditions, such as [4+2], [3+2] and [5+2], have been achieved at the 4,5-position of the dienamine intermediate. In this review, we outline and discuss the state of the art in the emerging field of dienamine-mediated catalysis of unsaturated aldehyde derivatives, including the catalytic systems reported so far in this area, grouping them by the reactivity pathway followed by the dienamine intermediate (1,3-, 1,5-, 2,5- and 4,5-functionalisation, Fig. 2) and the type of reaction developed. Furthermore, we briefly comment on more complex cascade processes involving dienamine intermediates and we conclude by considering the outlook of this area in the future.
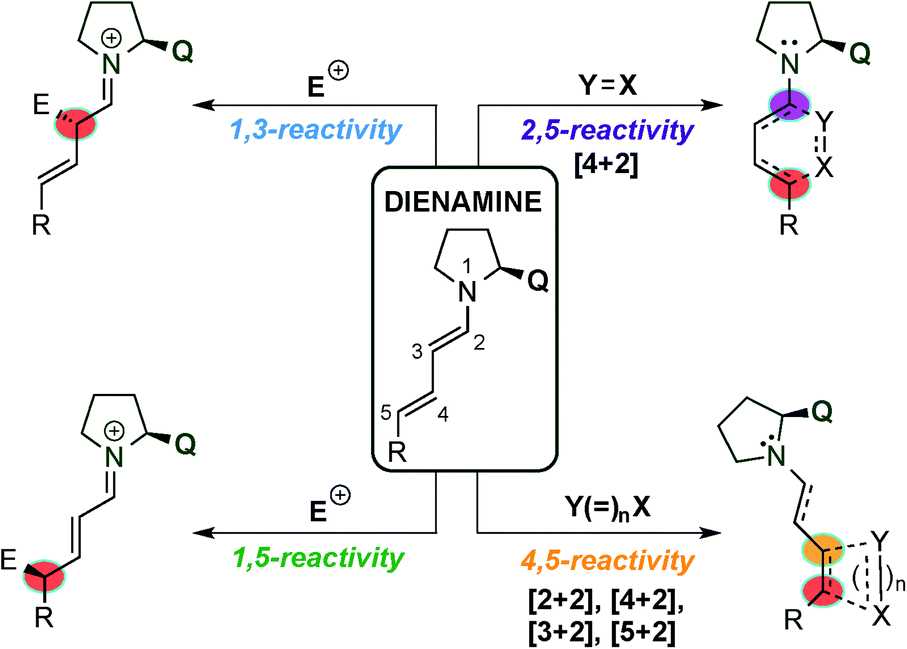 | ||
| Fig. 2 Classification of different reactivity pathways of dienamine intermediates used in the present review. | ||
2. 1,3-Reactivity: α-functionalisation
The early example of in situ generation of dienamine intermediates demonstrated by Serebryakov et al.10 has inspired others to make use of these intermediates for the α-functionalisation of α,β-unsaturated aldehydes in a wide variety of reactions.
In 2005, Hong and colleagues described the first proline-catalysed enantioselective intramolecular Baylis–Hillman (BH) reaction of hept-2-enedial (9) through a dienamine intermediate from poor to good yield and from poor to moderate enantioselectivity (right, Scheme 4) depending on the reaction conditions.15 Of interest is the reported inversion and enhancement of the stereoselectivity in the presence of imidazole due to the formation of an enamine intermediate (left, Scheme 4). In the proline catalysed reaction, enal 9 is activated with the catalyst to give the corresponding iminium ion, which evolves to form the dienamine intermediate. The enamine-aldol reaction proceeds preferentially through a Zimmerman–Traxler transition state (C), furnishing (S)-10. In contrast, the addition of a catalytic amount of imidazole proceeds from the Si-face of the iminium-ion intermediate, which facilitates the formation of the enamine intermediate D that suffers from the presence of an axial imidazolium group, leading to the formation of (R)-10.
 | ||
| Scheme 4 Intramolecular BH reaction through a dienamine (C)-or enamine (D) intermediate. | ||
Barbas III and co-workers reported a highly enantioselective organocatalytic aza-Morita–Baylis–Hillman (aza-MBH) reaction of β-mono- and di-substituted α,β-unsaturated aldehydes (11) and N-PMP-protected α-imino esters 12 (Scheme 5)16 catalysed by proline and imidazole, affording the corresponding E-β-amino aldehydes 13 as major isomers with moderate to good yields and excellent enantioselectivities. Through several conclusive 1H-NMR experiments, the authors demonstrated that this reaction proceeds through a Mannich-type mechanism via the dienamine intermediate (E transition state) followed by isomerisation of the double bond (E′). In the same year, Córdova's group described a similar approach for the catalytic enantioselective formation of aza-MBH-type products via the dienamine intermediate using L-proline/DABCO as a catalytic system. The authors explored the reaction of β-substituted α,β-unsaturated aldehydes with N-Boc-protected imines which afforded the corresponding amino aldehydes with moderate to good yields, E/Z ratios and excellent enantioselectivities.17
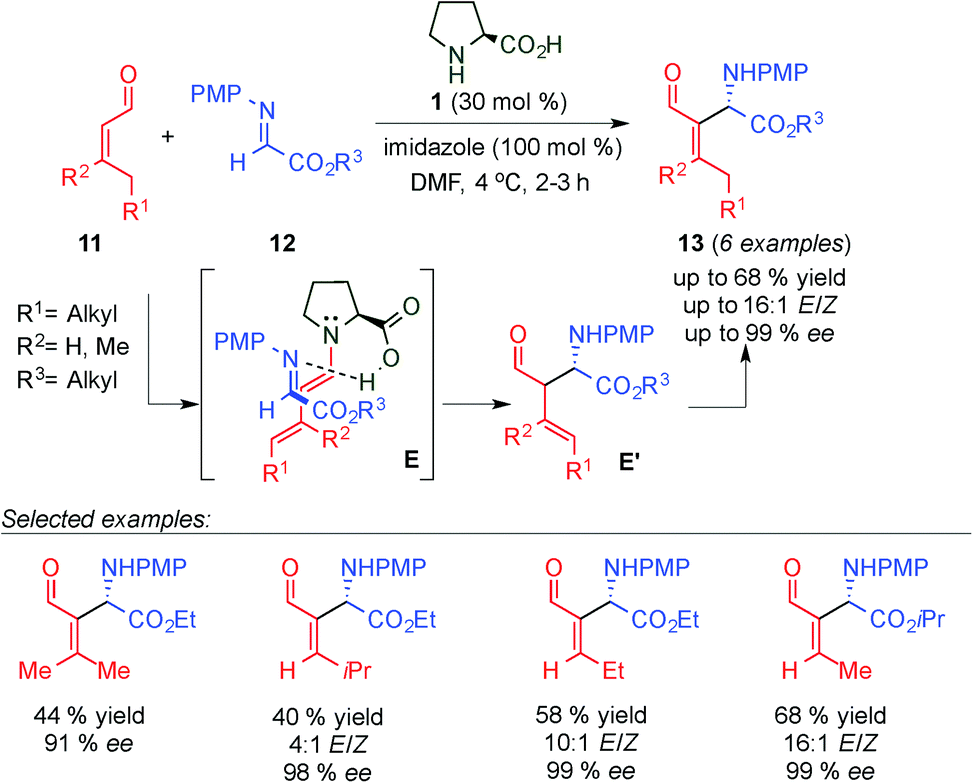 | ||
| Scheme 5 Intramolecular aza-MBH reaction through a dienamine activation pathway. | ||
Christmann et al. successfully applied this activation pathway to the formation of chiral cyclopentenals (16) with moderate to good yields and enantioselectivities. These compounds were obtained by an intramolecular Rauhut–Currier cyclisation reaction of acyclic precursors (15) which bear within their structure an α,β-unsaturated aldehyde and a Michael acceptor (Scheme 6).18 The authors proposed a catalytic cycle that starts with the activation of the enal and the Michael acceptor units of 15 with the catalyst and acetic acid, respectively, leading to the formation of a carbon–carbon bond through the electron rich dienamine intermediate F. Compound 16 was obtained by protonation of F′ at the γ-position followed by the release of the catalyst upon hydrolysis of the corresponding iminium ion intermediate. With the aid of sterics the reaction proceeds with complete α-regioselectivity.
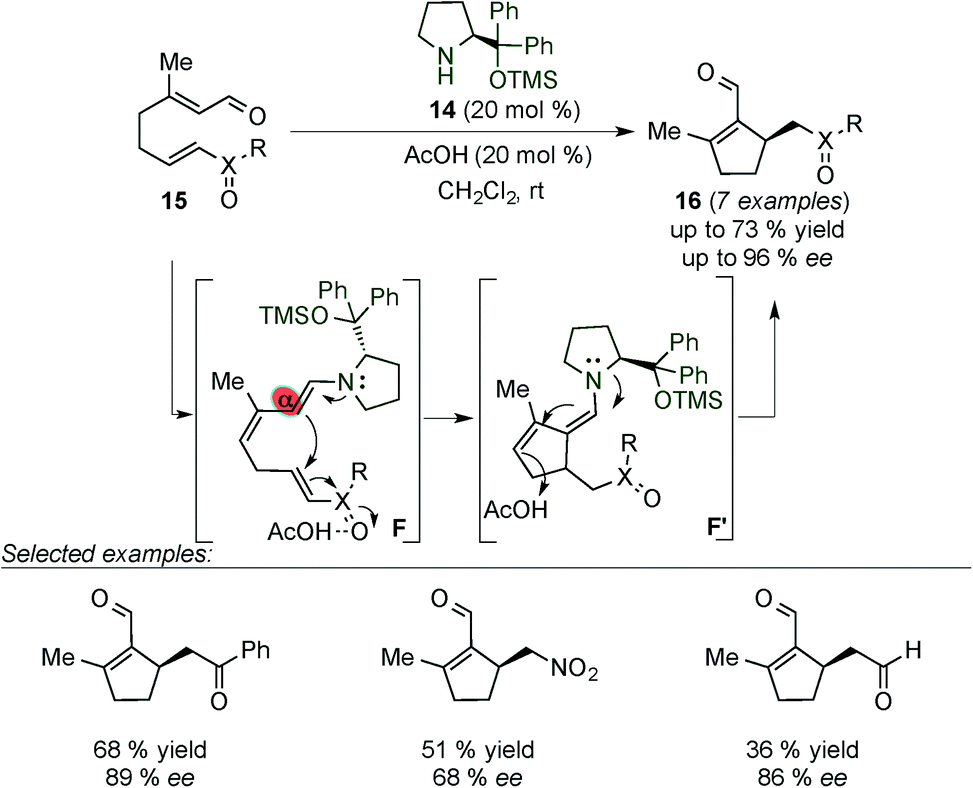 | ||
| Scheme 6 Intramolecular Rauhut–Currier cyclisation via the dienamine intermediate. | ||
In 2009, Chen and colleagues reported the first direct chemo-, regio-, and stereoselective Michael addition of γ,γ-disubstituted α,β-unsaturated aldehydes (17) to nitroolefins (18) through in situ generation of the dienamine intermediate (G transition state) using the Jørgensen–Hayashi catalyst 14 (Scheme 7).19 The corresponding alcohols 20 were obtained with excellent yields, diastereo- and enantioselectivities in a broad substrate scope after in situ reduction using NaBH4. The total α-regiocontrol is due to steric hindrance at the γ-position of the enals. Moreover, the authors demonstrated the utility of this methodology for the synthesis of optically pure six-member cyclic frameworks with versatile scaffold diversity, such as cyclohexenes (21) and piperidines (22) obtained by postsynthetic modifications involving tandem iminium–enamine and nitro-Mannich reactions of the Michael adducts 19, respectively. They further explored the synthetic utility of the methodology in affording a tetrahydrofuran (23) framework by an iodine-mediated cyclisation reaction of 20.
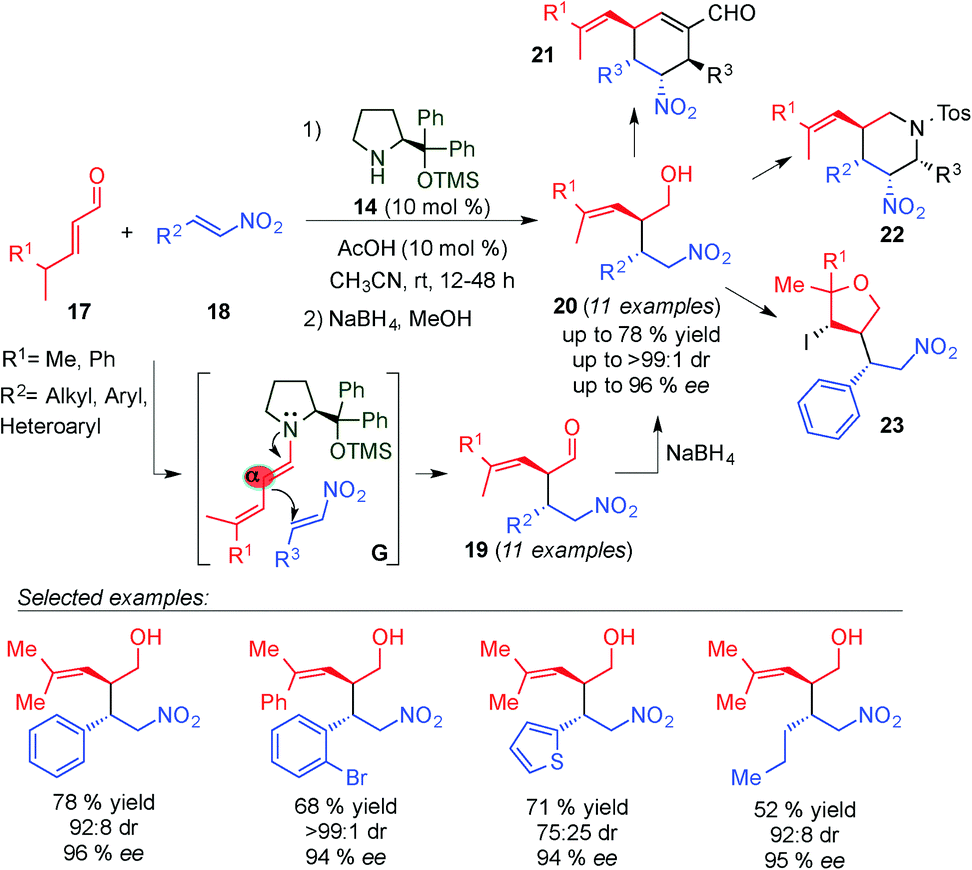 | ||
| Scheme 7 Dienamine-catalysed Michael addition and its application to the synthesis of cyclic compounds. | ||
Following a similar approach, the Enders group described the organocatalytic highly stereoselective synthesis of cis-3,4-difunctionalised chromans (27) and dihydrocoumarins (26) in moderate to good yields and high stereoselectivities via a domino Michael–hemiacetalisation process of ortho-nitrovinylphenols (24) and 4-methylpent-2-enal (17a), followed either by dehydroxylation or oxidation of the cis-3,4-disubstituted chromanols (25) (Scheme 8).20 Furthermore, using this protocol, the authors provided an efficient and stereoselective approach to the synthesis of a benzopyrano pyrrolidine which is a tricyclic heterocycle present as a subunit in fiduxosin, a potential drug for the treatment of benign prostatic hyperplasia.21
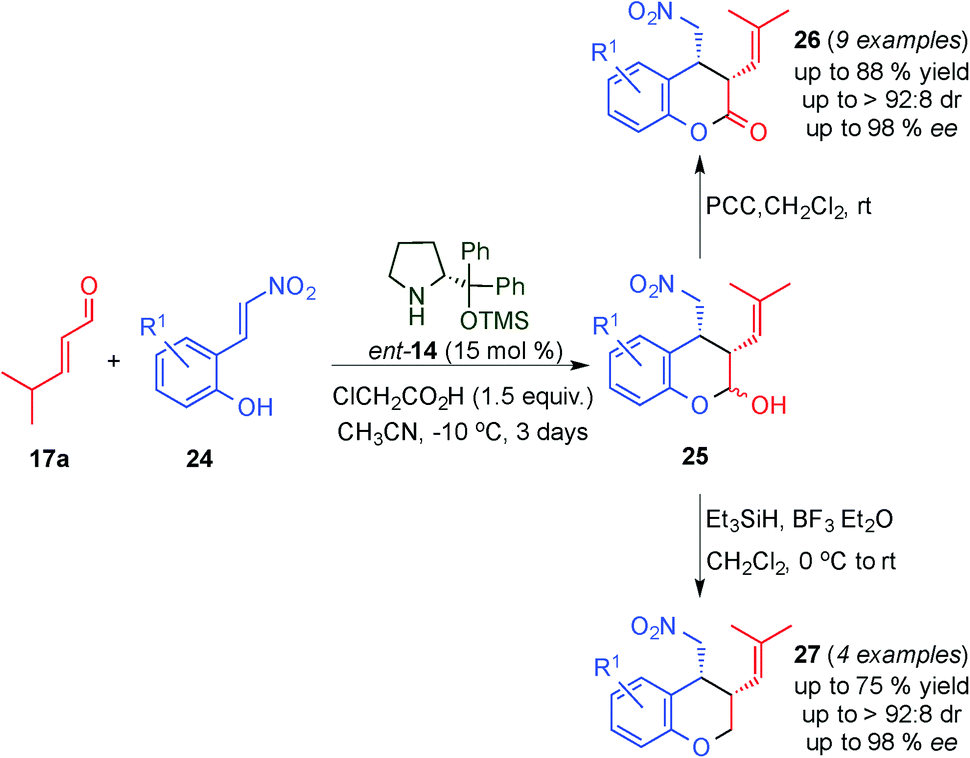 | ||
| Scheme 8 Tandem Michael–hemiacetalisation reaction through dienamine activation followed by either oxidation or dehydroxylation reaction. | ||
In a related study, Hong and colleagues reported formal [3+2] cycloadditions of 4-hydroxybut-2-enal (28) and nitroalkenes (18) for the synthesis of cyclopentanecarbaldehydes (29), containing four consecutive stereogenic centres in good yields and high enantioselectivities through a tandem Michael–Henry reaction (Scheme 9).22 To explain the results obtained, the authors proposed a plausible mechanism based on the iminium ion activation of 28 with the catalyst, followed by deprotonation to give a dienamine intermediate which then undergoes nucleophilic attack by the nitroalkene (H). Subsequently, tautomerization of H′ and a Henry reaction (H′′) lead to 29 as a major diastereomer of the reaction. This work represents a noteworthy example of the use of a succinaldehyde surrogate in dienamine-mediated catalytic reactions.
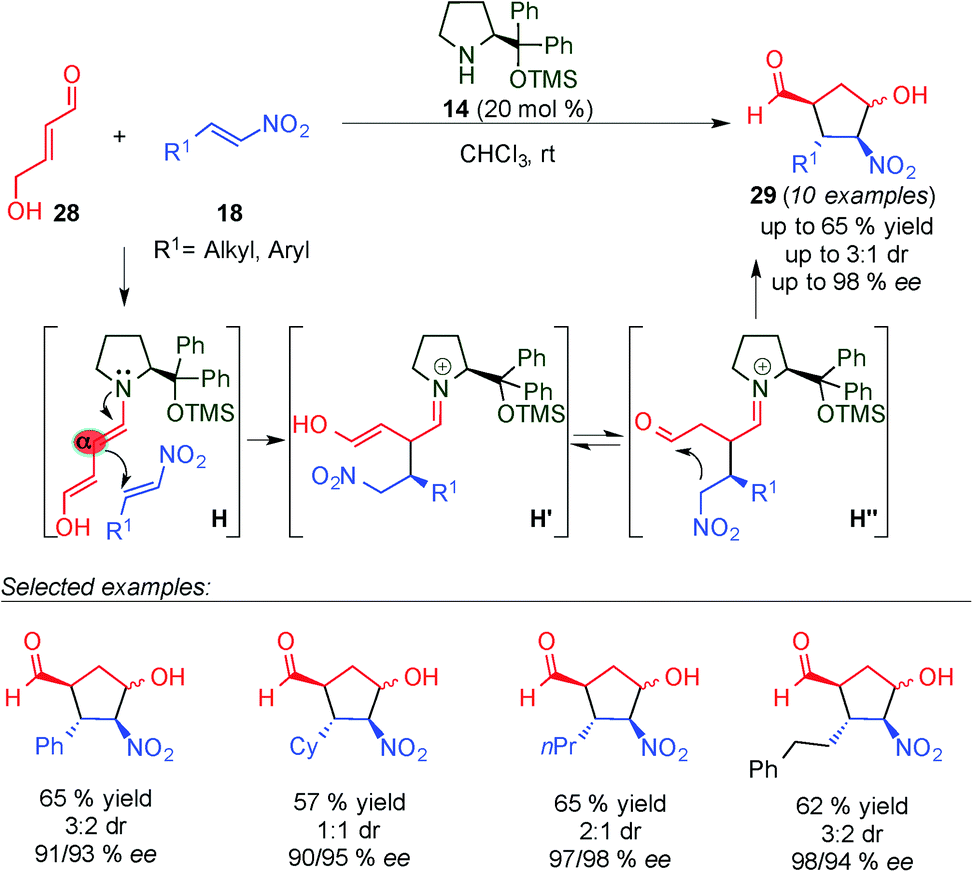 | ||
| Scheme 9 Organocatalytic formal [3+2] cycloaddition by a tandem Michel–Henry process. | ||
Chen and colleagues reported the first alkylation reaction between 3-indolylmethanols (31) and γ,γ-disubstituted α,β-unsaturated aldehydes (30) through a dienamine intermediate (I transition state), using 14 and AcOH as the catalytic system (Scheme 10).23 The corresponding Michael adducts 32 were obtained in a broad substrate scope with excellent diastereo- and enantioselectivity. Complete control at the α-position was achieved with the use of γ,γ-disubstituted enals. Subsequently, in the presence of a Lewis acid (AlCl3), the corresponding indole products (32) evolved to cyclopentyl[b]indoline derivatives (33) through an unusual intramolecular imino-ene reaction with excellent yields and enantioselectivities. This novel reaction proceeded through a key Lewis acid catalysed enamine–imine isomerisation step, which followed the Lewis acid mediated ene cyclisation.
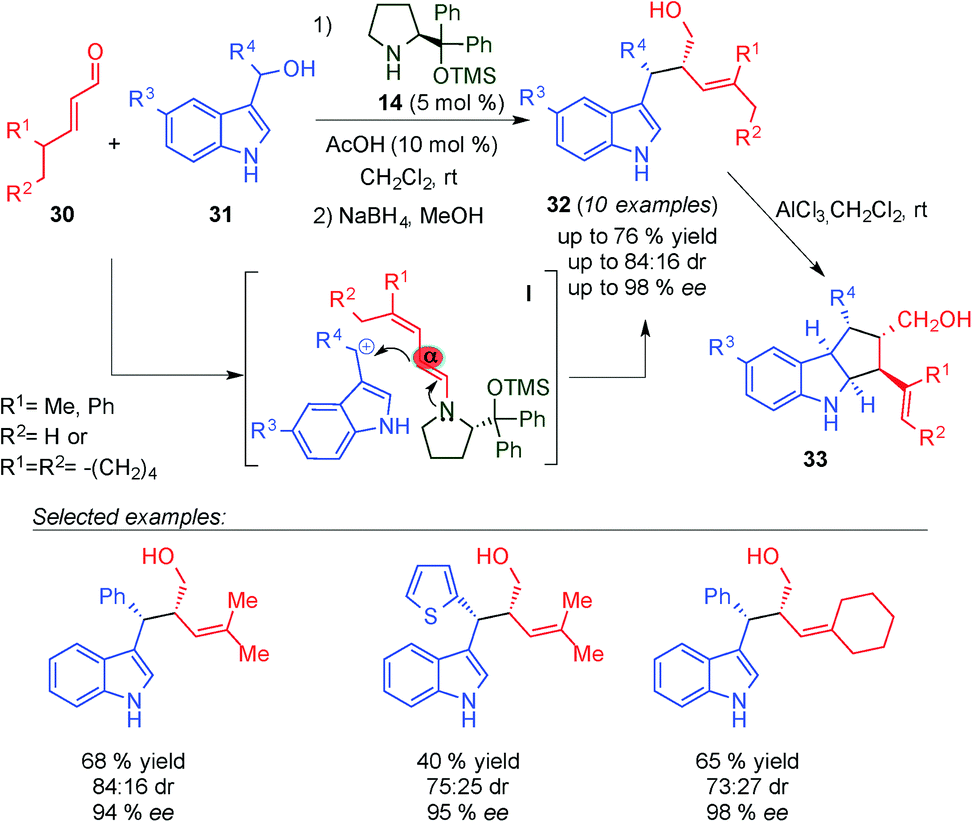 | ||
| Scheme 10 α-Site selective SN1-alkylation of γ,γ-disubstituted α,β-unsaturated aldehydes and 3-indolylmethanols. | ||
Soon afterwards, Christmann's group investigated the asymmetric SN1-alkylation of different γ,γ-disubstituted α,β-unsaturated aldehydes (35) through the dienamine intermediate with a stabilized carbocation derived from bis[4-(dimethylamino)methanol] (36) as an electrophile with catalyst 34 (Scheme 11).24 The authors showed a substrate dependent α- and γ-alkylation procedure with moderate to excellent regio- and enantioselectivity. Using γ,γ-disubstituted α,β-unsaturated aldehydes, the reaction proceeded with either complete or high α-regioselectivity (products 37) due to steric and electronic factors (J, Scheme 11). By contrast and as expected, linear α,β-unsaturated aldehydes afforded predominantly the corresponding γ-product (38) (K, Scheme 11). Almost simultaneously, Melchiorre et al. by using a different approach for the same reaction achieved the completely selective alkylation at the γ-position (see below, 1,5 Reactivity section).25
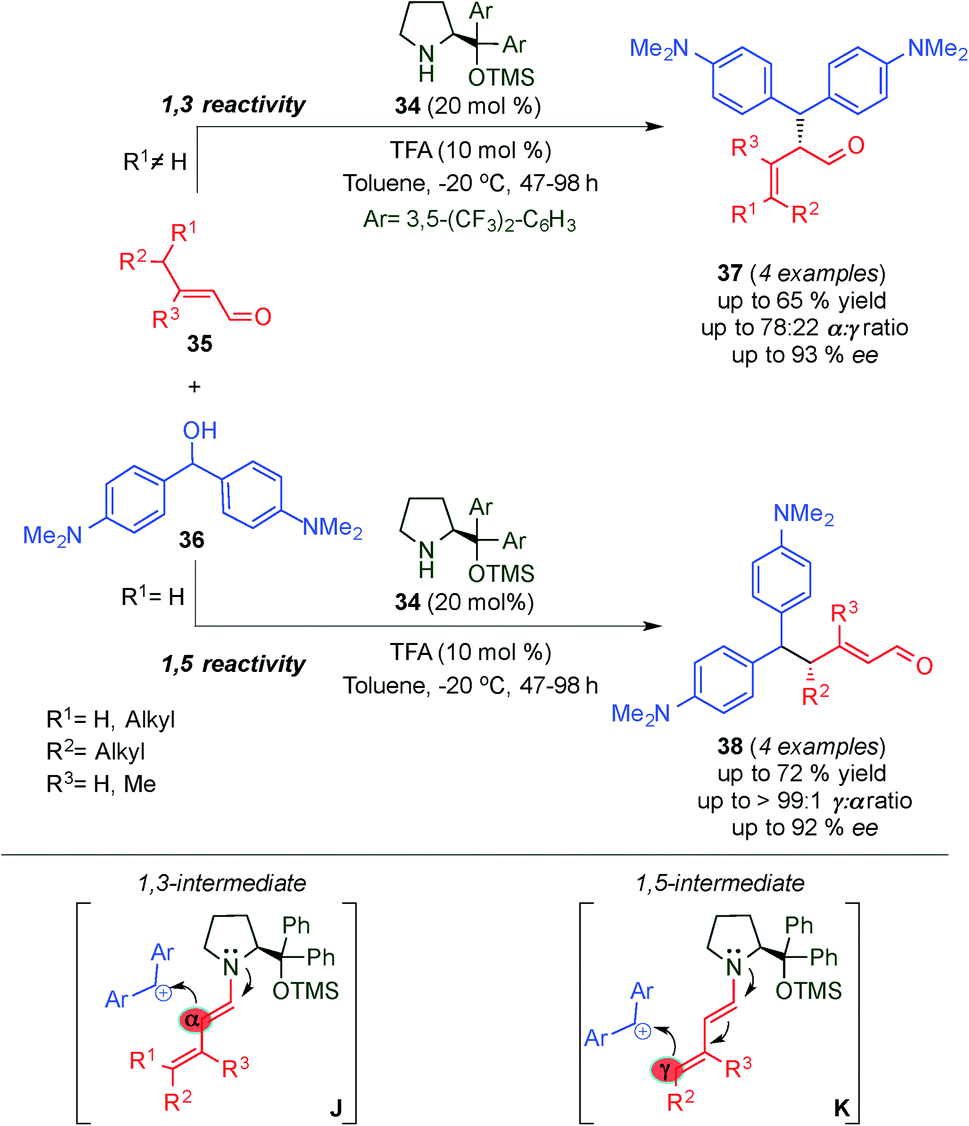 | ||
| Scheme 11 α- vs. γ-site selective SN1-alkylation of γ,γ-disubstituted α,β-unsaturated aldehydes. | ||
In 2009, a striking alternative approach to the normal electron demand Diels–Alder via dienamine activation was developed by Chen et al. They demonstrated the use of the formed dienamine intermediate as the dienophile, thus reporting the first highly regioselective and stereoselective inverse-electron-demand aza-Diels–Alder reaction between α,β-unsaturated aldehydes (40) and N-tosyl-1-aza-1,3-butadienes (41) for the asymmetric synthesis of piperidines 42 (Scheme 12).26 The authors successfully demonstrated the possibility of using the HOMO-energetic destabilization of the in situ generated dienamine intermediate as a dienophile, with a broad scope of α,β-unsaturated aldehydes and N-tosyl-1-aza-1,3-butadienes, affording exclusively α-regioselective Diels–Alder adducts (42) with good yields and enantioselectivities through a 1,3-intermediate (L transition state, Scheme 12). The great preference towards the α-position is easily explained as the more proximal, regarding the aminocatalyst, of the dienamine double bonds, is the most reactive alkene. Intriguingly, crotonaldehyde exclusively afforded the corresponding γ-regioselective adduct 43 with moderate enantioselectivity using catalysts 14 or imidazolidinone catalyst 39 through a 1,5-intermediate (M transition state, Scheme 12), most likely due to steric factors. However, only poor or moderate selectivities were obtained in this example, the unique reactivity shown by crotonaldehyde laid the foundations for further developments of dienamine-mediated catalytic inverse-electron-demand Diels–Alder reactions, which will be covered in detail in Section 5.2.
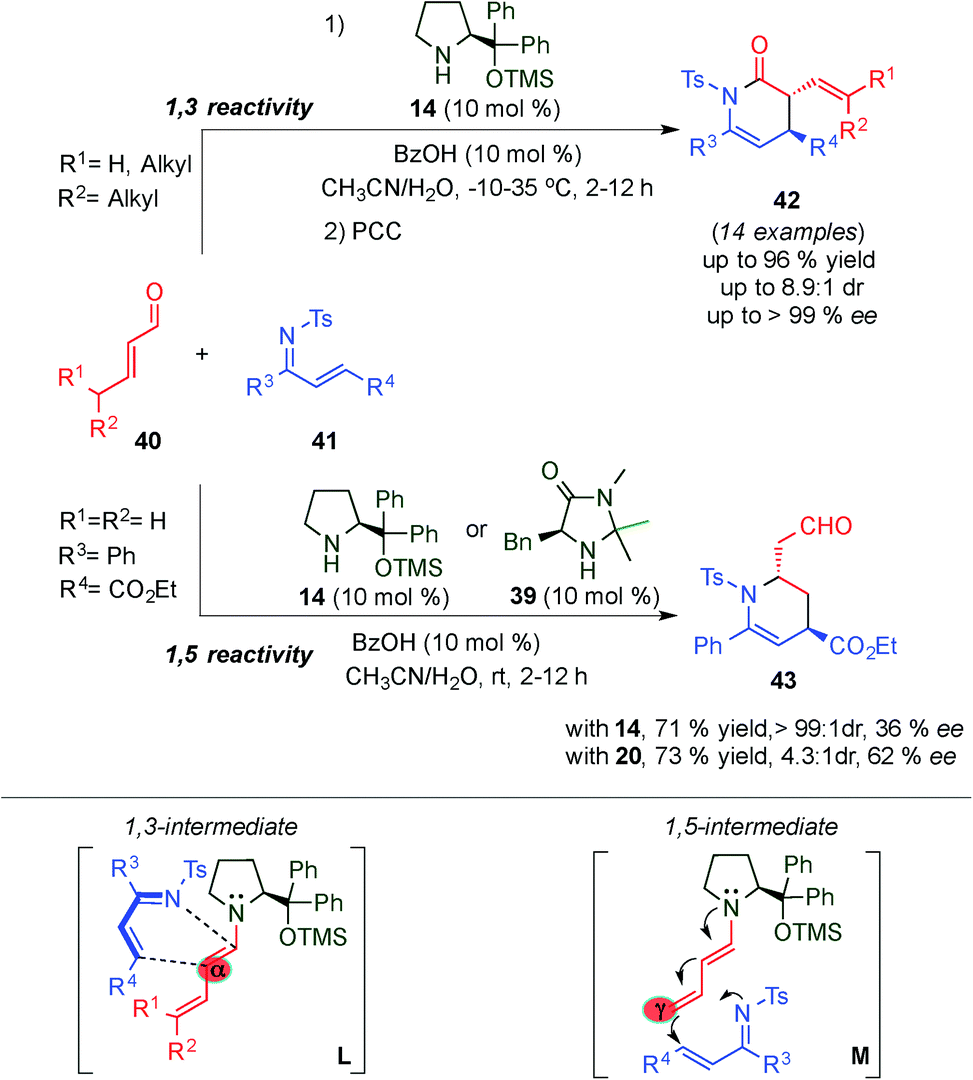 | ||
| Scheme 12 1,3- and 1,5-site-selective inverse-electron-demand aza-Diels–Alder reaction of α,β-unsaturated aldehydes and N-tosyl-1-aza-1,3-butadienes via dienamine intermediates. | ||
3. 1,5-Reactivity: γ-remote asymmetric functionalisation
In 2006, Jørgensen and co-workers reported the first example of an asymmetric vinylogous catalytic process through dienamine activation of γ-enolisable unsaturated aldehydes (top, Scheme 13).11 They developed the γ-amination of different α,β-unsaturated aldehydes (44) with diethyl azodicarboxylate (45) under aminocatalysis using 34 with moderate yields and high enantioselectivities and perfect site selectivity for the γ position. To support the excellent stereo- and selectivity observed in the reaction, the authors reported experimental and theoretical studies that suggested a [4+2] cycloaddition path as the most probable reaction mechanism of the reaction. In this concerted mechanism, under these reaction conditions, the generated hetero-Diels–Alder adduct N′ evolves into the final product 46 and the concerted transition state N proceeds via a dienamine E,s-cis,E, which is supported by the experimental stereochemical outcome obtained in the reaction. As further evidence of the Diels–Alder type pathway, the authors carried out reaction between 2-pentenal (44a) and N-methylmaleimide (47) in the presence of 1 equivalent of the aminocatalyst 34, which afforded the Diels–Alder cycloadduct (48), leading to a dead end of a possible catalytic process (bottom, Scheme 13).
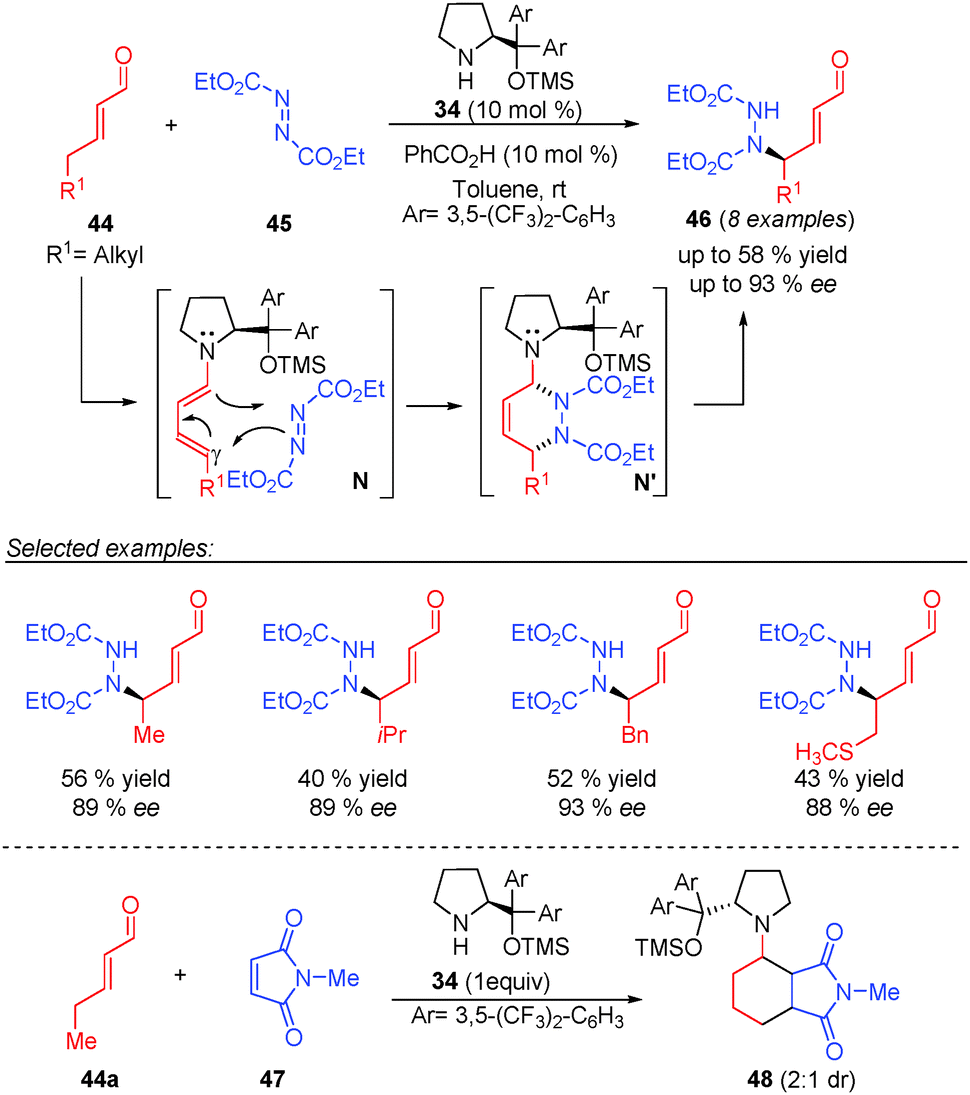 | ||
| Scheme 13 γ-Amination of α,β-unsaturated aldehydes with diethyl azodicarboxylate. | ||
Afterwards, Brenner-Moyer et al. employed the above methodology to the synthesis of enantioenriched γ-amino alcohols and β-functionalised-γ-amino alcohols through a dienamine-iminium cascade reaction, which consisted of the γ-amination of α,β-unsaturated aldehydes followed by either conjugated reduction or nucleophilic addition of an oxime to 46.27
This seminal work reported by Jørgensen's group inspired other groups to explore the potential of dienamine activation as a powerful tool for the direct asymmetric 1,5-functionalisation of α,β-unsaturated aldehydes. In 2008, Woggon and colleagues reported an aldol/oxa-Michael cascade process via dienamine-iminium ion intermediates (O, O′), as a key step in the total synthesis of α-tocopherol (53), a member of the vitamin E family (Scheme 14).28 The product obtained via the cascade sequence is subjected to a three step synthetic process based on ring opening via hydrogenation, decarboxylation and demethylation leading to the target molecule 53. The dienamine catalysed reaction proceeded with a perfect γ-regioselectivity and can be considered an astonishing demonstration of the potential of the dienamine activation as a tool in the total synthesis of natural products.
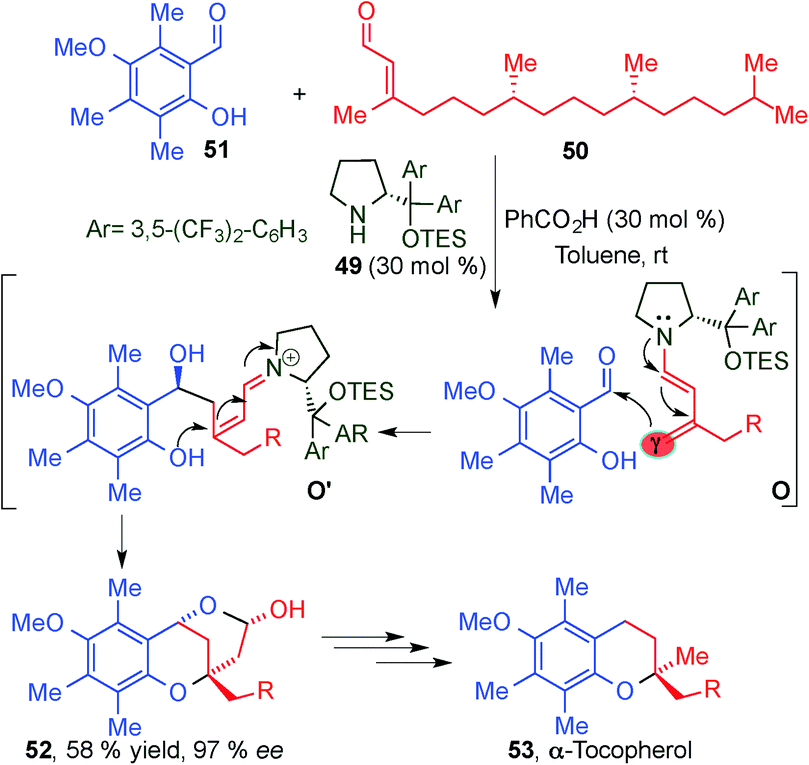 | ||
| Scheme 14 Asymmetric total synthesis of α-tocopherol through an aldol dienamine mediated catalysed reaction. | ||
Targeting to develop discrete intermolecular nucleophilic additions of the remote γ-position of enals, concurrently and independent of Christmann's work (see Scheme 11, Section 2),24 Melchiorre's group developed a cooperative catalytic system based on a combination of chiral primary amine 54 and chiral phosphoric acid 55, which is able to promote the γ-site-selective alkylation of α-branched enals 56via an SN1 pathway (Scheme 15).25a The authors proposed that the reaction between α-branched enals and bis[4-(dimethylaminophenyl)methanol] (36) proceeds through a cooperative activation path that combines dienamine- and Brønsted-acid activation modes simultaneously. In the transition states (P), 55 participates in the formation of a chiral contact ion pair with 36, which react with the chiral dienamine intermediate generated from condensation with 54. Thus, through an interwoven of non-covalent interaction of the cooperative catalytic system and the reagents, the reaction furnishes exclusively 57 with excellent yields and enantioselectivities. As a following work, the same authors optimized the methodology using a more effective catalytic system based on a chiral secondary cyclic amine and saccharin, as an achiral acid additive, achieving better yields and enantioselectivities.25b
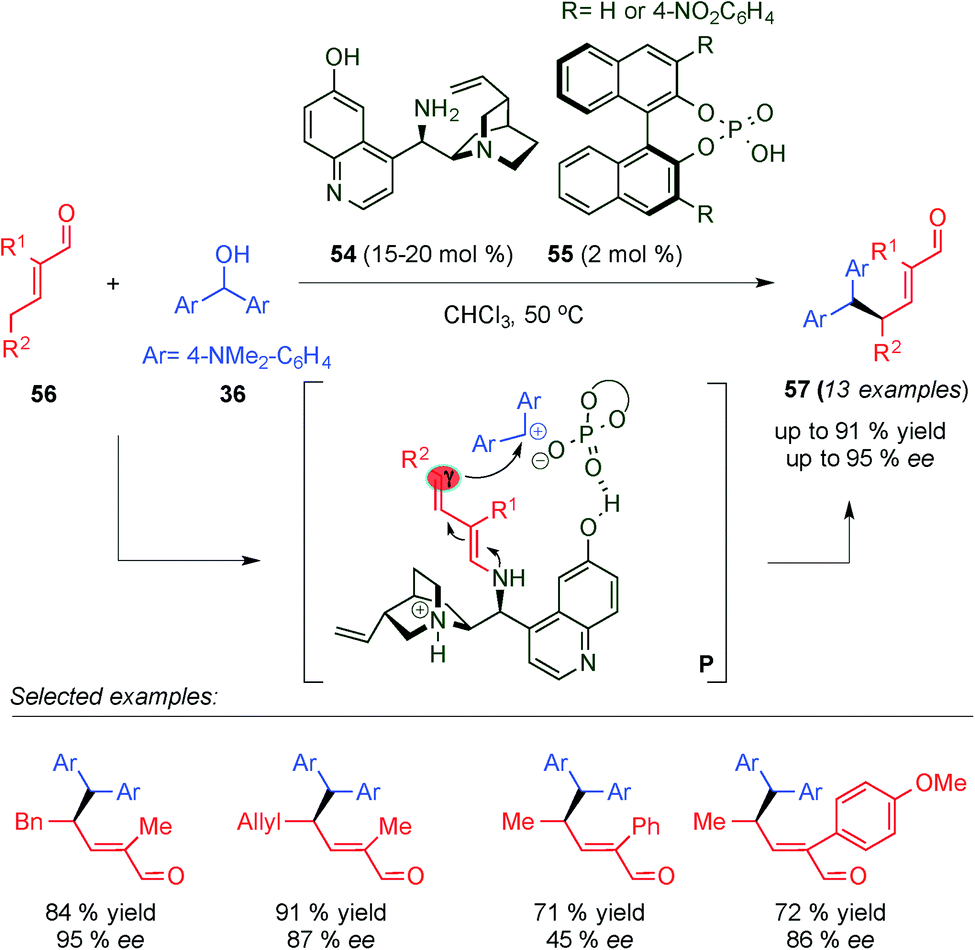 | ||
| Scheme 15 γ-Site selective SN1-alkylation of α-branched enals through dienamine catalysis. | ||
Seeking to extend the nucleophilic addition at the γ-position of the in situ generated dienamine intermediates with other electrophilic species, the same group reported the high stereoselective γ-site-selective aldol reaction between α-branched enals (60 and 60′) and isatin (59) (Scheme 16) via a dienamine intermediate using the sterically congested diphenylprolinol silyl ether 58 as a catalyst.29 The authors observed that the nature of the α-branched enal's substituents led to different product outcomes, postulating two plausible divergent mechanisms for the reaction. Using α-alkyl substituted enals (60), the 3-substituted 3-hydroxyoxindole derivatives 61 were obtained with high stereocontrol and selectivity, which could be rationalized through a “steric control” transition state (a pathway, Scheme 16), which is based on the addition of isatin from the unshielded face of intermediate Q at the γ-position. In contrast, α-aryl substituted enals (60′) provided access to spirocyclic oxindole scaffolds 62, with moderate diastereoselectivity, high enantioselectivity (pathway b, Scheme 16) and opposite configuration of the γ-stereocenter, which is indicative of a [4+2] cycloaddition transition state (R). To prove the two different transition states, Q and R, involved, the authors mixed enal 60a′ with a stoichiometric amount of catalyst 14 and nitrostyrene 63, a dienophile that can react in a concerted pericycle reaction catalysed by 14, leading to the Diels–Alder adduct 64, with great stereocontrol. In contrast, under the same reaction conditions, enal 60 remains unreacted.
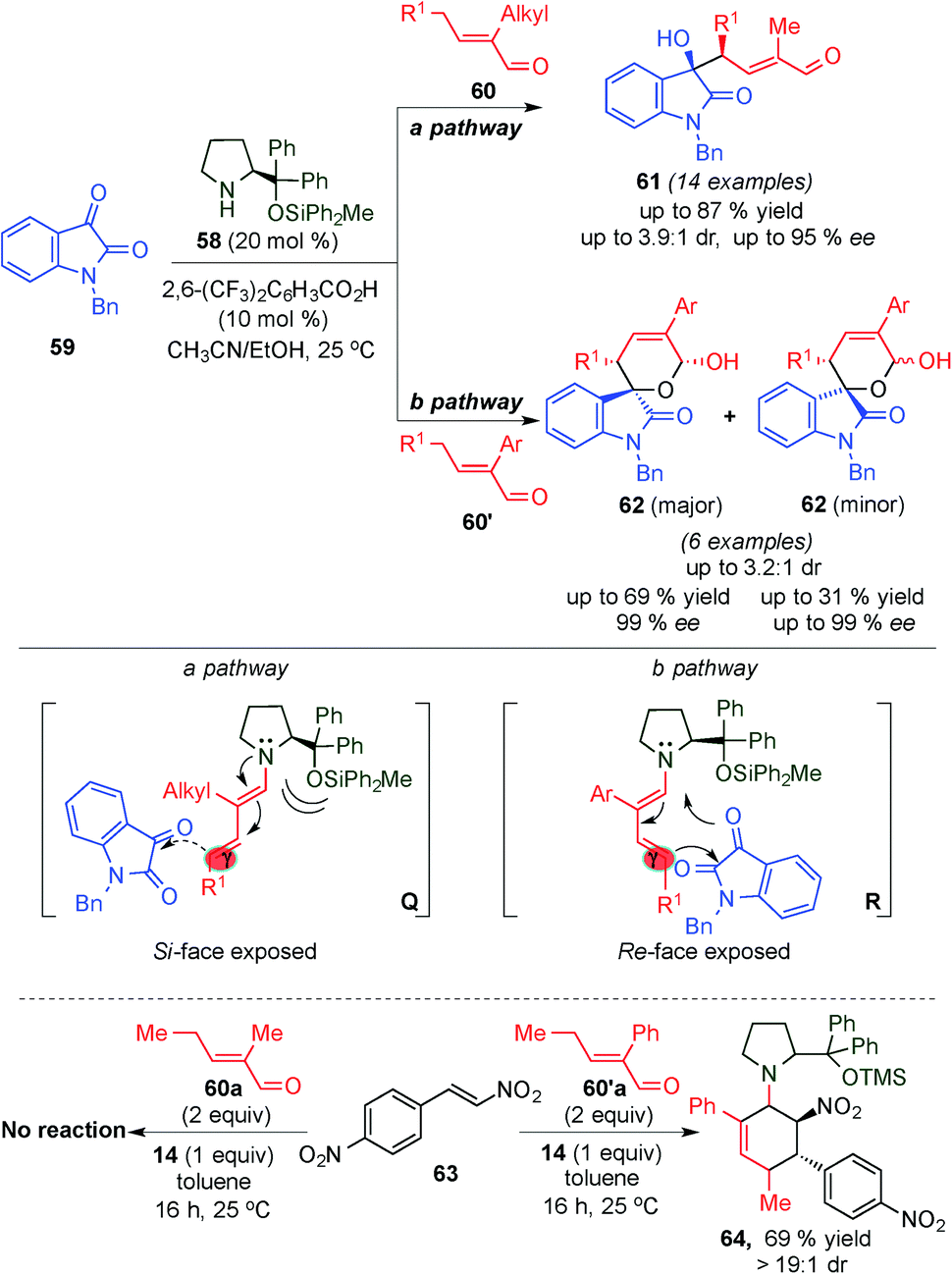 | ||
| Scheme 16 γ-Site-selective aldol reaction between α-branched enals and isatin via dienamine activation. | ||
Very recently, Brenner-Moyer and colleagues reported the first γ-site-selective catalytic reaction to directly introduce nitrone functionality to α,β-unsaturated aldehydes via dienamine intermediate S using catalyst 14 (Scheme 17).30 This work represents an unprecedented and useful example of an organocatalytic redox reaction, in which the alkyl and aryl substituted enal (65) is oxidized to the corresponding γ-nitrone (67) with moderate yields and diastereoselectivities, thereby reducing the corresponding nitrosoaryl derivative to N-arylhydroxylamine. Supported by experimental and theoretical studies, the authors proposed a plausible mechanism, in which the reaction starts with the condensation of the enal 65 with catalyst 14, to form an iminium ion intermediate, which is in equilibrium with the dienamine intermediate (S) via the loss or gain of a proton. Subsequently, the in situ generated dienamine reacts with 2 equivalents of the nitrosoaryl compound (66), furnishing intermediate S′. However, there is no evidence to confirm whether an asynchronous or concerted transition state is involved. Finally, deprotonation of S′ liberates the nitrone 67, along with 1 equivalent of the product of reduction of 66.
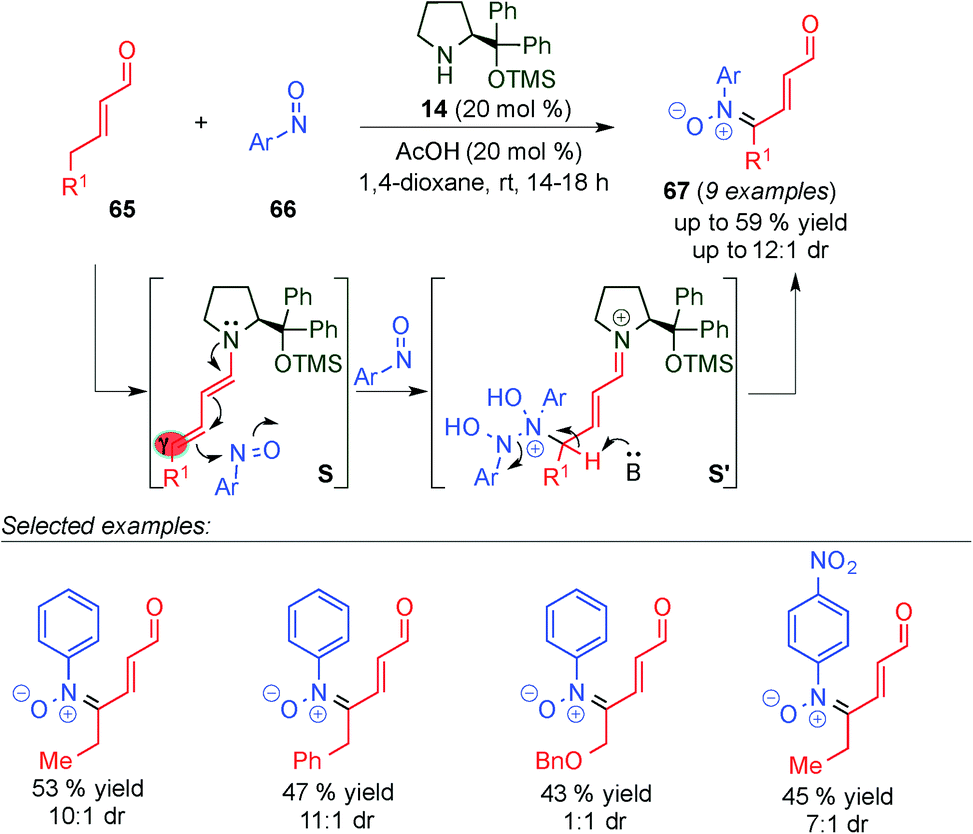 | ||
| Scheme 17 γ-Site-selective addition of nitrone to α,β-unsaturated aldehydes. | ||
The extension of the asymmetric functionalisation at remote centers of enals by cooperative catalytic processes, involving the combination of dienamine activation with transition-metal catalysis, constitutes an exciting approach to provide a general synthetic technology for designing vinylogous reactions. Pursuing this prospect, the Nishibayashi group developed the γ-site-selective alkylation of γ-enolisable unsaturated aldehydes through cooperative dienamine–metal catalysis (Scheme 18).31 The diruthenium complex 68 in combination with the chiral secondary amine 34 promoted the γ-selective propargylation of different α,β-unsaturated aldehydes (69) with a broad diversity of propargylic alcohols (70), with good yields, poor diastereoselectivities and moderate enantioselectivities. The author proposed a plausible reaction pathway which starts with the addition of the dienamine intermediate to the allenylidene complex formed by the reaction of the ruthenium complex with the propargyl alcohol (T, Scheme 18), leading to an alkynyl vinylidene complex intermediate via an alkynyl complex intermediate, which evolves into the γ-alkylated enal by ligand exchange with another propargylic alcohol (70). Compounds (71) were obtained by in situ reduction of the aldehydes using NaBH4. Despite the poor diastereoselectivities and moderate enantioselectivities obtained, this example laid the foundations for further development of cooperative dienamine–metal catalysed methodologies.
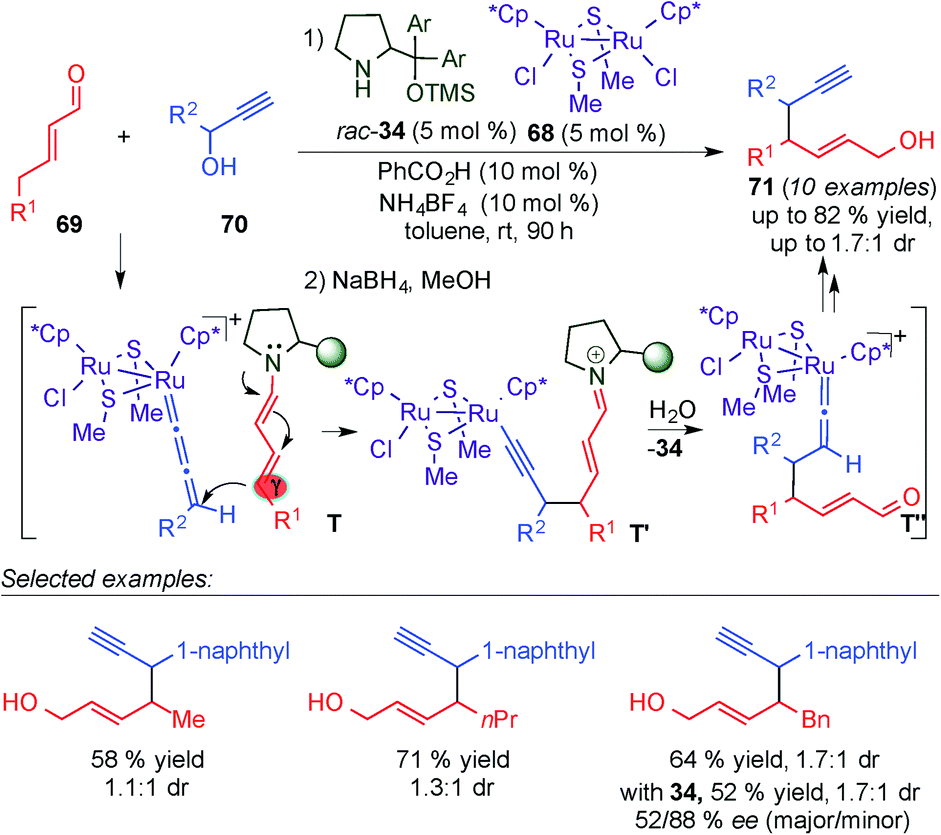 | ||
| Scheme 18 γ-Propargylation reaction of α,β-unsaturated aldehydes through cooperative ruthenium-dienamine catalysis. | ||
In 2014, a second example of dual dienamine–metal catalysis was reported by Jang and colleagues.32 They developed a multicatalytic reaction where a copper catalyst (CuBr) combined with aminocatalyst 14 was used to promote both the γ-oxyamination of α,β-unsaturated aldehydes (72) (top, Scheme 19). They also developed the one pot aerobic oxidation of allylic alcohols (75)/γ-oxyamination of α,β-unsaturated aldehydes (bottom, Scheme 19) using TEMPO (73) as an oxidative reagent. Various allylic alcohols and α,β-unsaturated aldehydes were converted to γ-oxyaminated α,β-unsaturated aldehydes with moderate to good yields. A plausible reaction mechanism for the tandem copper catalysed oxidation and copper–aminocatalysed oxyamination of α,β-unsaturated aldehydes was proposed. In the first step, copper–TEMPO complexes are used to oxidize 75. The corresponding aldehyde (73) condensates with the aminocatalyst (14), forming the dienamine intermediate. Upon addition of the copper–TEMPO complex to the dienamine intermediate (U, Scheme 19), an iminium intermediate is formed, which due to the acidity of the γ-carbon isomerised to a dienamine intermediate, preventing further addition of nucleophiles at the β-position of the enal. After hydrolysis the desired product 74 is obtained. Although a chiral aminocatalyst (14) was used, stereocontrol was not observed, presumably, due to racemization at the γ-position of 74 during the reaction.
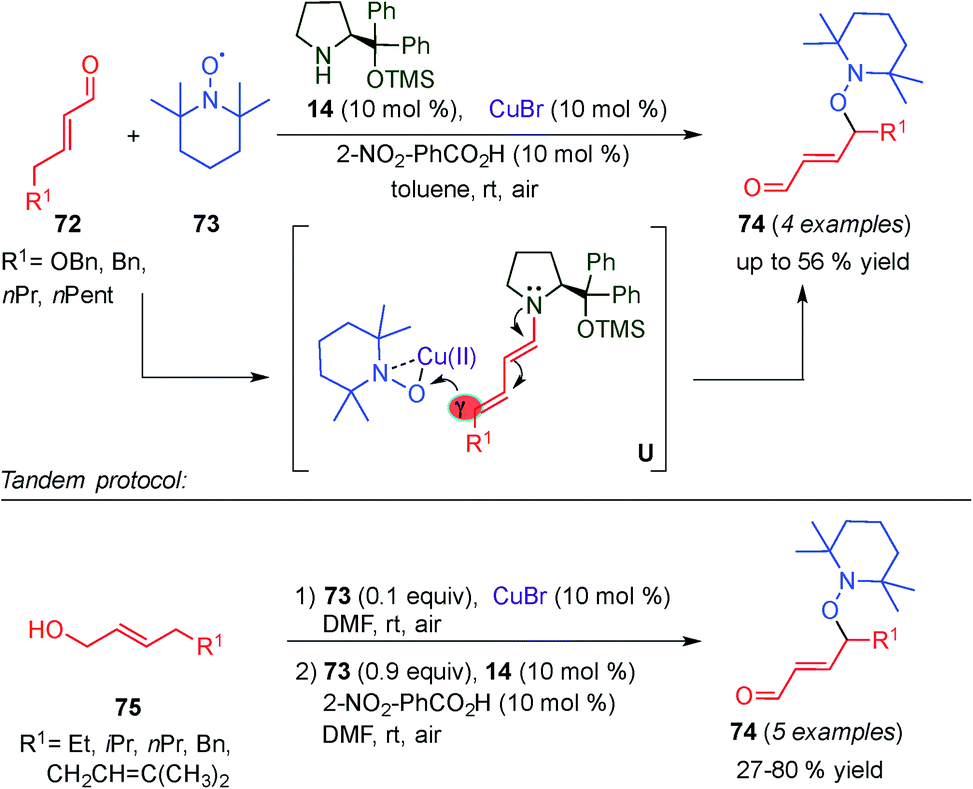 | ||
| Scheme 19 γ-Oxyamination of α,β-unsaturated aldehydes and tandem oxidation/γ-oxyamination of allylic alcohols via copper-dienamine catalysis. | ||
Recently, Jørgensen's group developed the asymmetric γ-allylation of α,β-unsaturated aldehydes based on the combination of dienamine-mediated catalysis and transition-metal catalysis (Scheme 20).33 This transformation is associated with several potential selectivity issues; the regioselectivity (α- versus γ-allylation) of the α,β-unsaturated aldehyde due to the reactive dienamine intermediate contains two nucleophilic sites. Likewise, the activated π-allyl system has two electrophilic sites and the regioselectivity (branched versus linear products) of this intermediate also needs to be controlled. Moreover, the control of the E/Z ratio, the diastereomeric ratio (for branched products), and enantiomeric excess of the products are a challenge. To overcome the mentioned problems the authors developed a methodology, which provides access to all six isomers of the γ-allylated product (4 stereoisomers of branched products and 2 enantiomers of linear products) in a divergent fashion, in excellent selectivity, by choosing the appropriate combination of aminocatalysts, transition-metal catalysts, and ligands. By the use of aminocatalysts (76) in combination with an iridium catalyst, selective access to branched γ-allylated products (80) was achieved in excellent diastereo- and enantioselectivity. This approach is based on stereodivergent dual catalysis, and thus allows selective access to both diastereomers of 80 by using both enantiomers of 76. Furthermore, by replacing the iridium catalyst with a palladium catalyst under otherwise similar reaction conditions (aminocatalyst 14 instead of 76) the linear products of the γ-allylation (81) were obtained in excellent enantioselectivity.
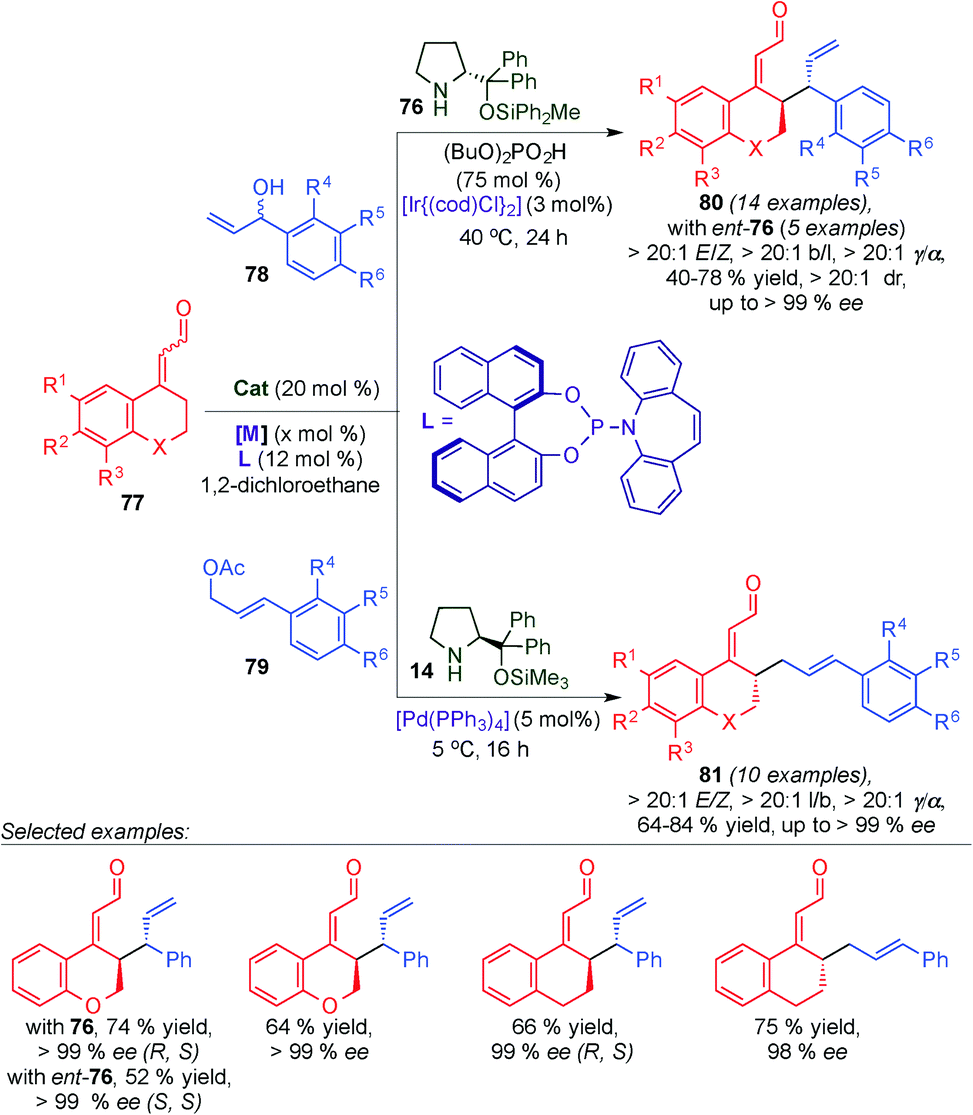 | ||
| Scheme 20 γ-Allylation of cyclic α,β-unsaturated aldehydes by dual dienamine-transition metal catalysis. | ||
In 2015, Alemán and co-workers reported the synthesis of chiral diheteroarylalkanals (84) by aldol/Friedel–Crafts one-pot reaction (Scheme 21).34 The one-pot process starts with the condensation of dialdehyde 82 with the catalyst (14) to form the dienamine intermediate (V) via isomerisation of the iminium ion intermediate, which then undergoes intramolecular aldol reaction (V, Scheme 21). Subsequently, the generated iminium ion intermediate (V′) reacts with the indole (83), leading to the diheteroarylalkanals (84) in good yields and enantioselectivities due to the classical “steric control approach”. In addition, the authors carried out antiproliferative activity studies of these new diheteroarylalkanals in representative cancer tumor cell lines. Their structure-activity relationship indicated that with the appropriate substitution pattern, these compounds are as cytotoxic as Cisplatin.
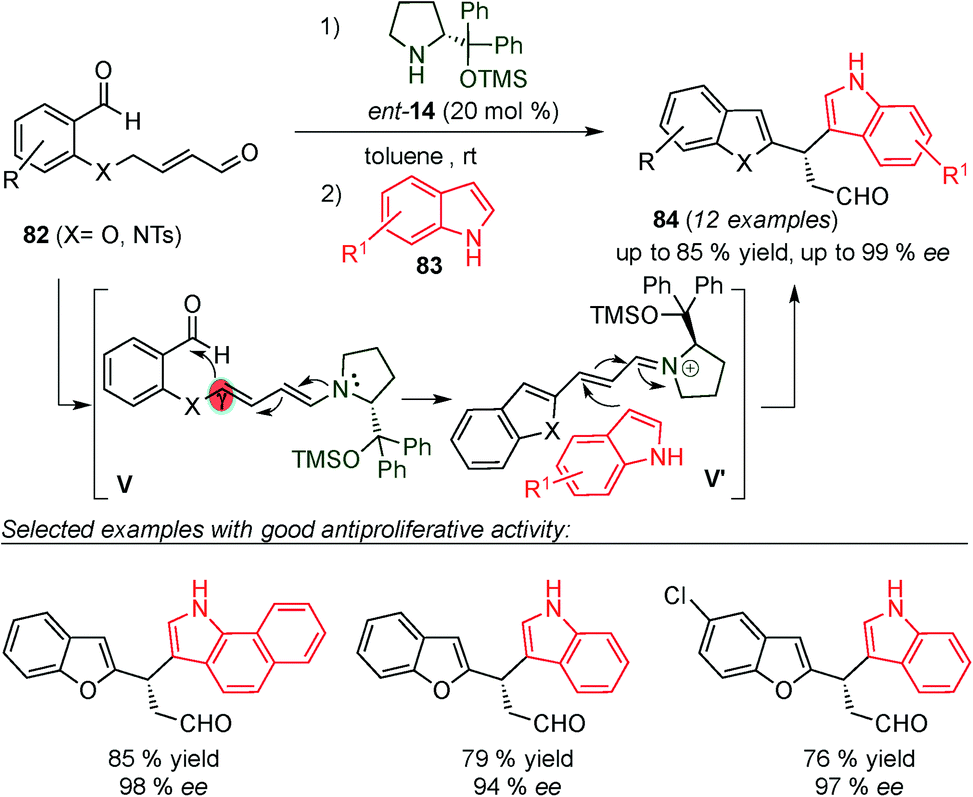 | ||
| Scheme 21 Synthesis of diheteroarylalkanals through a one-pot dienamine-iminium mediated catalytic process. | ||
Very recently, Chen's group developed an asymmetric formal α,γ-regioselective [3+3] cycloaddition reaction of α,β-unsaturated aldehydes (86) and 2-nitroallylic acetates (87) using chiral bifunctional secondary amine–thiourea 85 as a catalyst via a cascade dienamine–dienamine mediated catalytic reaction (Scheme 22).35 This cascade reaction proceeds through the generation of the dienamine intermediate (by condensation of 86 with the aminocatalyst 85), which reacts with the nitroolefin acceptor (87) with complete γ-regioselectivity. Thus, this first Michael addition step generates the required second acceptor, through the elimination of a molecule of acid, favouring the subsequent α-site selective dienamine-mediated intramolecular catalytic Michael addition to furnish the cyclohexene structures. The formal [3+3] cycloaddition reaction produced cyclohexanes 88 in moderate yields, excellent enantioselectivities and poor diastereomeric ratios (cis/trans). Both diastereomers were obtained with identical ee values, indicating that the enantiocontrol is determined in the first γ-regioselective and the poor diastereocontrol is due to the protonation step. After a simple basic treatment second step, a trans-88 diastereomer was obtained as a sole product of the reaction through epimerization of the chiral center adjacent to the NO2 group of the major diastereomer (cis-88).
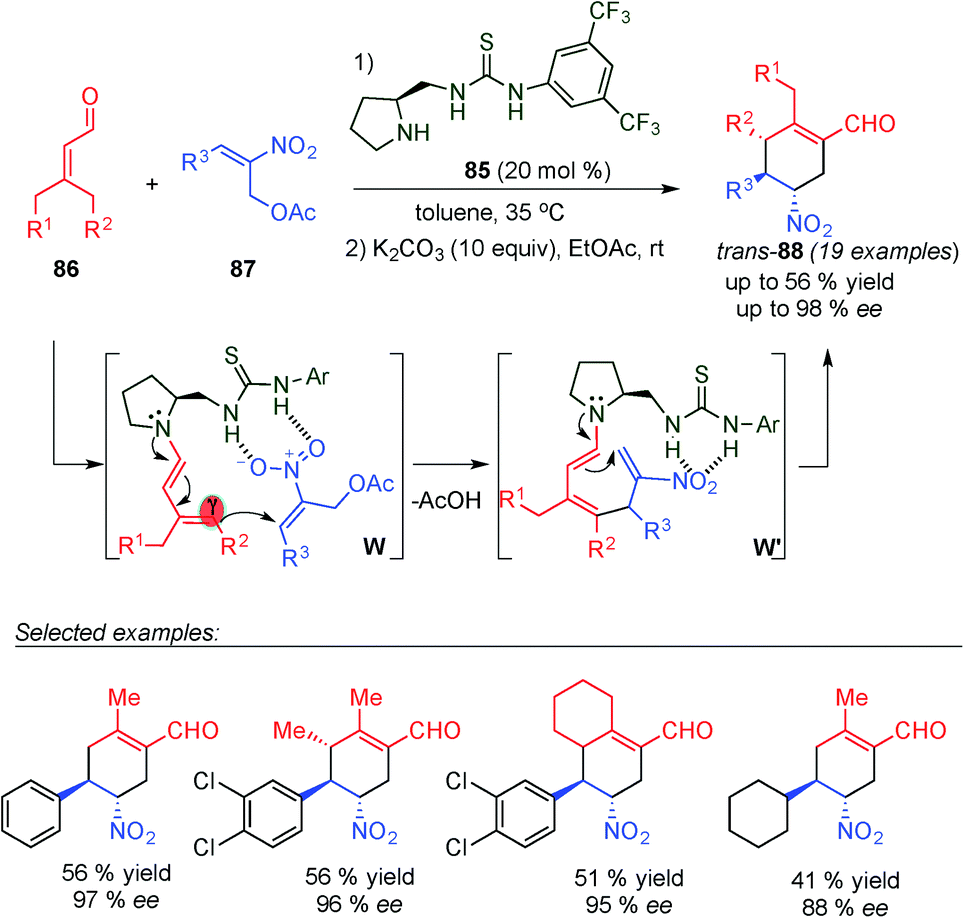 | ||
| Scheme 22 Synthesis of trans-cyclohexene derivatives via a dienamine–dienamine mediated catalytic process. | ||
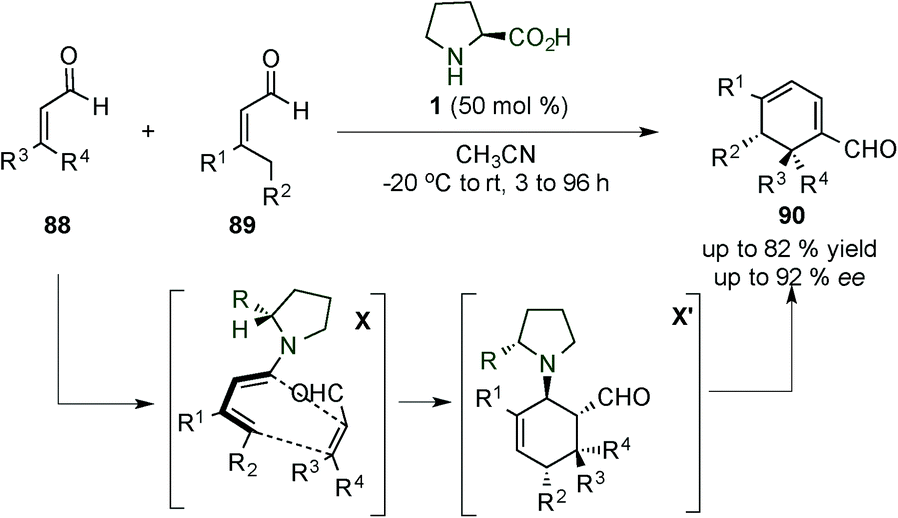
4. 2,5-Reactivity: [4+2]-Diels–Alder cycloadditions of electron-rich dienes
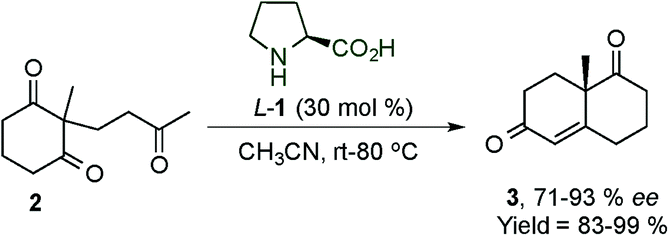
After the rediscovered reaction in the γ-functionalisation, Hong's group published the asymmetric Robinson annulation of α,β-unsaturated aldehydes36 and subsequently applied this methodology to the total synthesis of (+)-palitantin.37 In this work, the authors investigated eighteen different pyrrolidine-type catalysts and found that the best catalyst was the proline catalyst, obtaining a large number of six membered ring architectures (90) with, in some cases, excellent enantioselectivities (ee > 99%) (Scheme 23). Interestingly, they proposed a mechanism for the formation of a [4+2] adduct via a cis-dienamine intermediate and an alternative via a stepwise mechanism (trans-dienamine addition to an iminium ion intermediate followed by an intramolecular aldol reaction).
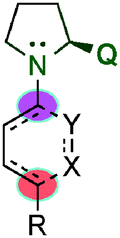
 | ||
| Scheme 23 First intermolecular [4+2] cycloaddition by Hong et al. | ||
After this work, in 2008, Christmann et al. published the synthesis of mono- and bicycle derivatives via dienamine activation of α,β-unsaturated aldehyde (91).38 The dienamine intermediate Y was easily formed using the Jørgensen–Hayashi catalyst (14), requiring benzoic acid as an additive. Once the intermediate Y was formed, an intramolecular Diels–Alder reaction can take place forming the bicyclic product (92) in good yield and enantioselectivity. The aldehyde was subsequently reduced to the alcohol 93 due to the easier isolation (95%) (Scheme 24). Based on this work, Gil Santo et al. studied Christmann's reaction development using DFT calculations.39 They proved that the use of an external acid is necessary for both the initial enamine formation and also the final catalyst elimination steps. The enantioselectivity is related to the orientation around the C–N enamine bond during the cyclisation step, whereas the diastereoselectivity depends on the syn/anti configuration of the substituents on the final ring.
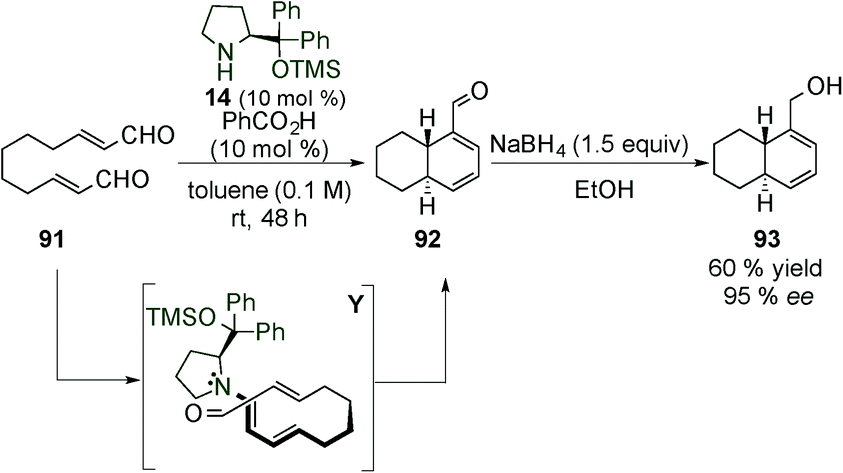 | ||
| Scheme 24 First intramolecular [4+2] cycloaddition by Christmann et al. | ||
Later on, Vicario's group studied the dynamic kinetic resolution of 5-acyloxydihydropyranones (96) through dienamine activation.40 Thus, they were able to react the racemic pyranones 95 with α,β-unsaturated aldehydes (94) in the presence of the Jørgensen–Hayashi catalyst (14, 20 mol%) and p-nitrobenzoic acid (20 mol%) (Scheme 25). The reaction proceeded through of a Diels–Alder/elimination cascade reaction, obtaining the final products (63) with excellent enantioselectivity.
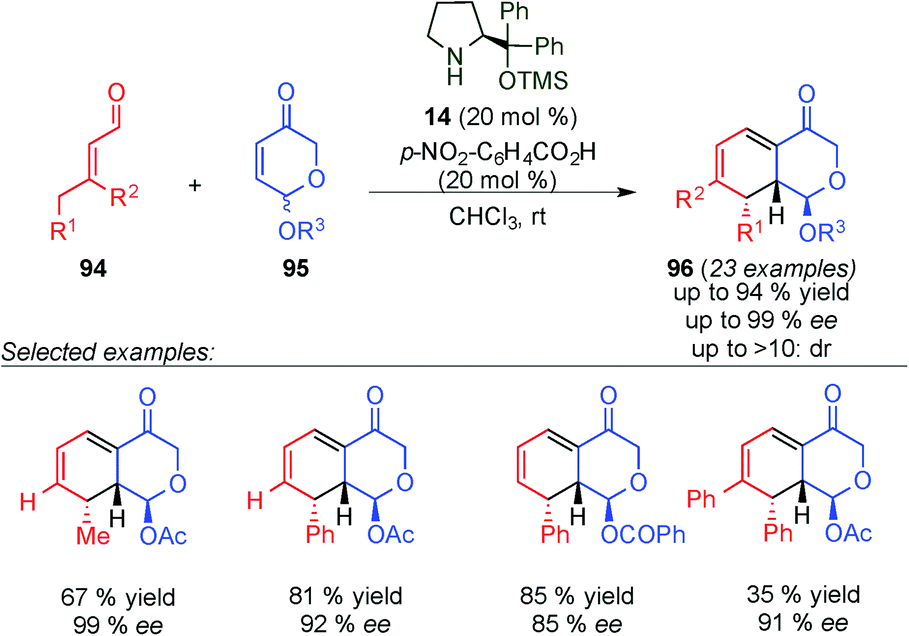 | ||
| Scheme 25 Kinetic dynamic resolution of pyranones 96 and synthesis of dihydrodibenzofurans via dienamine catalysis. | ||
Yang's group published the synthesis of dihydrodibenzofurans 99 by dienamine-mediated catalytic [4+2] cycloaddition reaction requiring a more sterically demanding catalyst 97 (Ar = naphthyl, Scheme 26).41 Under these conditions, the authors generated a large number of different substrates, most of them with moderate ee (ranging from 22–80% ee) and only two examples with excellent enantiocontrol (91% ee). Aldehyde-dihydrodibenzofuran products (99) were subsequently reduced with NaBH4 to their more stable corresponding alcohols 100 to ease isolation. The moderate enantiomeric excess achieved in most of the cases can likely be attributed to the highly strained concerted dienamine transition state (Z) (Scheme 26).
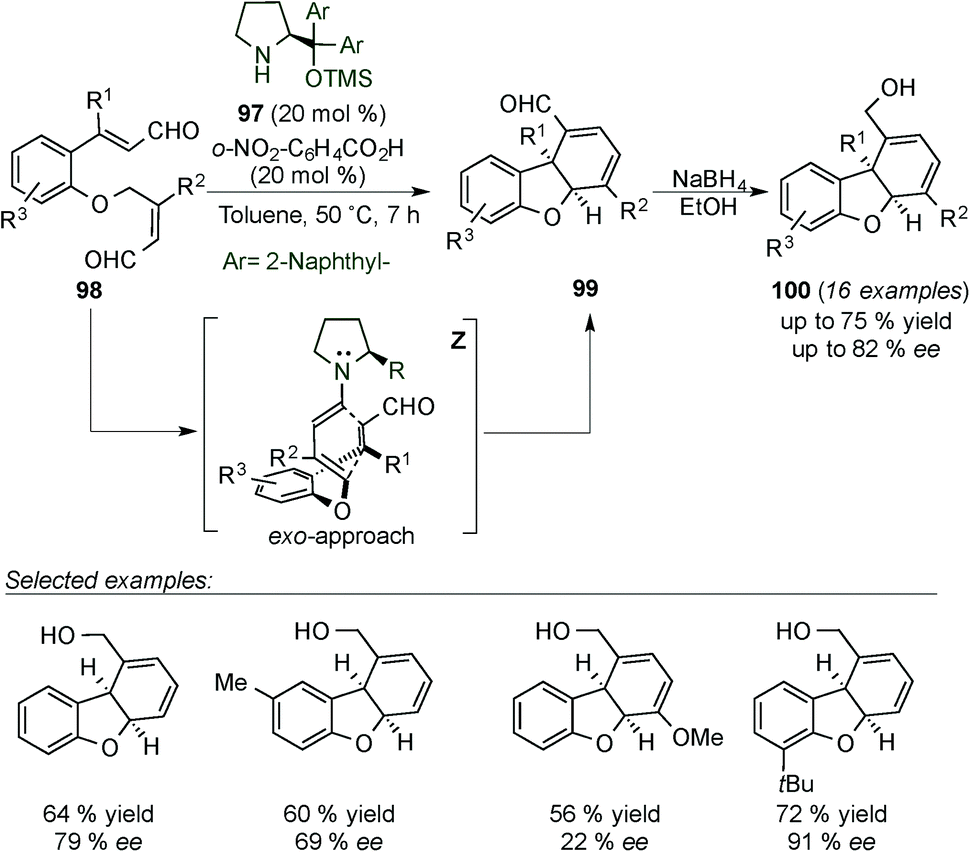 | ||
| Scheme 26 Intramolecular Diels–Alder reaction for the synthesis of dihydrodibenzofurans. | ||
In 2014, Albrecht, Jørgensen and co-workers described the multicomponent one-pot cascade approach for the synthesis of enantiomerically enriched methylene bridged benzo[1,5]oxazocines 105 (Scheme 27).42 It utilizes initial condensation of the corresponding aniline (101) with salicylaldehyde derivatives (102) followed by a dienamine-mediated γ-selective Mannich-initiated cycloaddition reaction. Subsequent cyclisation via oxy-Michael addition furnishes 105 in good yields and with high stereoselectivities. The developed cascade allowed for direct γ,β,ipso-functionalisation of the employed α,β-unsaturated aldehydes. Based on the absolute configuration assignments established through the single crystal X-ray analysis of 105 a plausible reaction mechanism is proposed. The reaction is initiated by condensation of 101 with 102 to afford N-aryl imine 103. Subsequently, a Mannich-initiated cyclisation reaction between 103 and the s-cis dienamine derived from the condensation of aminocatalyst 34 with 104 furnishes the cyclohexene framework (AA–AA′). The catalyst was eliminated to give the iminium ion intermediate and a subsequent intramolecular oxy-Michael addition (AA′′) was carried out to yield 105. In addition, the authors postulated that the employment of N-aryl imines possessing an extended electron-rich π-system might favour the [4+2]-cycloaddition pathway over a sequential reaction mechanism initiated by a γ-Mannich reaction involving an s-trans-configured dienamine intermediate.
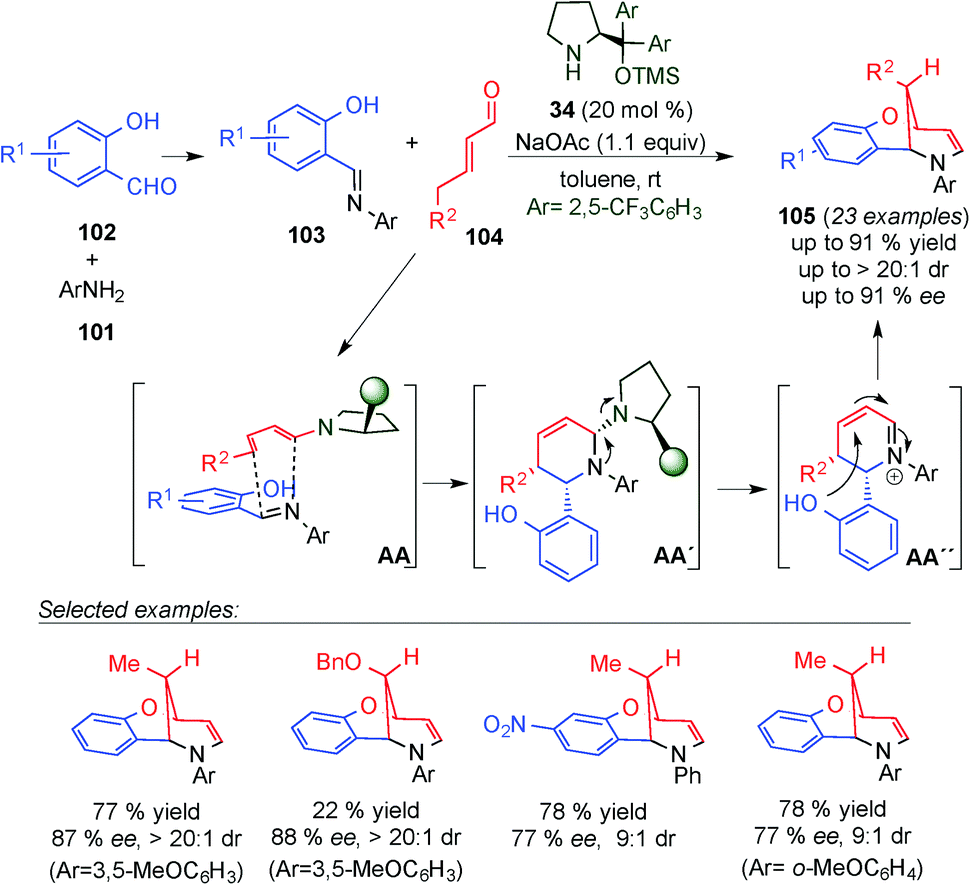 | ||
| Scheme 27 Synthesis of bridged benzoxazocines through a multicomponent one-pot cascade process. | ||
The same group showed the potential of the dienamine intermediates as electron-rich dienes for the asymmetric synthesis of steroids.43 This approach is based on the organocatalytic [4+2]-cycloaddition of cyclic enals (106) and diketone (107) using 14 and benzoic acid as a catalytic system. In most cases, the enantioselectivities were excellent (≥99% ee), providing β-steroid derivatives (108) with high yield in a broad substrate scope on the aromatic ring of the enal starting material (106). Furthermore, substrates with varying ring sizes and heteroatom incorporation were also synthesised (Scheme 28). In addition, post-synthetic transformation of 108 leading to the synthesis of Torgov's diene and its analogues was carried out in this work. This methodology was also applied to quinone-based dipolarophiles for the asymmetric synthesis of D-homosteroid products.
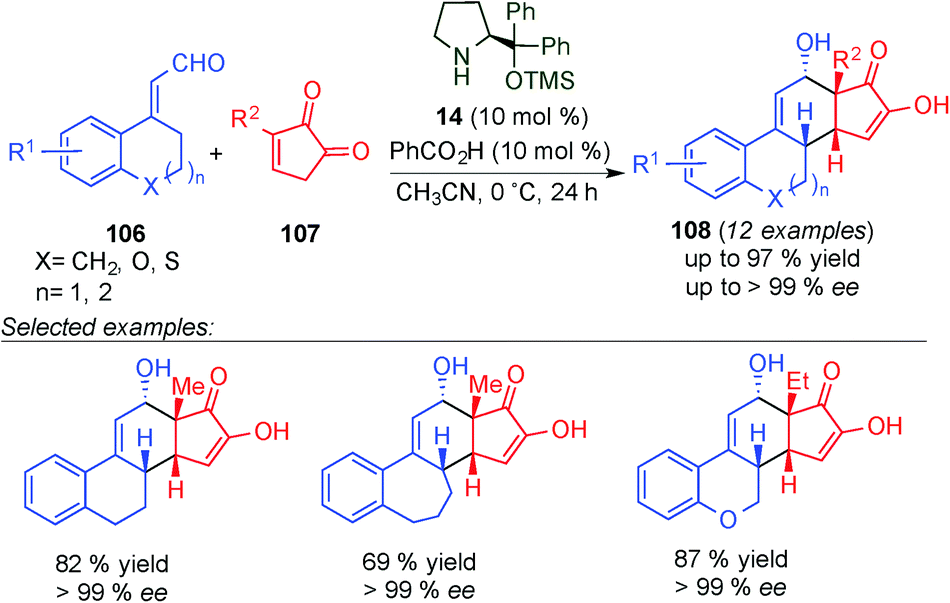 | ||
| Scheme 28 Synthesis of steroids via dienamine-mediated catalytic [4+2]-cycloaddition reaction. | ||
Wang's group also exploited the in situ generation of dienamine dienes for the synthesis of tricyclic benzopyrans 112,44 which can be found in important biologically complex natural products, such as cannabinoids and Murrayamine D, among others (Scheme 29). The synthesis is based on a two-step process: first, a [4+2] cycloaddition via dienamine activation, followed by a reduction/acid-catalysed intramolecular cyclisation. The first Diels–Alder reaction takes place by a [4+2]-cycloaddition pathway involving a cis-configured dienamine intermediate (AB), and it was proposed that the expulsion of CO2 promotes the cycloaddition process (AB′ intermediate) to form 111 in line with Serebryakov's original postulation in the 90's.10 The reaction proceeded quite well for a large variety of cases (19 examples) in good yields and enantioselectivities.
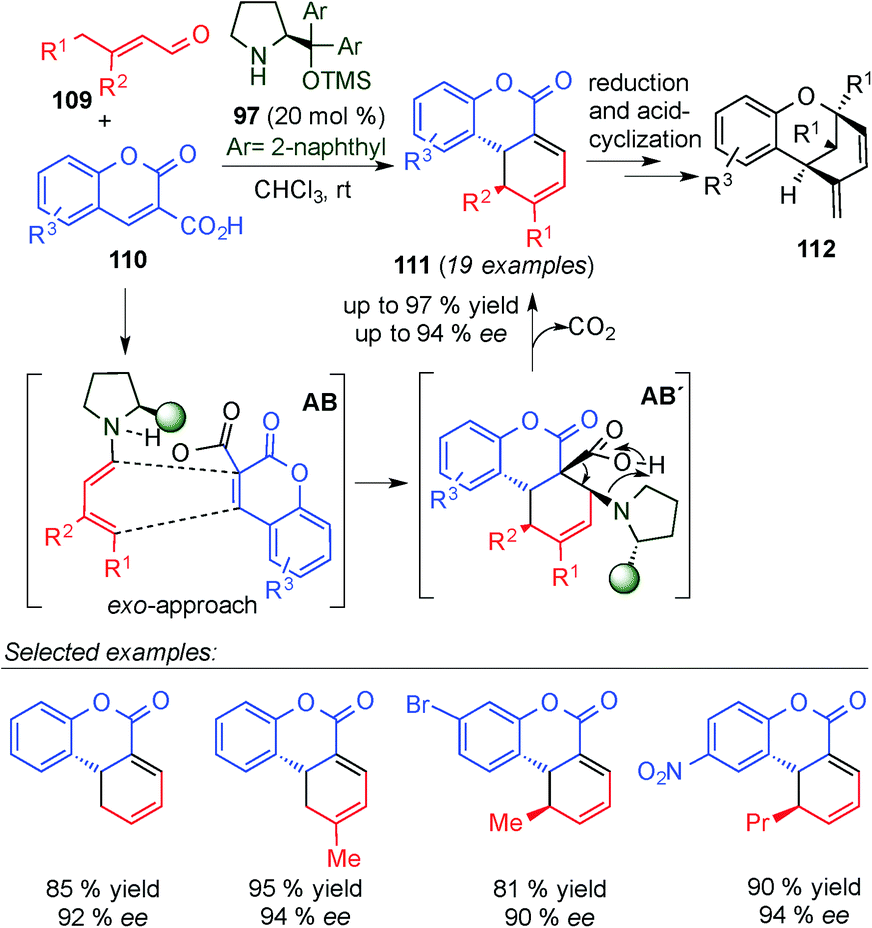 | ||
| Scheme 29 Synthesis of tricyclic benzopyrans by a tandem dienamine-mediated catalytic Diels–Alder/ one-pot reduction/acid-catalysed cyclisation. | ||
In 2014, Alemán's group presented the synthesis of tricyclic derivatives (114) (which are present in different natural and biological products, e.g. momilactone A) by desymmetrisation of cyclohexadienones 113via dienamine intermediate AC (Scheme 30).45 The reaction tolerated a large variety of substituents at different positions of the cyclohexadienone 113 and different heterocyclic ring sizes can be achieved. Mechanistic studies by DFT calculations have shown that the reaction proceeds via an asynchronous [4+2] cycloaddition and not a stepwise reaction (intermediate AC).
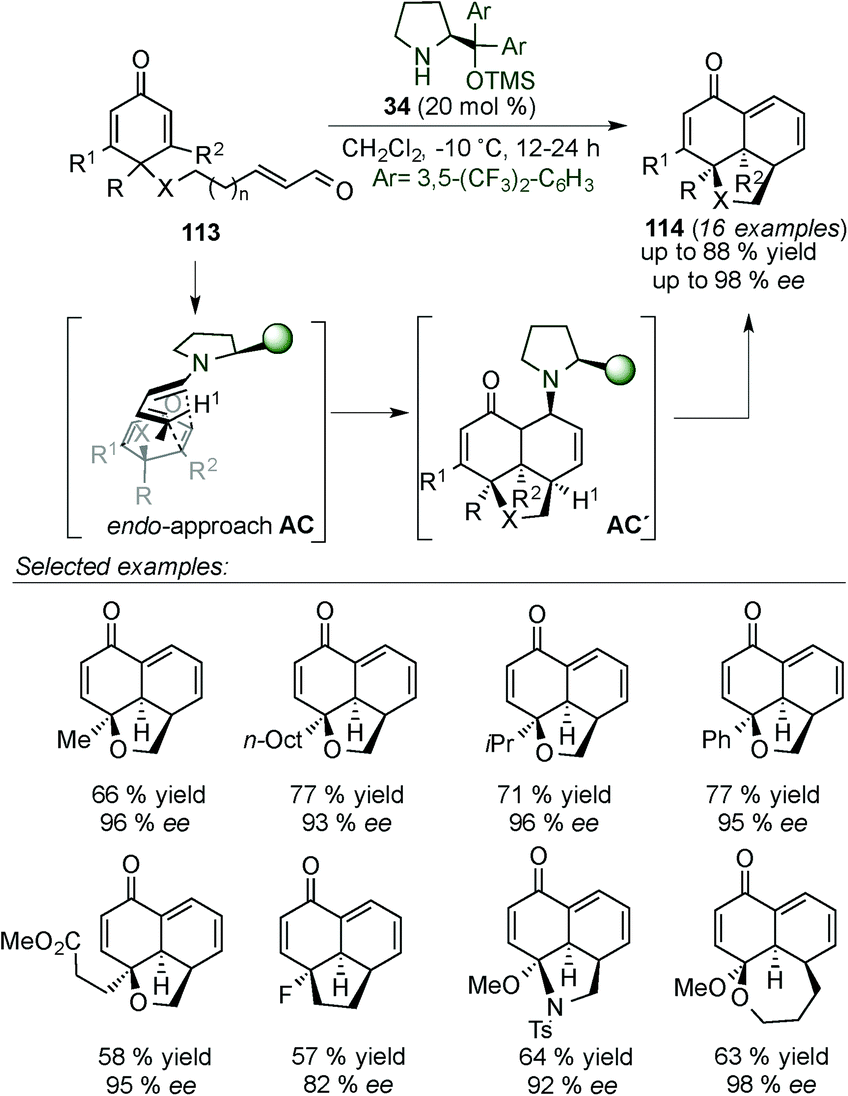 | ||
| Scheme 30 [4+2]-cycloaddition for the desymmetrisation of cyclohexadienones. | ||
In a related study, Jørgensen et al. described the intermolecular [4+2]-cycloaddition reaction between quinone derivatives 116 and β-disubstituted enals 115 (Scheme 31).46 The reaction tolerated different substituents at the β-position of the enals. Based on DFT calculations, the authors found that the Diels–Alder reaction proceeded in a two-step manner with endo-selectivity due to a favourable electrostatic interaction in the formed zwitterionic intermediate.
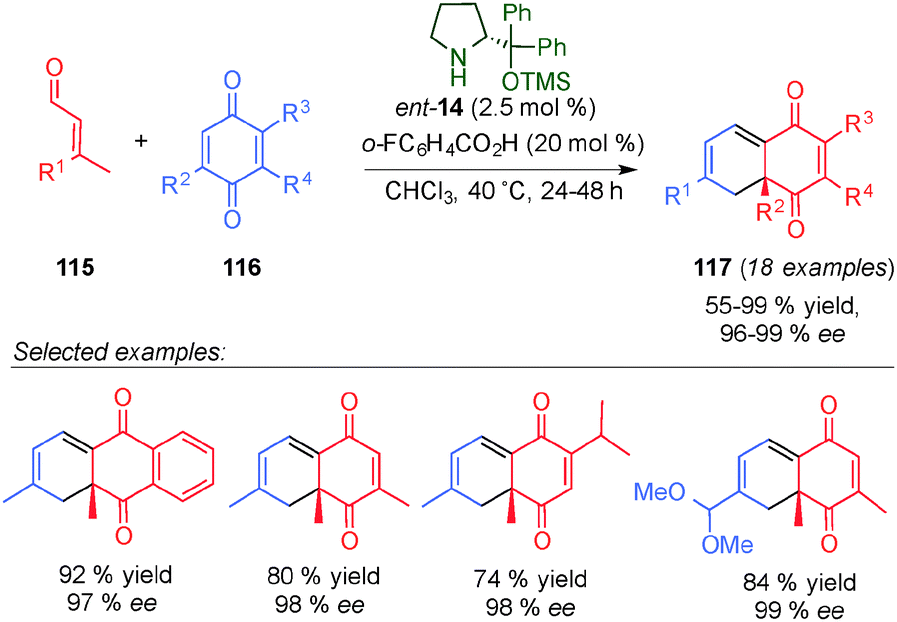 | ||
| Scheme 31 Diels–Alder reaction between quinone derivatives and enals. | ||
5. 4,5-Reactivity: [2+2], [4+2] and [3+2] cycloaddition
5.1. [2+2] cycloadditions: strained ring formation
With the aim of achieving higher levels of both reactivity and stereocontrol and, more importantly, overcoming the problem of site selectivity inherent to the remote functionalisation of enals through dienamine activation, Jørgensen et al. designed a bifunctional squaramide-based aminocatalyst (118), which simultaneously activates the two reaction partners (electrophile and enal) via H-bond and dienamine activations, respectively (Scheme 32).47 As a proof of concept, catalyst 118 was applied to a formal [2+2]-cycloaddition of linear α,β-unsaturated aldehydes (119) with nitroolefins (18) leading to cyclobutane derivatives (120) in high yields and with excellent stereocontrol. In this example, the H-bonding interaction of the electrophile (18a) with the squaramide moiety arranged the amine protons with a syn/syn configuration. This conformation of the squaramide allows for three H-bonding interactions with the nitro group and a staggered π-stacking interaction between the phenyl rings of the nitroolefin and the dienamine (AD), which provide the proper positioning of the electrophile, and therefore allow for complete regio- and stereocontrolled functionalisation at the γ-carbon. The results were further supported by computational studies, and the authors concluded that a stepwise process would be the most plausible mechanism for this transformation (AD′, Scheme 32). Various cyclobutanes were efficiently synthesised by a formal [2+2] cycloaddition from aryl, heteroaryl and alkyl nitroalkenes (18) as well as 5-aryl (R1 = H, R2 = Ar) and 5-alkyl (R1 = Me, R2 = Me) substituted aldehydes (11).
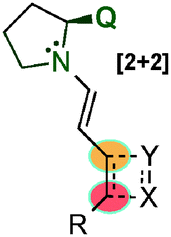
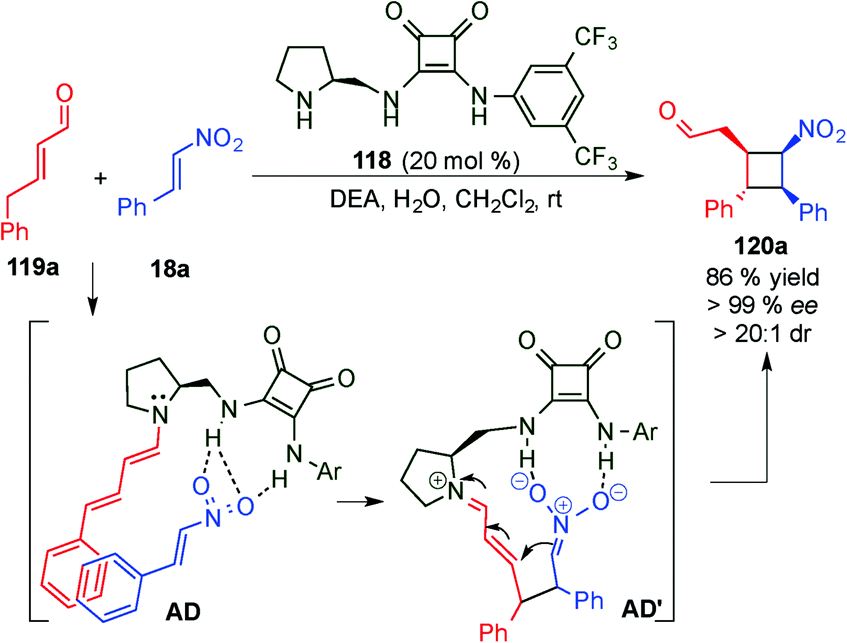 | ||
| Scheme 32 Bifunctional H-bond directed dienamine catalysis. | ||
Concurrently, Vicario et al. described a similar formal [2+2] reaction involving cooperative catalysis, utilising an aryl-prolinol ether (14) and a thiourea (121) that were used concomitantly to activate both reactants (Scheme 33).48 In this case, it was necessary to have an α-hydroxymethyl-substitution on the nitroalkene (123) in order to push the reaction towards the hemiacetal product (124) with full conversion. This reaction provided a general protocol for a series of aryl-bearing either electron-rich [EDG] or electron-poor [EWG] substituents as well as heteroaryl and 5-alkyl substituted α,β-unsaturated aldehydes 122. Whilst the presence of both EDGs and EWGs on the aryl ring of nitroalkene was tolerated, substitution with an alkyl chain was not described (Scheme 33).
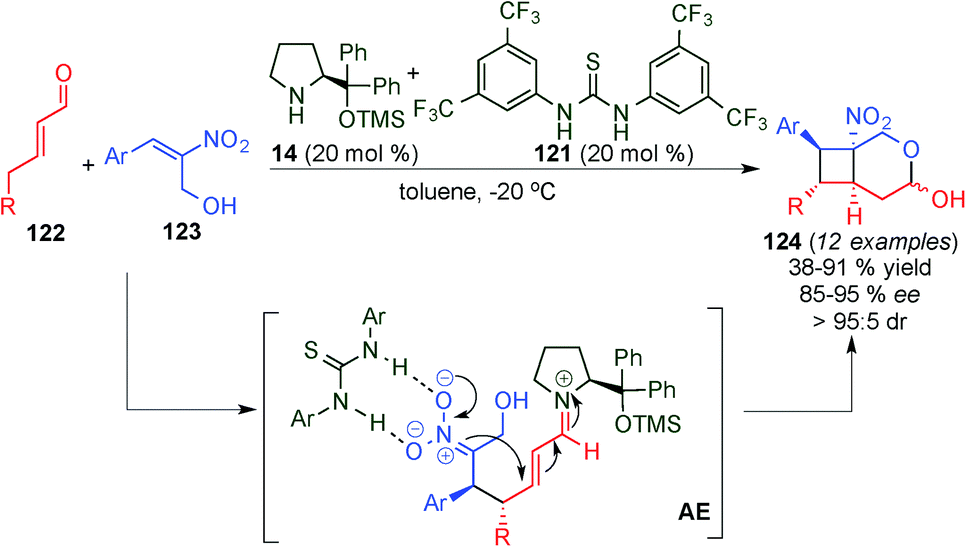 | ||
| Scheme 33 Cooperative catalysis in the formal [2+2] cycloaddition. | ||
In 2014, Wang et al. described the synthesis of spirooxindole skeletons incorporating a cyclobutane moiety (127), which are quite common scaffolds in natural and pharmaceutical products, by using H-bond-directing dienamine activation.49 By contrast, they found that the best catalyst was the diphenylprolinol 4 (20 mol%) in contrast to the one used by Jørgensen (squaramide 118vs. prolinol 4, Scheme 34). The reaction tolerated different groups at the dienamine functionality, however alkyl chains were not reactive enough to give the final product. In addition, the method allowed different aryl- and nitrogen-substitution groups on 126.
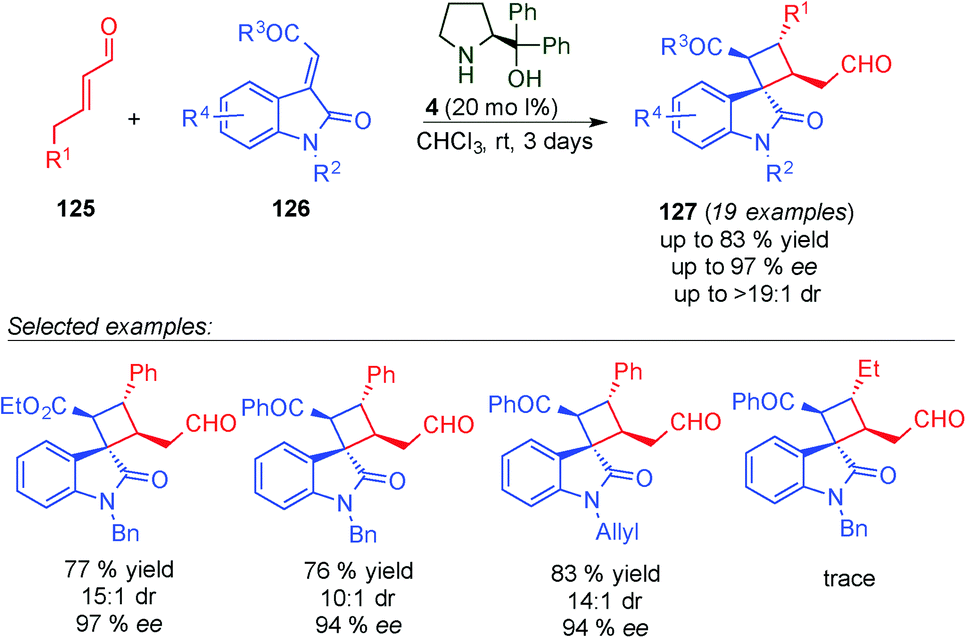 | ||
| Scheme 34 Wang's [2+2] cycloaddition for the synthesis of spirooxindole skeletons. | ||
One year later, Jørgensen's group described an alternative reaction for the synthesis of spirooxindole skeletons incorporating a cyclobutane moiety with a diverse substitution pattern (130) by using a novel approach to generate the dienamine intermediate (Scheme 35).50 They found that cyclopropylacetaldehydes (128) can be condensed with aminocatalyst 14 to generate dienamine intermediate AF. This process is facilitated by a favourable orbital interaction between the π-orbital of the enamine and the σ* C–C orbital of the cyclopropyl ring. The latter intermediate (AF) reacted in a [2+2] manner to give similar cyclobutanes as Wang (compare Schemes 34 and 35).49 Another plausible mechanism described by the authors is a [3+2] reaction followed by a ring-contracting rearrangement. However, based on the precedents in the literature, and the proof described by them, the [2+2] seems to be the more plausible one. As in Wang's case, the reaction tolerated a large number of substituents at the indol group (129), but in this case it was more restricted in the aldehyde reagent (128), as it required gem diester groups to achieve the ring opening (Scheme 35).
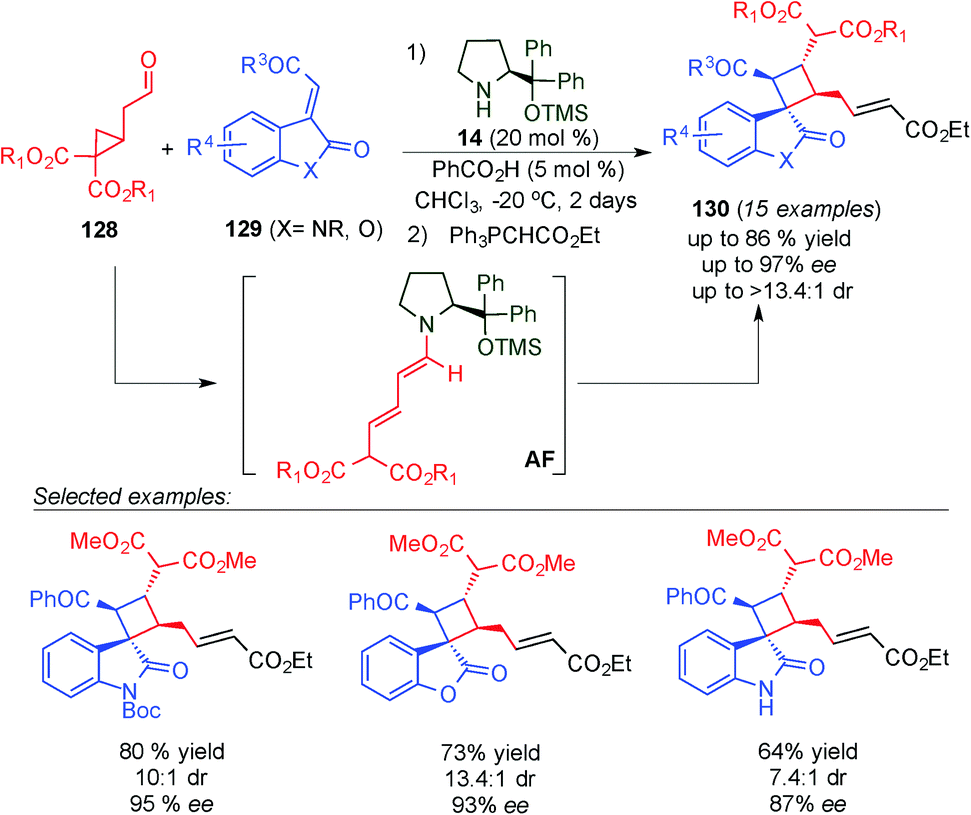 | ||
| Scheme 35 Jørgensen's [2+2] cycloaddition for the synthesis of spirooxindole skeletons. | ||
5.2. [4+2] cycloadditions of electron-rich dienophiles
In 2010 Chen's group developed a new regio- and stereoselective dienamine-mediated catalytic inverse-electron demand Diels–Alder reaction using α,β-unsaturated aldehydes as dienophiles.51 This is the first case of using the double bond at the 4,5-position as a dienophile through a dienamine-activation strategy. It is of particular interest that even when six positions were removed from the chiral centre of the catalyst, the obtained enantiomeric excess was excellent (Scheme 36). The reaction tolerated a large number of variations at the diene (131), different aromatic and alkyl derivatives at the δ-position, and also some alkyl substitutions at β- and γ-positions with excellent enantioselectivities and good diastereomeric ratios in all cases (see selected examples, Scheme 36). However, in the case of the aldehyde, the scope was extremely limited and only crotonaldehyde (40a) was used for this study.
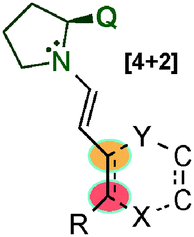
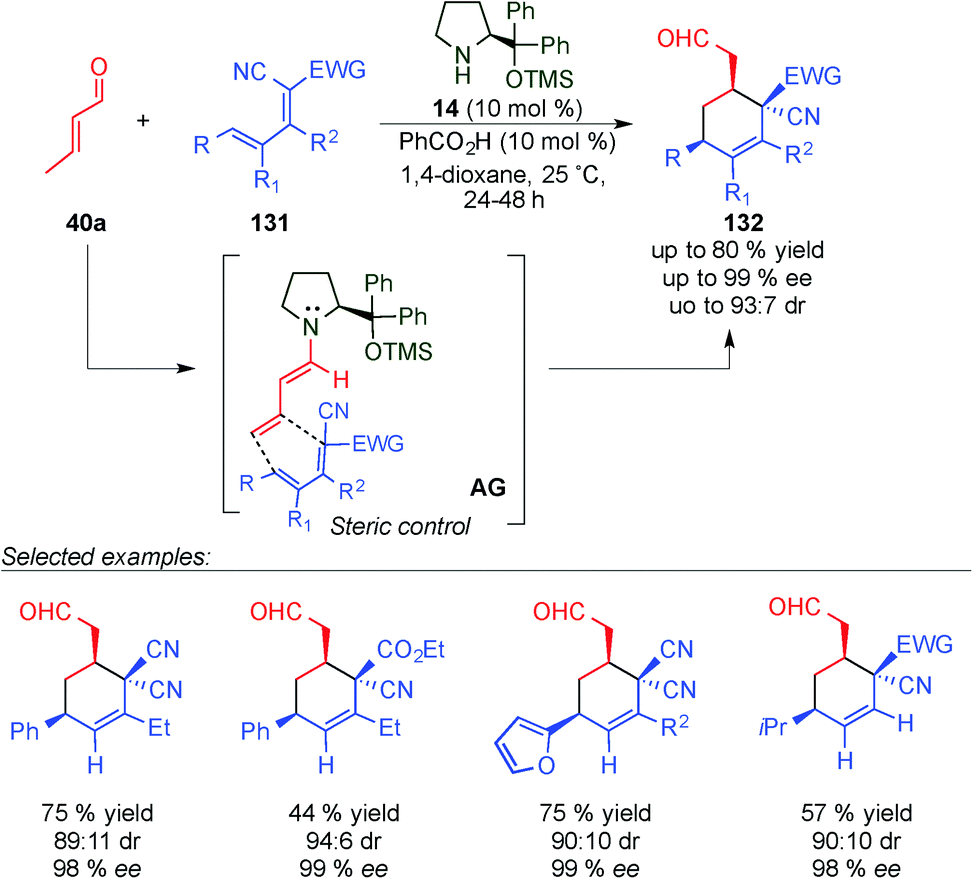 | ||
| Scheme 36 First dienamine-mediated catalytic inverse-electron demand Diels–Alder reaction with crotonaldehyde. | ||
After two years, Jørgensen et al. described a similar dienamine-mediated catalytic inverse Diels Alder reaction pathway but instead of using a steric shielding strategy as Chen reported in his work, they described an aminocatalytic H-bond directing strategy (intermediate AH, Scheme 37).52 Thus, the limitations that Chen found, such as the use of only crotonaldehyde as a nucleophile, were solved using this strategy. Only 4,5-reactivity was observed, high stereocontrol was found, and a broader scope was achieved, giving access to chiral dihydropyran frameworks (135). Thus, the reaction tolerated a large number of substituents at the 1,2-dicarbonyl electrophilic acceptors (134) with excellent ee values and diastereoselectivity. However, the scope at the aldehyde (133) was more limited and enantioselectivities from moderate to good were obtained (75% to 90% ee).
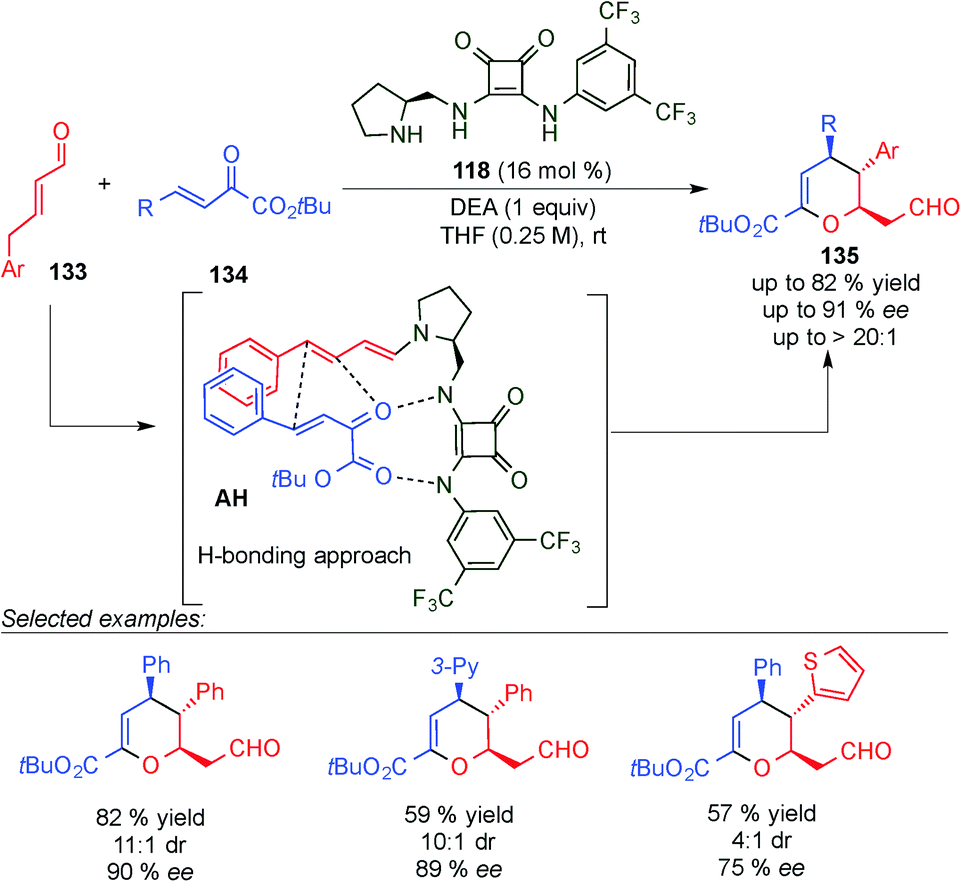 | ||
| Scheme 37 First H-bond controlled dienamine-mediated catalytic inverse-electron demand Diels–Alder reaction. | ||
In an associated study, the same group developed the synthesis of enantioenriched dihydropyran phosphonates (137)53 using acyl phosphonates (136) instead of the 1,2-dicarbonyl compounds (134) (compare Schemes 37 and 38). In this case, the products were also obtained with good ee values and yields, allowing even the use of alkyl acyl phosphonate derivatives.
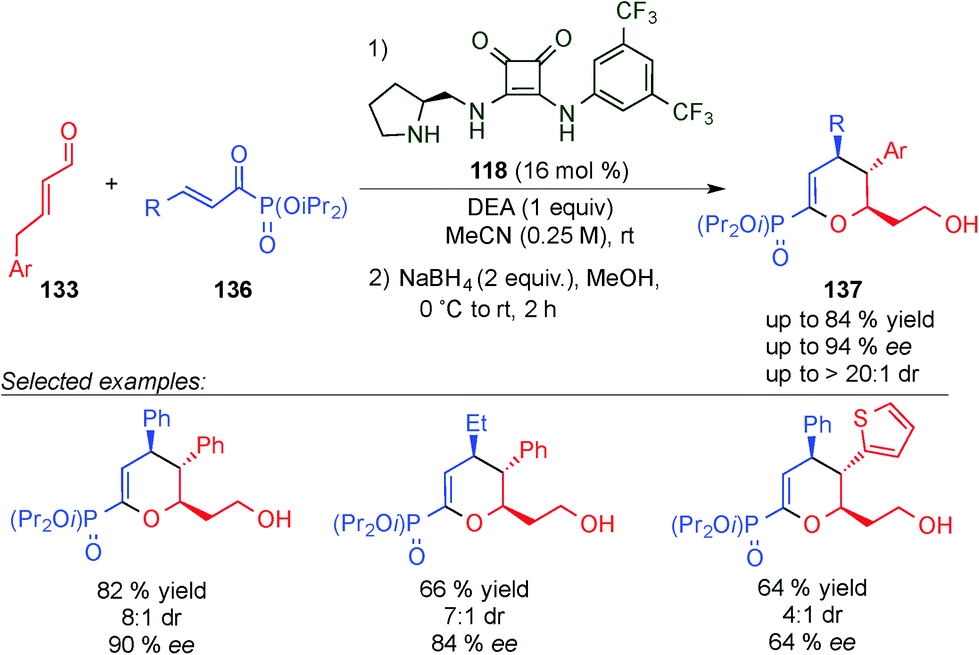 | ||
| Scheme 38 H-bond controlled dienamine-mediated catalytic inverse-electron demand Diels–Alder reaction for the synthesis of dihydropyran phosphonates. | ||
Later on, Chen described an asymmetric Diels–Alder reaction of β,β-disubstituted enals (139) and chromone-fused dienes 140 which gave access to different chiral tetrahydroxanthone based natural products 141 (Scheme 39).54 This strategy is based on the HOMO-raising of the 4,5-double bond and represents the first use of β,β-disubstituted enals in dienamine activation. The Diels–Alder reaction was followed by a domino deprotonation-isomerisation-vinylogous aldol reaction to give the final bicyclo derivatives 141 (Scheme 39). The reaction tolerates a large number of aldehydes with different substitution (even at the α-position) and different chromone substrates. In all cases, only one diastereoisomer was detected and good yields and ee values were obtained.
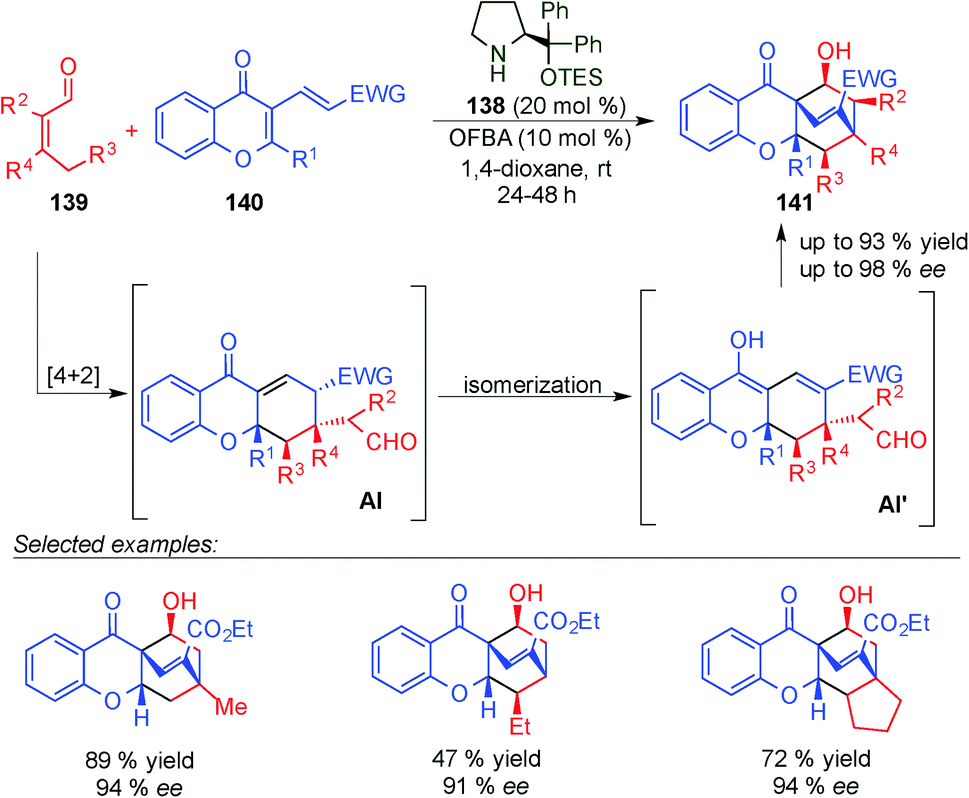 | ||
| Scheme 39 Dienamine-mediated catalytic inverse-electron demand Diels–Alder reaction for the synthesis of tetrahydroxanthone based natural products. | ||
Very recently, Jørgensen and colleagues reported the stereoselective formation of highly substituted CF3-dihydropyrans (144),55 which are important motifs in medical chemistry and in bioactive compounds, such as the antiviral agent zanamivir,56 risugacin A5057 and swertiamarin58 (Scheme 40). Therefore, they developed the inverse-electron demand hetero-Diels–Alder reaction with a wide substrate scope of substituted 5-bromo-6-CF3-dyhydropyrans 143 and γ-aryl substituted α,β-unsaturated aldehydes 133, catalysed by C2-symmetric 2,5-diphenylpyrrolidine catalyst 142 with moderate to good yields and diastereoselectivities and excellent enantioselectivity. Additionally, the authors showed a further derivatisation of the Diels–Alder products (see the bottom, Scheme 40) by cross-coupling reactions and lithiation reactions, as well as an unexpected cyclisation protocol, which furnished optically active tetracyclic compounds.
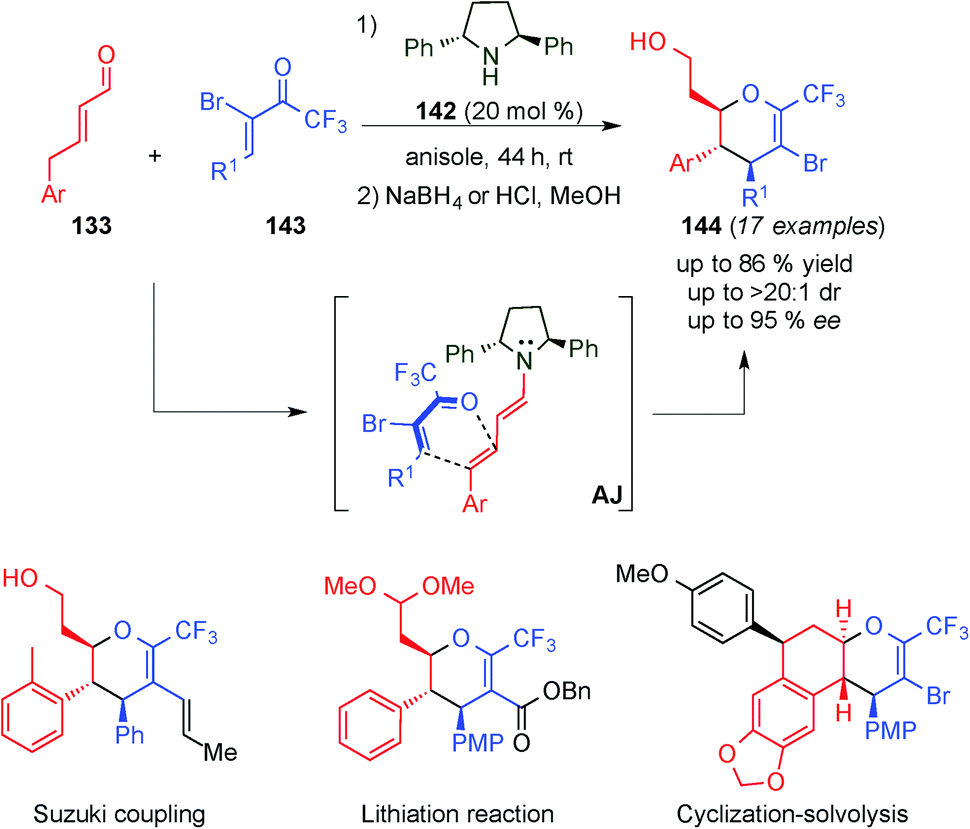 | ||
| Scheme 40 Inverse-electron demand hetero-Diels–Alder reaction. | ||
5.3. [3+2] cycloadditions
In 2014, Wang's59 and Alemán's60 groups independently developed the [3+2] cycloaddition reaction between azomethine imines and α,β-unsaturated aldehydes through dienamine activation (Scheme 41). Despite the fact that the iminium ion is the previous intermediate for the formation of the corresponding dienamine and that both species are in equilibrium, there were no studies investigating the reactivity control of these two intermediates with their differing nucleophilic and electrophilic natures. In order to have complete control over the plausible equilibrium between the iminium–dienamine (k1vs. k2) or over the reactivity of both reactions (k3vs. k4), different reaction conditions were explored (Scheme 41).
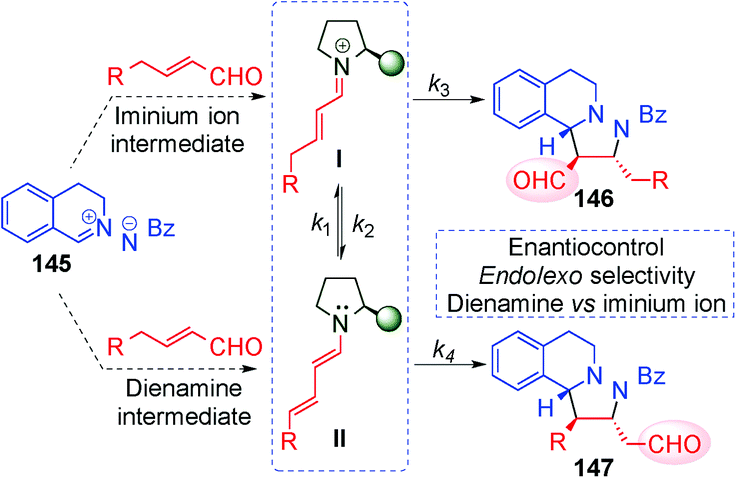 | ||
| Scheme 41 [3+2] cycloaddition reaction: dienamine vs. iminium ion intermediates. | ||
After screening conditions, Alemán's group found that complete control of the dienamine or iminium intermediates could be achieved by employing appropriate conditions.60a The use of catalyst 34, TBAB as an additive and toluene as a solvent was the best conditions for iminium type products 146 (top Scheme 41). By contrast, the use of the hydrated dipole 145, catalyst 14 and CH2Cl2 as a solvent is the best conditions so far to obtain dienamine products 147 (Scheme 42). With this work, Alemán's group was able to describe the first example of regio-control of an iminium ion intermediate versus a dienamine intermediate in the [3+2] cycloaddition of azomethine imines. Dienamine products (147) were achieved with good yield and good enantioselectivity in most cases, regardless of the nature of the aromatic ring used in the aldehydes (133) or the substituent on the aryl group of the azomethine imine (145) (Scheme 42).
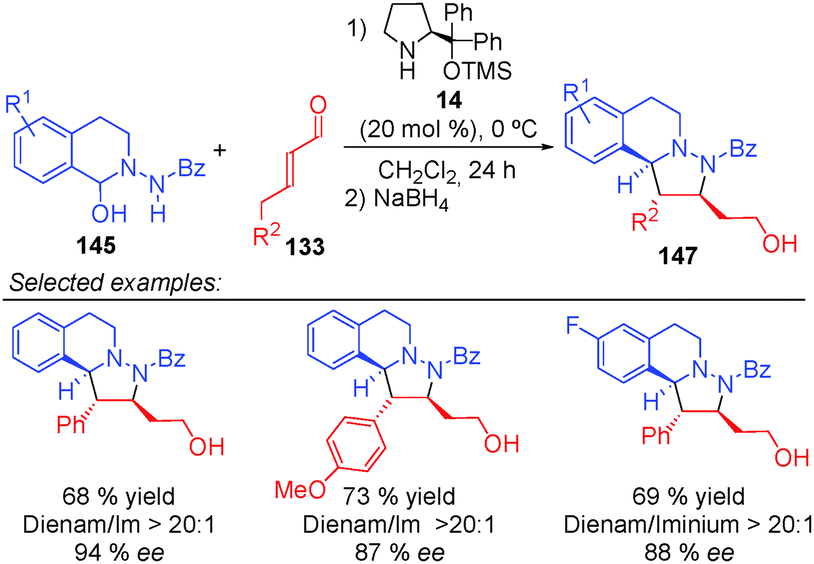 | ||
| Scheme 42 First examples of [3+2] cycloaddition via dienamine activation. | ||
5.4. [5+2] cycloaddition
Vicario's group published the catalytic [5+2] cyclisation between oxidopyrylium ylides and enals (148) by dual hydrogen-bond dienamine activation (Scheme 43).61 Thus, benzopyrylium ylides were generated from the acetoxy chromanone derivative 149 and, mediated by a squaramide secondary amine catalyst 118, were able to react with the dienamine intermediate (AK). The reaction proceeds with moderate to excellent yields and good ee values in most cases, but with a low diastereomeric ratio in some cases. Unlike with [3+2] cycloadditions, the dienamine intermediate showed exclusively 4,5-reactivity, most likely due to the H-bond interactions of the bifunctional secondary-amine/squaramide catalyst, thus furnishing excellent regiocontrol.
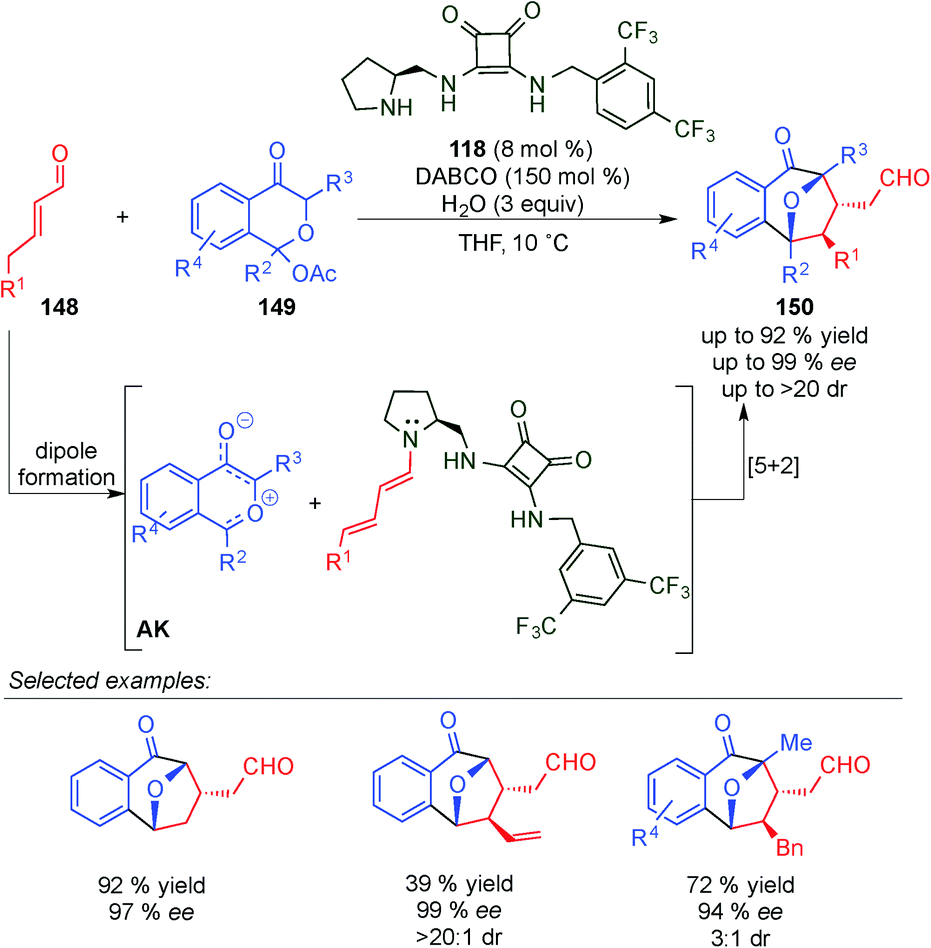 | ||
| Scheme 43 [5+2] cycloaddition of oxidopyrylium ylides via dienamine activation. | ||
6. Related dienamine and tandem processes
In 2014, Jørgensen's group used a tetraenamine intermediate (AL) to functionalise cycloheptatriene derivatives 151 (Scheme 44).62 Once the tetraenamine AL is formed, it reacts with electrophile 129via the dienamine intermediate from the less hindered prochiral face to afford intermediate AL′. The final cyclisation step of the anion to the iminium ion intermediate (AL′) takes place and leads to the corresponding spiro-compound 152. Through DFT calculations the authors explained the high diastereomeric ratios that the authors obtained in the reaction.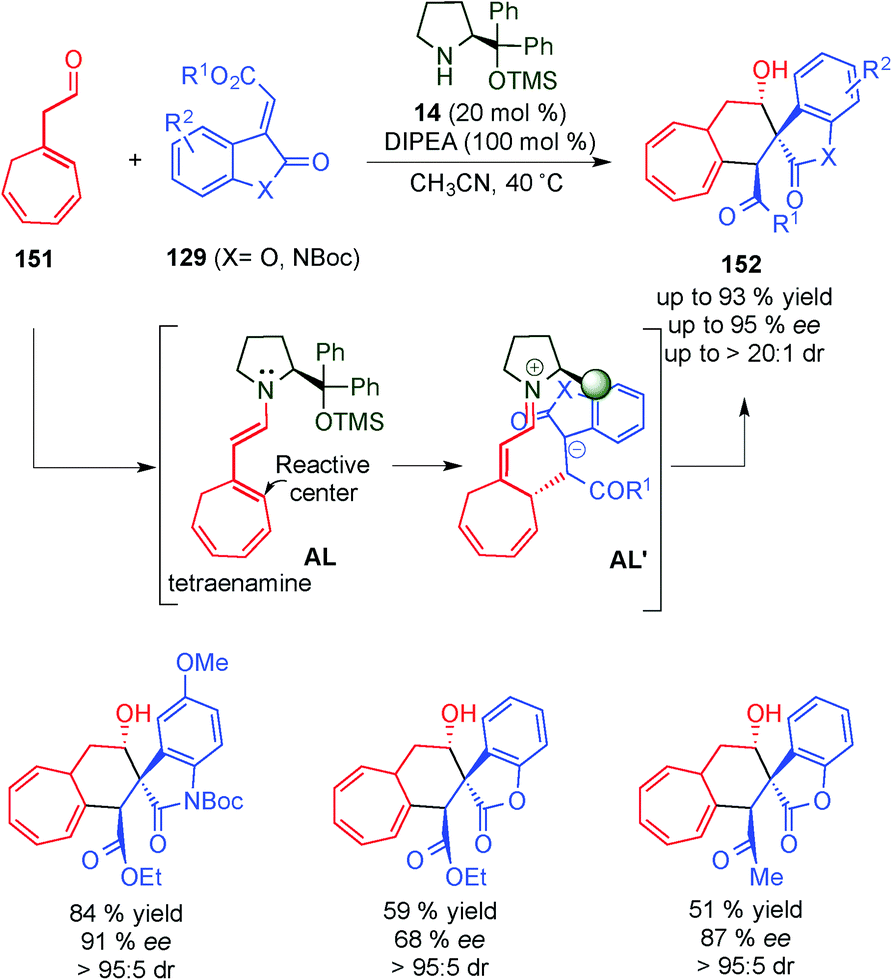 | ||
| Scheme 44 Functionalisation of cycloheptatriene derivatives via the tetraenamine intermediate. | ||
In 2013, the same group carried out the remote aziridination of 2,4-dienals (153) based on a vinylogous iminium ion/dienamine intermediate strategy (Scheme 45).63 In this work, an iminium ion intermediate AM is attacked (via 1,6) by 154 to furnish a dienamine intermediate AM′ that evolves through cyclisation (TSO− as a leaving group) to 155 in good yields and excellent enantioselectivities.
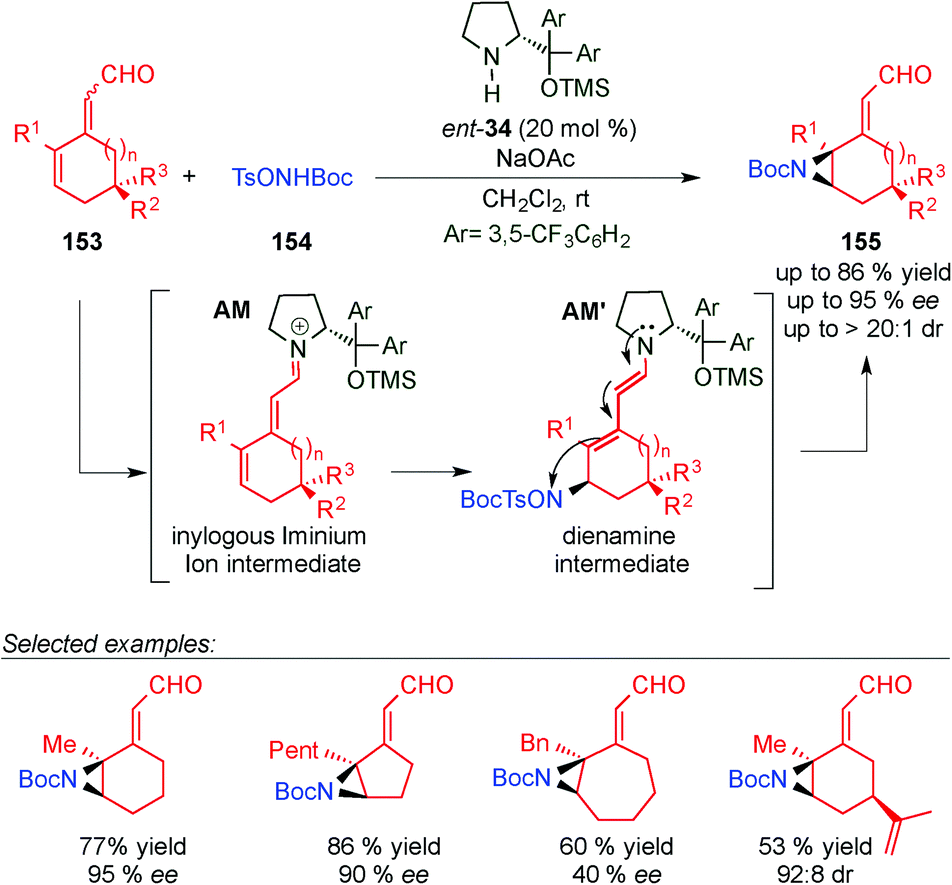 | ||
| Scheme 45 Remote aziridination through dienamine intermediate AM. | ||
The same concept was used by Melchiorre et al. in the 1,6-addition of an ambivalent compound 156 (a compound with both nucleophilic and electrophilic characteristics) to the vinylogous iminium ion 157, to give an intermediate AN (Scheme 46).64 The formed dienamine intermediate cyclises, via substitution of the chlorine atom at the benzylic position, and led to the final spiro-cyclohexane derivatives 158 in high enantiomeric excesses and good yields. Using dienamine intermediates, the same group published a triple vinylogous cascade reaction, via a Michael/1,6-addition/vinylogous aldol sequence (Scheme 47).65 In this case, the key step was the formation of the dienamine intermediate AO, which presents the appropriate groups to promote the attack of the dienamine intermediate by the aldehyde to form the final alcohol 161 after reduction with NaBH4.
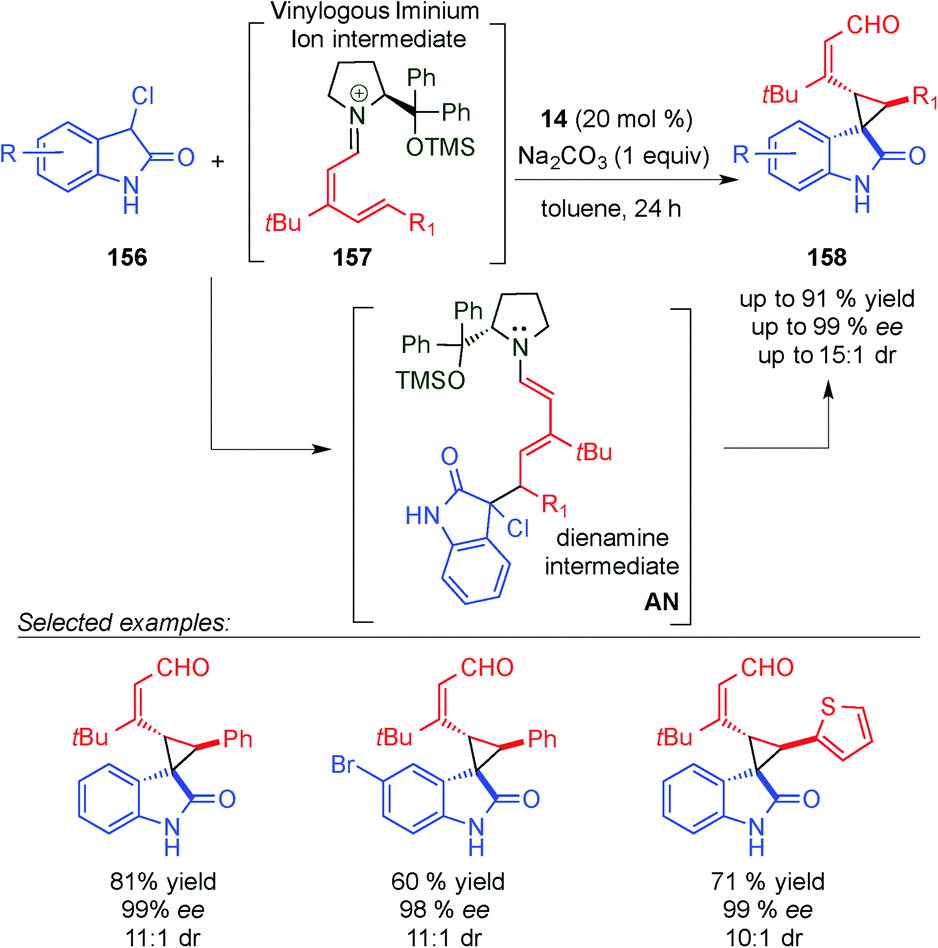 | ||
| Scheme 46 Remote cyclopropanation towards dienamine intermediate AN. | ||
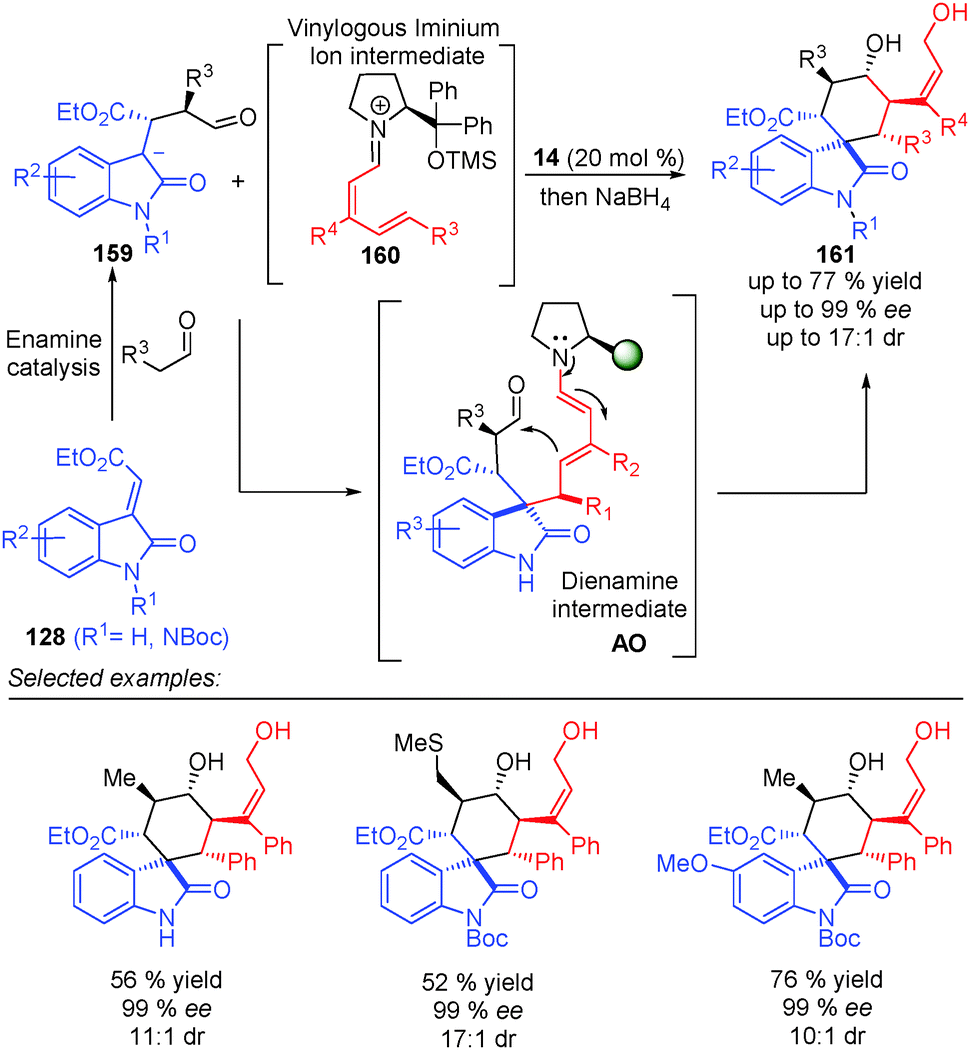 | ||
| Scheme 47 Triple cascade reaction in the synthesis of six spiro-oxindolic cyclohexanes via a dienamine intermediate AO. | ||
In 2015, Melchiorre et al. described the direct alkylation of enals 162 using various photocatalytic methodologies. Chiral dienamines, as well as enamines, have the ability to act as photosensitisers upon under light irradiation.66 In this outstanding work, the reaction took place with complete γ-selectivity, but only α-substituted enals and tertiary bromo-alkanes were used as starting materials and four examples (164) were synthesised (Scheme 48).
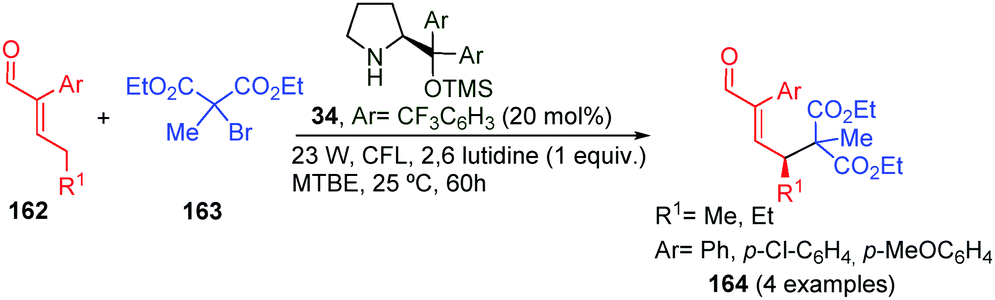 | ||
| Scheme 48 Organocatalytic alkylation of enal by direct photoexcitation. | ||
7. Conclusions and outlook
As highlighted in this review, since the early examples of Serebryakov dienamines to their evolution during the organocatalytic age, dienamine activation has established itself as a powerful and reliable strategy for the functionalisation of α,β-unsaturated aldehydes. Several reactive dienamine intermediates have enabled the development of a large variety of new methodologies for the synthesis of complex molecules with excellent control over the regio- and stereoselectivity. By exploiting the broad variety of possible reactive dienamine intermediates, formed upon condensation of an aminocatalyst with an α,β-unsaturated aldehyde, the highly regio- and stereoselective generation of new stereogenic centres located at the 3- and 5-positions of the α,β-unsaturated aldehydes (α- and γ-functionalisation, respectively) has been successfully demonstrated.With regard to regioselectivity, 1,3 vs. 1,5 reactivity, three main strategies have been used for regio-control: (i) In general terms, a delicate balance of electronic and steric effects is found in which the steric effect plays a predominant role in the reactivity of the dienamine intermediate with the electrophile. Therefore, the use of double substitution at C-5 (R2 and R3 ≠ H) means that only 1,3 reactivity can take place (left, Fig. 3; see e.g.Scheme 11). (ii) The appropriate substitution at the α-position of the aldehyde (R1 ≠ H) changes the selectivity to the γ-position (middle; see e.g.Scheme 15). (iii) The third and final strategy is based on the design of the catalyst. Thus, the use of a bifunctional catalyst that approaches the electrophile at the second double bond (C-4 and C-5) provokes a large selectivity at this γ-position, with squaramide proline catalysts 118 being the most often employed (right Fig. 3; see e.g.Scheme 43).
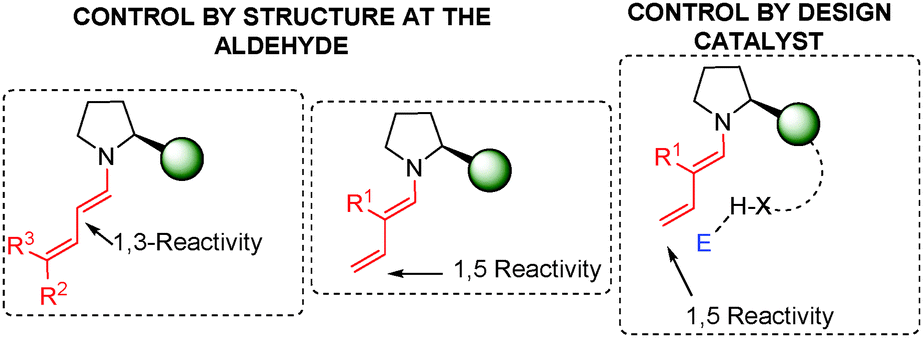 | ||
| Fig. 3 Strategies for regiocontrol in the dienamine intermediate. | ||
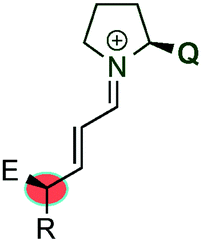
Very recently, during the evaluation of this manuscript, Gschwind et al. have described a mechanistic study in the remote stereocontrol of the dienamine-mediated catalytic nucleophilic substitution reaction based on NMR and computational studies.67 In this work, the authors reveal a plausible pathway in the Z/E dilemma, describing in detail the kinetics/thermodynamics in the formation of the dienamine and the latter reaction with an electrophile (SN1 reaction with bis(4-dimethylamino-phenyl)(methanol) in the presence of acid). The enantioselectivity is directly correlated with the configuration of the second double bond (Z and E, Scheme 49). Thus, the Z configuration leads to the R-enantiomer whereas the E configuration yields the S-enantiomer. The study reveals the kinetic preference in the formation of the Z-isomer of the second double bond (Z![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :E ≈ 2:1), which is independent of the structure of the catalyst (catalysts e.g.14 and 34). However, the reaction of the Z or E dienamine intermediate with the carbocation is catalyst dependent, and different interactions (CH-π, stacking) are the main factors responsible for the kinetic preference and faster reaction of Z-over the E-isomer. This is also corroborated by calculations and kinetic experiments.
:E ≈ 2:1), which is independent of the structure of the catalyst (catalysts e.g.14 and 34). However, the reaction of the Z or E dienamine intermediate with the carbocation is catalyst dependent, and different interactions (CH-π, stacking) are the main factors responsible for the kinetic preference and faster reaction of Z-over the E-isomer. This is also corroborated by calculations and kinetic experiments.
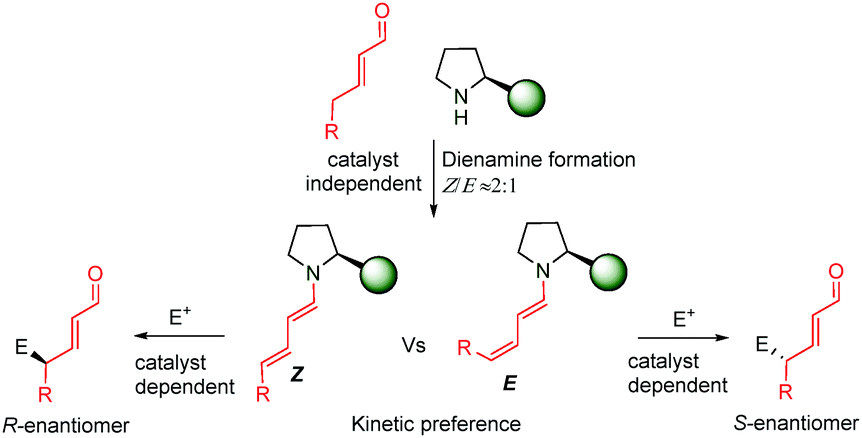 | ||
| Scheme 49 Origin of the enantioselectivity in the dienamine-mediated catalysis. | ||
Despite the great potential offered by dienamine catalysis in recent years, there are still challenging problems to solve and new potentially useful methodologies waiting to be discovered. For these reasons, the activation of α,β-unsaturated aldehydes via dienamine intermediates is expected to develop further in the near future with more and more diverse and ingenious findings. In our opinion, three main roads should be considered in the future: (i) New reactions, especially in the photocatalytic field, should be explored with different reagents (only one publication has been reported using photocatalytic conditions). (ii) New regioselective strategies for the control in 1,3 vs. 1,5 reactivity should be studied, especially concerning the use of bifunctional catalysts that could approach the electrophile at C-3 or C-5 regardless of the substitution of the enal. (iii) The combination of metal catalysis and organocatalysis in the dienamine functionalisation has not been fully explored and only a limited number of examples have been reported thus far.
Acknowledgements
J. A. thanks the MICINN for a “Ramon y Cajal” contract. Financial support from the Spanish Government (CTQ2015-64561-R, MINECO/FEDER) and the European Research Council (ERC-CG, contract number: 647550) is gratefully acknowledged. The authors wish to thank T. A. Singleton who assisted in the proof-reading of the manuscript.Notes and references
- For reviews on organocatalysis, see: (a) P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2001, 40, 3726 CrossRef CAS; (b) P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2004, 43, 5138 CrossRef CAS PubMed; (c) A. Berkessel and H. Groger, Asymmetric Organocatalysis: From Biomimetic Concepts to Applications in Asymmetric Synthesis, Wiley-VCH, Weinheim, 2005 Search PubMed; (d) P. I. Dalko, Enantioselective Organocatalysis, Wiley-VCH, Weinheim, 2007 Search PubMed; (e) D. W. C. MacMillan, Nature, 2008, 455, 304 CrossRef CAS PubMed; (f) A. Mielgo and C. Palomo, Chem. – Asian J., 2008, 3, 922 CrossRef CAS PubMed; (g) P. Melchiorre, Angew. Chem., Int. Ed., 2009, 48, 1360 CrossRef CAS PubMed; (h) S. Bertelsen and K. A. Jørgensen, Chem. Soc. Rev., 2009, 38, 2178 RSC; (i) C. Grondal, M. Jeanty and D. Enders, Nat. Chem., 2010, 2, 167 CrossRef CAS PubMed; (j) E. Marqués-López, R. P. Herrera and M. Christmann, Nat. Prod. Rep., 2010, 27, 1138 RSC; (k) P. H. Y. Cheong, C. Y. Legault, J. M. Um, N. Celebi-Olecm and K. N. Houk, Chem. Rev., 2011, 111, 5042 CrossRef CAS PubMed; (l) K. L. Jensen, G. Dickmeiss, H. Jiang, Ł. Albrecht and K. A. Jørgensen, Acc. Chem. Res., 2012, 45, 248 CrossRef CAS PubMed; (m) S. Meninno and A. Lattanzi, Chem. Commun., 2013, 49, 3821 RSC; (n) J. Alemán and S. Cabrera, Chem. Soc. Rev., 2013, 42, 774 RSC; (o) Ł. Albrecht, H. Jiang and K. A. Jørgensen, Chem. – Eur. J., 2014, 20, 358 CrossRef PubMed.
- For selected reviews on HOMO catalysis, see: Reviews on enamine catalysis: (a) W. Notz, F. Tanaka and C. F. Barbas, III, Acc. Chem. Res., 2004, 37, 580 CrossRef CAS PubMed; (b) S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471 CrossRef CAS PubMed; (c) G. Guillena, C. Nájera and D. J. Ramon, Tetrahedron: Asymmetry, 2007, 18, 2249 CrossRef CAS; (d) T. Kano and K. Maruoka, Chem. Sci., 2013, 4, 907 RSC; (e) D. Deng, S. Kumar and H. Wang, Chem. Commun., 2014, 50, 4272 RSC; (f) A. Desmarchelier, V. Coeffard, X. Moreau and C. Greck, Tetrahedron, 2014, 70, 2491 CrossRef CAS . Reviews on dienamine catalysis: ; (g) D. B. Ramachary and Y. V. Reddy, Eur. J. Org. Chem., 2012, 865 CrossRef CAS; (h) M. Nielsen, D. Worgull, T. Zweifel, B. Gschwend, S. Bertelsen and K. A. Jørgensen, Chem. Commun., 2011, 47, 632 RSC; (i) A. Parra, S. Reboredo and J. Alemán, Angew. Chem., Int. Ed., 2015, 51, 9734 CrossRef PubMed . Reviews on trienamine catalysis: ; (j) H. Jiang, Ł. Albrecht and K. A. Jørgensen, Chem. Sci., 2013, 4, 2287 RSC; (k) I. Kumar, P. Ramaraju and N. A. Mir, Org. Biomol. Chem., 2013, 11, 709 RSC; (l) I. D. Jurberg, I. Chatterjee, R. Tanert and P. Melchiorre, Chem. Commun., 2013, 49, 4869 RSC; (m) S. Reboredo, A. Parra and J. Alemán, Asymmetric Catal., 2013, 1, 24 Search PubMed.
- For selected reviews on LUMO catalysis, see: (a) S. B. Tsogoeva, Eur. J. Org. Chem., 2007, 1701 CrossRef CAS; (b) D. Almasi, D. A. Alonso and C. Nájera, Tetrahedron: Asymmetry, 2007, 18, 299 CrossRef CAS; (c) J. L. Vicario, D. Badía and L. Carrillo, Synthesis, 2007, 2065 CrossRef CAS; (d) A. Erkkilä, I. Majander and P. M. Pihko, Chem. Rev., 2007, 107, 5416 CrossRef PubMed; (e) M. Shimizu, I. Hachiya and I. Mizota, Chem. Commun., 2009, 874 RSC.
- For selected examples in SOMO-catalysis, see: (a) T. D. Beeson, A. Mastracchio, J. Hong, K. Ashton and D. W. C. MacMillan, Science, 2007, 316, 582 CrossRef CAS PubMed; (b) S. Bertelsen, M. Nielsen and K. A. Jørgensen, Angew. Chem., Int. Ed., 2007, 46, 7356 CrossRef CAS PubMed; (c) M. P. Sibi and M. Hasegawa, J. Am. Chem. Soc., 2007, 129, 4124 CrossRef CAS PubMed; (d) D. A. Nicewicz and D. W. C. MacMillan, Science, 2008, 322, 77 CrossRef CAS PubMed; (e) H. Kim and D. W. C. MacMillan, J. Am. Chem. Soc., 2008, 130, 39 Search PubMed; (f) D. A. Nagib, M. E. Scott and D. W. C. MacMillan, J. Am. Chem. Soc., 2009, 131, 10875 CrossRef CAS PubMed; (g) J. F. Van Humbeck, S. P. Simonovich, R. R. Knowles and D. W. C. MacMillan, J. Am. Chem. Soc., 2010, 132, 10012 CrossRef CAS PubMed; (h) P. V. Pham, K. Ashton and D. W. C. MacMillan, Chem. Sci., 2011, 2, 1470 RSC; (i) P. Gentili and S. Pedetti, Chem. Commun., 2012, 48, 5358 RSC.
- K. A. Ahrendt, C. J. Borths and D. W. C. MacMillan, J. Am. Chem. Soc., 2000, 122, 4243 CrossRef CAS.
- For remote functionalisation under aminocatalytic conditions, see: (a) J.-L. Li, T.-Y. Liu and Y.-C. Chen, Acc. Chem. Res., 2012, 45, 1491 CrossRef CAS PubMed; (b) H. Jiang, L. Albrecht and K. A. Jørgensen, Chem. Sci., 2013, 4, 2287 RSC; (c) I. D. Jurberg, I. Chatterjee, R. Tannert and P. Melchiore, Chem. Commun., 2013, 49, 4869 RSC.
- (a) M. C. Whisler, S. MacNeil, V. Snieckus and P. Beak, Angew. Chem., Int. Ed., 2004, 43, 2206 CrossRef CAS PubMed; (b) K. Mikami, M. Shimizu. H.-C. Zhang and B. E. Maryanoff, Tetrahedron, 2001, 57, 2917 CrossRef CAS.
- C. Mannich and H. Davidsen, Ber. Dtsch. Chem. Ges., 1936, 69, 2106 CrossRef.
- (a) U. Eder, G. Sauer and R. Wiechert, Angew. Chem., Int. Ed., 1971, 10, 496 CrossRef CAS; (b) Z. G. Hajos and D. R. Parrish, J. Org. Chem., 1974, 39, 1615 CrossRef CAS.
- (a) A. G. Nigmatov and E. P. Serebryakov, Russ. Chem. Bull., 1993, 42, 213 CrossRef; (b) A. G. Nigmatov and E. P. Serebryakov, Russ. Chem. Bull., 1996, 45, 623 CrossRef; (c) E. P. Serebryakov, A. G. Nigmatov, M. A. Shcherbakov and M. I. Struchkova, Russ. Chem. Bull., 1998, 47, 82 CrossRef CAS.
- S. Bertelsen, M. Marigo, S. Brandes, P. Dinér and K. A. Jørgensen, J. Am. Chem. Soc., 2006, 128, 12973 CrossRef CAS PubMed.
- (a) R. Thayumanavan, D. B. Ramachary, K. Sakthivel, F. Tanaka and C. F. Barbas III, Tetrahedron Lett., 2002, 43, 3817 CrossRef CAS; (b) D. B. Ramachary, N. S. Chowdari and C. F. Barbas III, Tetrahedron Lett., 2002, 43, 6743 CrossRef CAS; (c) D. B. Ramachary, N. S. Chowdari and C. F. Barbas III, Synlett, 2003, 1910 CAS; (d) D. B. Ramachary, K. Anebouselvy, N. S. Chowdari and C. F. Barbas III, J. Org. Chem., 2004, 69, 5838 CrossRef CAS PubMed; (e) D. B. Ramachary, N. S. Chowdari and C. F. Barbas III, Angew. Chem., Int. Ed., 2003, 42, 4233 CrossRef CAS PubMed.
- For dienamine activation of unsaturated ketones, see: (a) D. B. Ramachary and Y. V. Reddy, Eur. J. Org. Chem., 2012, 865 CrossRef CAS; (b) H. B. Hepburn, L. Dell'Amico and P. Melchiorre, Chem. Rec., 2016, 16, 1787 CrossRef CAS PubMed . See also ref. 2g and 2h.
- (a) K. Patora-Komisarska, M. Benohoud, H. Ishikawa, D. Seebach and Y. Hayashi, Helv. Chim. Acta, 2011, 94, 719 CrossRef CAS , and references therein; (b) J. Burs, A. Armstrong and D. G. Blackmond, J. Am. Chem. Soc., 2011, 133, 8822 CrossRef PubMed; (c) J. Burs, A. Armstrong and D. G. Blackmond, J. Am. Chem. Soc., 2012, 134, 6741 CrossRef PubMed.
- S.-H. Chen, B.-C. Hong, C.-F. Su and S. Sarshar, Tetrahedron Lett., 2005, 46, 8899 CrossRef CAS.
- N. Utsumi, H. Zhang, F. Tanaka and C. F. Barbas III, Angew. Chem., Int. Ed., 2007, 46, 1878 CrossRef CAS PubMed.
- J. Vesely, P. Dziedzic and A. Córdova, Tetrahedron Lett., 2007, 48, 6900 CrossRef CAS.
- E. Marqués-López, R. P. Herrera, T. Marks, W. C. Jacobs, D. Könning, R. M. de Figueiredo and M. Christmann, Org. Lett., 2009, 11, 4116 CrossRef PubMed.
- B. Han, Y.-C. Xiao, Z.-Q. He and Y.-C. Chen, Org. Lett., 2009, 11, 4660 CrossRef CAS PubMed.
- D. Enders, X. Yang, C. Wang, G. Raabe and J. Runsik, Chem. – Asian J., 2011, 6, 2255 CrossRef CAS PubMed.
- A. R. Haight, A. E. Bailey, W. S. Baker, M. H. Cain, R. R. Copp, J. A. DeMattei, K. L. Ford, R. F. Henry, M. C. Hsu, R. F. Keyes, S. A. King, M. A. McLaughlin, L. M. Melcher, W. R. Nadler, P. A. Oliver, S. I. Parekh, H. H. Patel, L. S. Seif, M. A. Staeger, G. S. Wayne, S. J. Wittenberger and W. Zhang, Org. Process Res. Dev., 2004, 8, 897 CrossRef CAS.
- B. C. Hong, P.-Y. Chen, P. Kotame, P.-Y. Lu, G.-H. Lee and J.-H. Liao, Chem. Commun., 2012, 48, 7790 RSC.
- B. Han, Y.-C. Xiao, Y. Yao and Y.-C. Chen, Angew. Chem., Int. Ed., 2010, 49, 10189 CrossRef CAS PubMed.
- J. Stiller, E. Marqués-López, R. P. Herrera, R. Fröhlich, C. Strohmann and M. Christmann, Org. Lett., 2011, 13, 70 CrossRef CAS PubMed.
- (a) G. Bergonzini, S. Vera and P. Melchiorre, Angew. Chem., Int. Ed., 2010, 49, 9685 CrossRef CAS PubMed; (b) M. Silvi, C. Cassani, A. Moran and P. Melchiorre, Helv. Chim. Acta, 2012, 95, 1985 CrossRef CAS.
- B. Han, Z.-Q. He, J.-L. Li, R. Li, K. Jiang, T.-Y. Liu and Y.-C. Chen, Angew. Chem., Int. Ed., 2009, 48, 5474 CrossRef CAS PubMed.
- C. Appayee, A. J. Fraboni and S. E. Brenner-Moyer, J. Org. Chem., 2012, 77, 8828 CrossRef CAS PubMed.
- K. Liu, A. Chougnet and W.-D. Woggon, Angew. Chem., Int. Ed., 2008, 47, 5287 CrossRef PubMed.
- C. Cassani and P. Melchiorre, Org. Lett., 2012, 14, 5590 CrossRef CAS PubMed.
- A. J. Fraboni and S. E. Brenner-Moyer, Org. Lett., 2016, 18, 2146 CrossRef CAS PubMed.
- M. Ikeda, Y. Miyake and Y. Nishibayashi, Organometallics, 2012, 31, 3810 CrossRef CAS.
- X.-H. Ho, W.-J. Jung, P. K. Shyam and H.-Y. Jang, Catal. Sci. Technol., 2014, 4, 1914 CAS.
- L. Næsborg, K. S. Halskov, F. Tur, S. M. N. Mønsted and K. A. Jørgensen, Angew. Chem., Int. Ed., 2015, 54, 10193 CrossRef PubMed.
- M. Frías, J. M. Padrón and J. Alemán, Chem. – Eur. J., 2015, 21, 8237 CrossRef PubMed.
- W. Xiao, X. Yin, Z. Zhou, W. Du and Y.-C. Chen, Org. Lett., 2016, 18, 116 CrossRef CAS PubMed.
- (a) B. J. Bench, C. Liu, C. R. Evett and C. M. H. Watanabe, J. Org. Chem., 2006, 71, 9458 CrossRef CAS PubMed; (b) B.-C. Hong, H.-C. Tseng and S.-H. Chen, Tetrahedron, 2007, 63, 2840 CrossRef CAS.
- B.-C. Hong, M.-F. Wu, H.-C. Tseng and J.-H. Liao, Org. Lett., 2006, 8, 2217 CrossRef CAS PubMed.
- R. M. de Figueiredo, R. Fröhlich and M. Christmann, Angew. Chem., Int. Ed., 2008, 47, 1450 CrossRef CAS PubMed.
- F. J. S. Duarte and A. Gil Santos, J. Org. Chem., 2012, 77, 3252 CrossRef CAS PubMed.
- A. Orue, E. Reyes, J. L. Vicario, L. Carrillo and U. Uria, Org. Lett., 2012, 14, 3740 CrossRef CAS PubMed.
- Z.-Y. Wang, W.-T. Wong and D. Yang, Org. Lett., 2013, 15, 4980 CrossRef CAS PubMed.
- L. K. Ransborg, M. Overgaard, J. Hejmanowska, S. Barfüsser, K. A. Jørgensen and Ł. Albrecht, Org. Lett., 2014, 16, 4182 CrossRef CAS PubMed.
- K. S. Halskov, B. S. Donslund, S. Barfüsser and K. A. Jørgensen, Angew. Chem., Int. Ed., 2014, 53, 4137 CrossRef CAS PubMed.
- A. Song, X. Zhang, X. Song, X. Chen, C. Yu, H. Huang, H. Li and W. Wang, Angew. Chem., Int. Ed., 2014, 53, 4940 CrossRef CAS PubMed.
- C. Martín-Santos, C. Jarava-Barrera, S. del Pozo, A. Parra, S. Díaz-Tendero, R. Mas-Ballesté, S. Cabrera and J. Alemán, Angew. Chem., Int. Ed., 2014, 53, 8184 CrossRef PubMed.
- T. K. Johansen, C. Villegas Gómez, J. R. Bak, R. L. Davis and K. A. Jørgensen, Chem. – Eur. J., 2013, 19, 16518 CrossRef CAS PubMed.
- Ł. Albrecht, G. Dickmeiss, F. Cruz Acosta, C. Rodríguez-Escrich, R. L. Davis and K. A. Jørgensen, J. Am. Chem. Soc., 2012, 134, 2543 CrossRef PubMed.
- G. Talavera, E. Reyes, J. L. Vicario and L. Carrillo, Angew. Chem., Int. Ed., 2012, 51, 4104 CrossRef CAS PubMed.
- L.-W. Qi, Y. Yang, Y.-Y. Gui, Y. Zhang, F. Chen, F. Tian, L. Peng and L.-X. Wang, Org. Lett., 2014, 16, 6436 CrossRef CAS PubMed.
- K. S. Halskov, F. Kniep, V. H. Lauridsen, E. H. Iversen, B. S. Donslund and K. A. Jørgensen, J. Am. Chem. Soc., 2015, 137, 1685 CrossRef CAS PubMed.
- J.-L. Li, T.-R. Kang, S.-L. Zhou, R. Li, L. Wu and Y.-C. Chen, Angew. Chem., Int. Ed., 2010, 49, 6418 CrossRef CAS PubMed.
- Ł. Albrecht, G. Dickmeiss, C. F. Weise, C. Rodríguez-Escrich and K. A. Jørgensen, Angew. Chem., Int. Ed., 2012, 51, 13109 CrossRef PubMed.
- C. F. Weise, V. H. Lauridsen, R. S. Rambo, E. H. Iversen, M.-L. Olsen and K. A. Jørgensen, J. Org. Chem., 2014, 79, 3537 CrossRef CAS PubMed.
- J. L. Li, S.-L. Zhou, P.-Q. Chen, L. Dong, T.-Y. Liu and Y.-C. Chen, Chem. Sci., 2012, 3, 1879 RSC.
- B. S. Donslund, A. Monleón, J. Larsen, L. Ibsen and K. A. Jørgensen, Chem. Commun., 2015, 51, 13666 RSC.
- P. Meindl, G. Bodo, P. Palese, J. Schulman and H. Tuppy, Virology, 1974, 58, 457 CrossRef CAS PubMed.
- T.-S. Jeong, S.-U. Kim, K.-H. Son, B.-M. Kwon, Y.-K. Kim, M.-U. Choi and S.-H. Bok, J. Antibiot., 1995, 48, 751 CrossRef CAS PubMed.
- N. Öztuörk, S. Korkmaz, Y. Öztuörk and K. H. C. Baser, Planta Med., 2005, 72, 289 Search PubMed.
- W. Li, J. Wei, Q. Jia, Z. Du, K. Zhang and J. Wang, Chem. – Eur. J., 2014, 20, 6592 CrossRef CAS PubMed.
- (a) C. Izquierdo, F. Esteban, A. Parra, R. Alfaro, J. Alemán, A. Fraile and J. L. García Ruano, J. Org. Chem., 2014, 79, 10417 CrossRef CAS PubMed; (b) J. Alemán and A. Fraile, Synlett, 2015, 1940 CrossRef.
- A. Orue, U. Uria, E. Reyes, L. Carrillo and J. L. Vicario, Angew. Chem., Int. Ed., 2015, 54, 3043 CrossRef CAS PubMed.
- J. Stiller, P. H. Poulsen, D. Cruz Cruz, J. Dourado, R. L. Davis and K. A. Jørgensen, Chem. Sci., 2014, 5, 2052 RSC.
- K. Halskov, T. Naicker, M. E. Jensen and K. A. Jørgensen, Chem. Commun., 2013, 49, 6382 RSC.
- R. C. da Silva, I. Chatterjee, E. Escudero-Adán, M. Weber Paixao and P. Melchiorre, Asian J. Org. Chem., 2014, 3, 466 CrossRef.
- I. Chatterjee, D. Bastida and P. Melchiorre, Adv. Synth. Catal., 2013, 355, 3124 CrossRef CAS.
- M. Silvi, E. Arceo, I. D. Jurberg, C. Cassani and P. Melchiorre, J. Am. Chem. Soc., 2015, 137, 6120 CrossRef CAS PubMed.
- A. Seegerer, J. Hioe, M. M. Hammer, F. Morana, P. J. W. Fuchs and R. M. Gschwind, J. Am. Chem. Soc., 2016, 138, 9864 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |