
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Investigation of structure-directing interactions within copper(I) thiocyanate complexes through X-ray analyses and non-covalent interaction (NCI) theoretical approach†
Khodayar
Gholivand
 *a,
Kaveh
Farshadfar
a,
S. Mark
Roe
b,
Mahdieh
Hosseini
a and
Akram
Gholami
a
*a,
Kaveh
Farshadfar
a,
S. Mark
Roe
b,
Mahdieh
Hosseini
a and
Akram
Gholami
a
aDepartment of Chemistry, Faculty of Science, Tarbiat Modares University, Tehran, Iran. E-mail: gholi_kh@modares.ac.ir
bDepartment of Chemistry, School of Life Sciences, University of Sussex, Brighton, BN1 9QJ, UK
First published on 10th August 2016
Abstract
Herein, we reported the synthesis of copper(I) thiocyanate complexes with ortho-pyridinyl carbohydrazones containing a thiophene (L1) or a furyl ring (L2) as a mixture of two different crystals for each compound, linkage isomers of C1N, [Cu(NCS)(L1)PPh3] and C1S, [Cu(SCN)(L1)PPh3], for L1, whereas monomeric and polymeric structures C2N, [Cu(NCS)(L2)PPh3], and C2P, [–(NCS)Cu(L2)–]n, for L2. Crystallographic information and theoretical calculations, mainly noncovalent interaction reduced density gradient (NCI-RDG) analyses, were pursued to generate a profound understanding of the structure-directing interactions in these complexes. The supramolecular assemblies are first driven by cooperative π⋯π interactions and hydrogen bonds followed by CH⋯π, S⋯S and S⋯π linkages. In the case of the linkage isomers, intermolecular interactions may have a significant role in the formation of the less stable S-bound isomer C1S.
Introduction
Copper(I) compounds have attracted a growing interest because of their high structural diversity,1–4 catalytic activity5–7 and photophysical properties.3,8–10 They have applications in different areas such as organic light-emitting diodes (OLEDs),11–22 supramolecular assemblies, oxygen sensors and biological probes. The rich structural features and utilitarian considerations have motivated researchers to focus on the synthesis and characterization of Cu(I) complexes with various donor ligands. The formation of structural variations is greatly influenced by several parameters such as the synthetic conditions or steric/electronic effects exerted by the ligand.The triatomic pseudohalide, thiocyanate anion (SCN−), is an excellent versatile ambidentate ligand with two donor atoms, S or N.23 It can coordinate to metal ions both in terminal and bridging coordination modes and potentially provide fascinating examples of linkage isomerism.24–26 When SCN− acts as a terminal ligand, it affords potential interaction sites to generate non-covalent intermolecular interactions and, accordingly, can direct the crystal packing. Controlling the self-assemblies in the solid state on the basis of molecular structures and through the use of weak interactions is a long-standing goal of supramolecular chemistry.27
Very recently in our previous work, cuprous halide complexes of ortho-, meta- and para-pyridinyl carbohydrazones were introduced.28 The influence of ligand structure and halide variations on the molecular structures and supramolecular arrays of the complexes were studied both experimentally and theoretically. In the following, we employed cuprous pseudohalide, CuSCN, for the synthesis of complexes with two ortho-pyridinyl carbohydrazones. Copper(I) thiocyanate compounds are very interesting in solar cell applications as a p-type semiconductor.29–31
In this contribution, we report the structural characteristics of complexes from the reaction of CuSCN with PPh3 and ortho-pyridinyl carbohydrazones containing a thiophene (L1) or a furyl ring (L2); see Scheme 1. The former resulted in two linkage isomers: C1N [Cu(NCS)(L1)PPh3] and C1S [Cu(SCN)(L1)PPh3], while the later afforded monomeric and polymeric complexes of C2N [Cu(NCS)(L2)PPh3] and C2P [–(NCS)Cu(L2)–]n. We have also used a recently introduced alternative interpretive technique, the non-covalent interaction (NCI) approach, to manifest the diverse NCIs at the crystal packing structures. This method is based on the analysis of the electron density and enables us to identify and visualize the interactions.32 Various non-covalent interactions, including hydrogen bonding,33 S⋯S, S⋯π,34 π⋯π35 and CH⋯π36 interactions, have been investigated in this work.
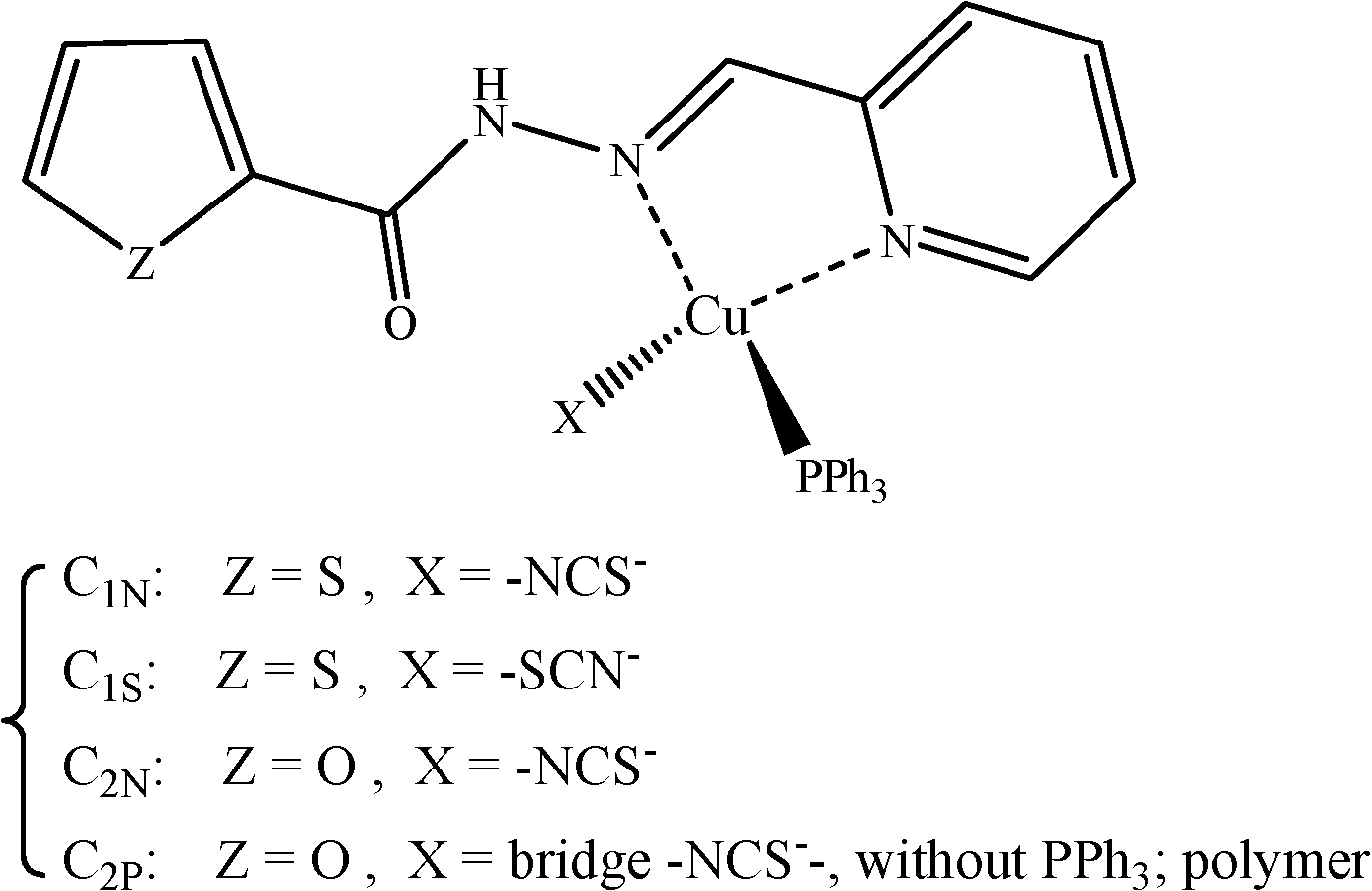 | ||
| Scheme 1 | ||
Results and discussion
Synthesis
Ligands L1 and L2 were prepared by mixing equivalent amounts of 2-thiophenecarboxylic acid hydrazide (1) or furoic hydrazide (2) and 2-pyridinecarboxaldehyde in methanol solution.A solution of the ligand in CHCl3 was added dropwise to a mixture of copper(I) thiocyanate and PPh3 while stirring in CH3CN and then the mixture was filtered off. After slow diffusion of diethyl ether in the filtered solutions, two different crystals were obtained for each compound including the light orange needle crystals (C1N) and clear light red irregular crystals (C1S) for L1 and orange needle crystals (C2N) and dark orange hexagonal crystals (C2P) for L2. A mixture of the isomeric crystals C1N and C1S is shown in Fig. 1.
 | ||
| Fig. 1 50× magnification photo of a mixture of orange rectangular cube C1N and red cube C1S. | ||
Various ratios of acetonitrile and chloroform solvents were assessed in the crystallization of C1. Upon using more chloroform, the percentage of C1N was dominant, whereas a higher amount of acetonitrile in the reaction pot increased the percentage of C1S. For C2, the formation of crystals depended on the concentration of the reaction mixture. At a high concentration, the polymeric compound precipitated fast and we only obtained crystals of C2N, but slower diffusion of diethyl ether in the more dilute solution afforded crystals of both C2P (as the dominant product) and C2N suitable for X-ray diffraction.
ORTEP diagrams of the molecular structures are shown in Fig. 2. The crystallographic data of the complexes are listed in Table 1. Selected bond distances and angles are summarized in Table 2.
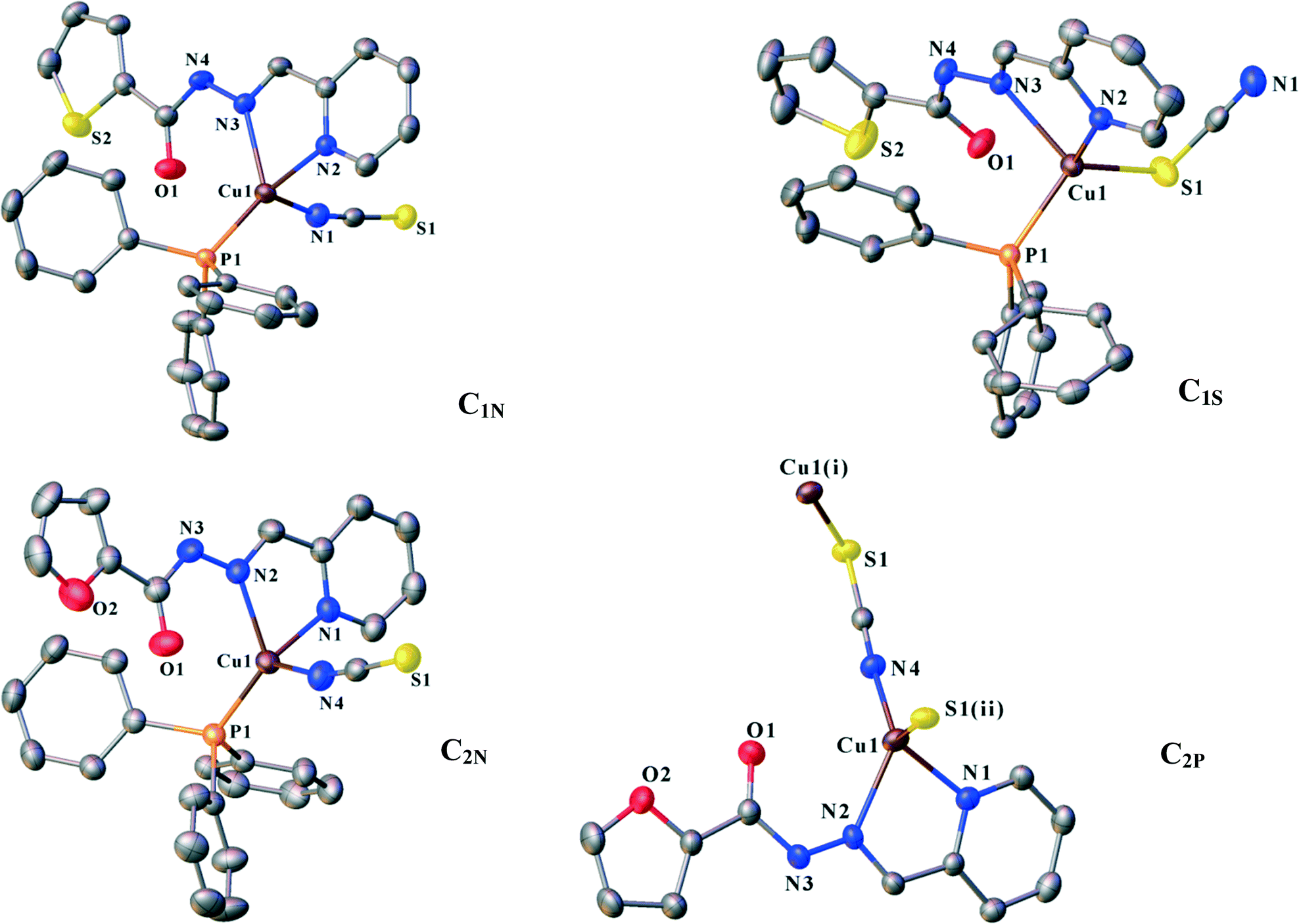 | ||
| Fig. 2 ORTEP diagrams of C1N, C1S, C2N and C2P with displacement ellipsoid at the 50% level. Hydrogen atoms were omitted for clarity. Symmetry codes: (i) x, 1/2 − y, 1/2 + z; (ii) x, 1/2 − y, −1/2 + z. | ||
| Compound | C1N | C1S | C2N | C2P |
|---|---|---|---|---|
| Formula | C30H24CuN4OPS2 | C30H24CuN4OPS2 | C30H24CuN4O2PS | C12H9CuN4O2S |
| Fw | 615.16 | 615.16 | 599.10 | 336.83 |
| λ/Å | 1.54184 | 0.71073 | 1.54184 | 1.54184 |
| T/K | 173 | 173 | 173 | 173 |
| Crystal system | Triclinic | Triclinic | Triclinic | Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P |
P21/c |
| a/Å | 9.8334(5) | 10.7768(9) | 9.8386(7) | 15.7005(7) |
| b/Å | 10.2673(6) | 11.0362(8) | 10.1356(6) | 8.0099(4) |
| c/Å | 15.0986(9) | 12.2584(5) | 14.9963(10) | 10.3633(5) |
| α/° | 87.742(5) | 85.195(5) | 90.004(5) | 90 |
| β/° | 89.510(4) | 78.024(5) | 90.051(6) | 94.374(4) |
| γ/° | 66.545(5) | 80.116(6) | 113.105(7) | 90 |
| V/Å3 | 1397.31(15) | 1403.30(17) | 1375.48(17) | 1299.49(11) |
| D calc/Mg m−3 | 1.462 | 1.456 | 1.447 | 1.722 |
| Z | 2 | 2 | 2 | 4 |
| μ/mm−1 | 3.292 | 1.015 | 2.666 | 3.948 |
| F(000) | 632 | 632 | 616 | 680 |
| 2θ/° | 142.128 | 59 | 143 | 142 |
| R(int) | 0.025 | 0.030 | 0.030 | 0.041 |
| GOOF | 1.022 | 1.11 | 1.04 | 1.03 |
| R 1 (I > 2σ(I)) | 0.0295 | 0.0444 | 0.0373 | 0.0356 |
| wR2 (I > 2σ(I)) | 0.0778 | 0.1072 | 0.1078 | 0.0955 |
| C1N | |||
| Cu1–P1 | 2.1880(6) Å | Cu1–N2 | 2.1367(16) Å |
| Cu1–N1 | 1.979(18) Å | Cu1–N3 | 2.1501(16) Å |
| P1–Cu1–N1 | 119.83(5)° | N1–Cu1–N2 | 99.81(7) |
| P1–Cu1–N2 | 121.68(5)° | N1–Cu1–N3 | 101.96(7) |
| P1–Cu1–N3 | 126.46(4)° | N2–Cu1–N3 | 77.56(6) |
| C1S | |||
| Cu1–S1 | 2.2936(9) Å | Cu1–N2 | 2.050(2) Å |
| Cu1–P1 | 2.1990(7) Å | Cu1–N3 | 2.181(2) Å |
| S1–Cu1–P1 | 117.60(3)° | P1–Cu1–N2 | 117.49(6)° |
| S1–Cu1–N2 | 107.38(6)° | P1–Cu1–N2 | 103.74(6)° |
| S1–Cu1–N3 | 127.28(6)° | N2–Cu1–N3 | 77.65(8)° |
| C2N | |||
| Cu1–P1 | 2.1893(6) Å | Cu1–N2 | 2.1547(19) Å |
| Cu1–N1 | 2.1411(19) Å | Cu1–N4 | 1.983(2) Å |
| P1–Cu1–N1 | 121.44(6)° | N1–Cu1–N2 | 77.23(7)° |
| P1–Cu1–N2 | 126.25(5)° | N1–Cu1–N4 | 98.52(8)° |
| P1–Cu1–N4 | 121.53(7)° | N2–Cu1–N4 | 101.32(8)° |
| C2P | |||
| Cu1–N1 | 2.105(2) Å | Cu1–N4 | 1.906(2) Å |
| Cu1–N2 | 2.131(2) Å | Cu1–S1 | 2.2971(8) Å |
| N1–Cu1–N2 | 77.74(9)° | N2–Cu1–N4 | 130.03(9)° |
| N1–Cu1–N4 | 109.43(9)° | N2–Cu1–S1 | 103.43(7)° |
| N1–Cu1–S1 | 111.59(7)° | N4–Cu1–S1 | 117.54(7)° |
Structural analysis
. Ligand L1 binds to the copper atom in a bidentate chelating manner via N2 (pyridine) and N3 (imine). An isothiocyanate anion (N-donor) and one PPh3 occupy the other coordination sites (Fig. 1). Houser and co-workers suggested an angular index (τ4) which determines the geometry of the four-coordinate metal centres as follows: τ4 = [360 − (α + β)]/141] (α and β are the two largest angles around a four-coordinate metal centre). The values of τ4 will range from 1.00 for a perfect tetrahedral geometry to zero for a perfect square planar environment. Intermediate structures including trigonal pyramid and seesaws fall within the range of 0 to 1.00.37 According to the τ4 value for C1N (0.79), the coordination polyhedra of the copper centre can be described as trigonal pyramid.
In the structure of C1N, each molecular unit of the complex is joined to the neighbouring unit by means of three 2-fold interactions including classical and non-classical hydrogen bonds N4–H4⋯S1 and C7–H7⋯S1, respectively (Table 3), and πpy–πthiophene interactions (Table 4).
| Structure | D–H⋯A | d D–H | d H⋯A | d D⋯A | ∠ D–H⋯A | Symm. codes |
|---|---|---|---|---|---|---|
| C1N | N4–H4⋯S1 | 0.880 | 2.6800 | 3.5042(18) | 157.00 | 1 − x, 1 − y, 1 − z |
| C7–H7⋯S1 | 0.950 | 2.8700 | 3.6680(2) | 143.00 | 1 − x, 1 − y, 1 − z | |
| C3–H3⋯S1 | 0.930 | 2.8754 | 3.7910(2) | 162.00 | −x, 2 − y, 1 − z | |
| C21–H21⋯S2 | 0.930 | 3.0230 | 3.5480(2) | 116.00 | 1 − x, 1 − y, 2 − z | |
| C1S | C10–H10⋯N1 | 0.950 | 2.5580 | 3.4540(4) | 157.30 | 1 − x, 1 − y, 1 − z |
| C7–H7⋯N1 | 0.950 | 2.7990 | 3.5690(5) | 138.80 | 1 − x, 1 − y, 1 − z | |
| N4–H4⋯N1 | 0.880 | 2.2330 | 3.0760(3) | 163.16 | 1 − x, 1 − y, 1 − z | |
| C2N | N3–H3⋯S1 | 0.879 | 2.6810 | 3.4930(2) | 153.90 | 1 − x, 1 − y, 1 − z |
| C6–H6⋯S1 | 0.949 | 2.8469 | 3.6410(3) | 141.80 | 1 − x, 1 − y, 1 − z | |
| C10–H10⋯N4 | 0.950 | 2.7230 | 3.5320(3) | 143.60 | 1 + x, 1 + y, z | |
| C2–H2⋯S1 | 0.950 | 2.8812 | 3.8020(2) | 163.60 | 2 − x, 2 − y, 1 − z | |
| C15–H15⋯S1 | 0.950 | 2.9839 | 3.8270(3) | 148.60 | x, −1 + y, z | |
| C2P | C9–H9⋯O2 | 0.950 | 2.4970 | 3.3940(4) | 157.40 | x, 1/2 − y, −1/2 + z |
| N3–H3⋯O1 | 0.880 | 2.3140 | 3.1000(3) | 148.70 | x, 1/2 − y, −1/2 + z | |
| C2–H2⋯N4 | 0.950 | 2.6850 | 3.5830(4) | 157.90 | 2 − x, 1/2 + y, 1/2 − z | |
| C11–H11⋯N4 | 0.950 | 2.5310 | 3.3480(4) | 144.10 | 1 − x, −1/2 + y, 1/2 − z | |
| C11–H11⋯O1 | 0.950 | 2.6870 | 3.1830(4) | 113.10 | 1 − x, −1/2 + y,1/2 − z |
| Structure | Interaction | C–C (Å) | P–P° | P–CC° | CH⋯CgI | C⋯Cg (Å) | C–H–Cg (°) | Symm. code |
|---|---|---|---|---|---|---|---|---|
| Cg stands for the centre of gravity of the mentioned ring: for C1N: Cg2: S2, C9–C12; Cg3: N2, C2–C6; Cg4: C13–C18; Cg6: C25–C30; for C1S: Cg7: C25–C30; for C2N: Cg2: O2, C8–C11; Cg4: C13–C18; Cg6: C25–C30; for C2P: Cg2: O2, C8–C11; Cg3: N1, C1–C5. | ||||||||
| C1N | πpy–πthiophene | 3.749 | 7.52 | 19.63 | — | — | — | 1 − x, 1 − y, 1 − z |
| 27.14 | ||||||||
| πpy–πpy | 3.486 | 0.0 | 7.88 | — | — | — | −x, 1 − y, 1 − z | |
| CH⋯πPPh3 | — | — | — | C4–H4A⋯Cg6 | 3.725 | 165.63 | −x, 1 − y, 1 − z | |
| CH⋯πPPh3 | — | — | — | C5–H5⋯Cg4 | 2.689 | 145.87 | −x, 1 − y, 1 − z | |
| CH⋯πpy | — | — | — | C11–H11⋯Cg3 | 3.737 | 134.12 | 1 + x, −1 + y, z | |
| CH⋯πthiophene | — | — | — | C14–H14⋯Cg2 | 3.134 | 145.84 | −1 + x, 1 + y, z | |
| CH⋯πthiophene | — | — | — | C2–H2⋯Cg2 | 3.592 | 134.97 | −1 + x, 1 + y, z | |
| C1S | Amide⋯πpy | 3.403 | — | — | — | — | — | 1 − x, 1 − y, 1 − z |
| CH⋯πPPh3 | — | — | — | C11–H11⋯Cg7 | 2.749 | 141.19 | 1 + x, y, z | |
| CH⋯πPPh3 | — | — | — | C17–H17⋯Cg7 | 3.275 | 132.77 | 1 − x, −y, 2 − z | |
| C2N | πpy–πfuryl | 3.719 | 11.74 | 17.28 | — | — | — | 1 − x, 1 − y, 1 − z |
| 28.73 | ||||||||
| πpy–πpy | 3.461 | 0.0 | 5.15 | — | — | — | 2 − x, 1 − y, 1 − z | |
| CH⋯πfuryl | — | — | — | C1–H1⋯Cg2 | 3.602 | 135.83 | 1 + x, 1 + y, z | |
| CH⋯πPPh3 | — | — | — | C4–H4⋯Cg6 | 2.721 | 145.84 | 2 − x, 1 − y, 1 − z | |
| CH⋯πPPh3 | — | — | — | C3–H3A⋯Cg4 | 3.761 | 166.03 | 2 − x, 1 − y, z | |
| C2P | S⋯πpy | 3.882 | — | 19.60 | — | — | — | x, y, 1 + z |
| πpy–πpy | 3.801 | 0.0 | 28.19 | — | — | — | 2 − x, 1 − y, −z | |
| πfuryl–πfuryl | 3.583 | 0.0 | 17.13 | — | — | — | 1 − x, −y, −z | |
The dimers are further connected to each other through πpy–πpy and C5–H5⋯πPPh3 interactions along the a-direction (Fig. 3a) to afford chains which are laterally linked together via various intermolecular interactions to generate a 3D network. The interactions which link the chains along the b-axis include (i) S1⋯S1, (ii) C3H3⋯S1, (iii) C11–H11⋯πpy, (iv) C2–H2⋯πthiophene and (v) C14–H14⋯πthiophene linkages (Fig. 3b). In addition, C21H21⋯S2 H-bonds plus weak C27–H27⋯πPPh3 interactions (C⋯Cg: 4.090 Å) connect them along the c-direction (Fig. 3c). The distance of the S⋯S interaction was found to be about 3.456 Å which is 4% shorter than the sum of the van der Waals radii of two sulfur atoms. A summary of the parameters for the other interactions mentioned above are presented in Tables 3 and 4.
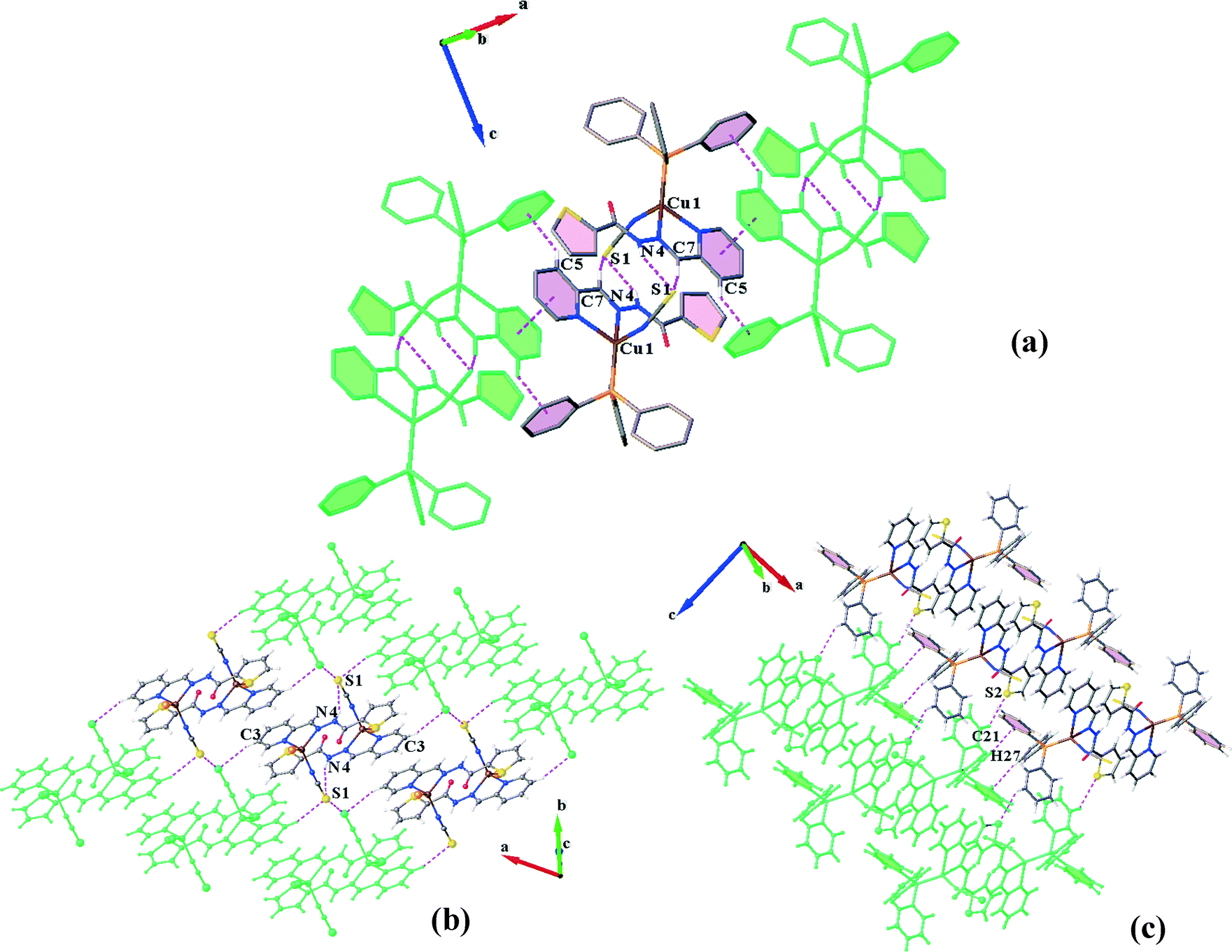 | ||
| Fig. 3 (a) Representation of dimeric units in C1N and their association through π–π and C–H⋯π interactions, creating the [100] chain; (b and c) side views of the crystal packing in the ac and ab planes, respectively, which show how the chains are connected in the 3D network. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of L1 and the mixture of cuprous thiocyanate and PPh3. X-ray diffraction analysis reveals that it crystallizes in the triclinic space group P. The central copper atoms are again in trigonal pyramidal environments (τ4 = 0.82, exactly equal to that for C1N) formed by Npy and Nim (from the chelating ligand), PPh3 moiety and thiocyanate anion (this time as an S-donor) (Fig. 1). Changing the coordination site of the ambidentate ligand, NCS−, alters the supramolecular architecture of C1S compared to that of the N-bound isomer C1N.
:1 molar ratio of L1 and the mixture of cuprous thiocyanate and PPh3. X-ray diffraction analysis reveals that it crystallizes in the triclinic space group P. The central copper atoms are again in trigonal pyramidal environments (τ4 = 0.82, exactly equal to that for C1N) formed by Npy and Nim (from the chelating ligand), PPh3 moiety and thiocyanate anion (this time as an S-donor) (Fig. 1). Changing the coordination site of the ambidentate ligand, NCS−, alters the supramolecular architecture of C1S compared to that of the N-bound isomer C1N.
The crystal structure of C1S contains hydrogen bonded dimers generated by three pairwise interactions, N4–H4⋯N1, C7–H7⋯N1 and C10–H10⋯N1 (Table 3 and Fig. 4a). It is worth noting that the coordination of sulfur to the Cu(I) atom and consequently the orientation of the N1 atom direct the formation of hydrogen bonds and lead to supernumerary slippage of molecules on each other. The offset of pyridine and thiophene rings prevents the formation of a πpy–πthiophene interaction, unlike in the structure C1N. Thus, instead of a πpy–πthiophene interaction, a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯π interaction is established between the discrete molecules in the dimers. In addition, S⋯S interactions connect the dimers to form [001] chains. The distance between two sulfur atoms is equal to 3.594 Å. These chains are held together by C11–C11⋯πPPh3 and C17–H17⋯πthiophene linkages (Table 4) along the a- and b-directions, respectively, which complete a 3D network (Fig. 4b–d).
O⋯π interaction is established between the discrete molecules in the dimers. In addition, S⋯S interactions connect the dimers to form [001] chains. The distance between two sulfur atoms is equal to 3.594 Å. These chains are held together by C11–C11⋯πPPh3 and C17–H17⋯πthiophene linkages (Table 4) along the a- and b-directions, respectively, which complete a 3D network (Fig. 4b–d).
 | ||
| Fig. 4 Self-assembly of C1S: (a) dimeric aggregates formed by pairwise H-bonds and CO⋯π linkages; (b) S⋯S interactions generating [001] chains; (c and d) C–H⋯π interactions which link the chains in the ac and ab planes, respectively. | ||
. A trigonal pyramidal configuration of the Cu(I) centres (τ4 = 0.80) has been formed by the bidentate chelating ligand, PPh3 and isothiocyanate (N-donor). C2N has a similar coordination environment to that of C1N leading to its being isostructural with this complex. The supramolecular organization of C2N is also disciplined by the formation of dimers through the same kind of interactions in C1N: N3–H3⋯S1, C6–H6⋯S1 and πpy–πfuryl stacking interactions. Pyridine rings participate in the other π–π interactions (πpy–πpy) which build up a chain of the dimers directed along the c-axis. The chains are further reinforced by C3–H3A⋯πPPh3 and C4–H4⋯πPPh3 contacts (see Table 4). On the other hand, S⋯S synthons accompanied by C2–H2⋯S1, C15–H15⋯S1 and C10–H10⋯N4 hydrogen bonds as well as C1–H1⋯πfuryl interactions link the chains to create (110) sheets. The third dimension of the supramolecular assembly results from the connection of the layers via C21–H21⋯πfuryl linkages. Crystal packing diagrams of C2N are presented in Fig. 5.
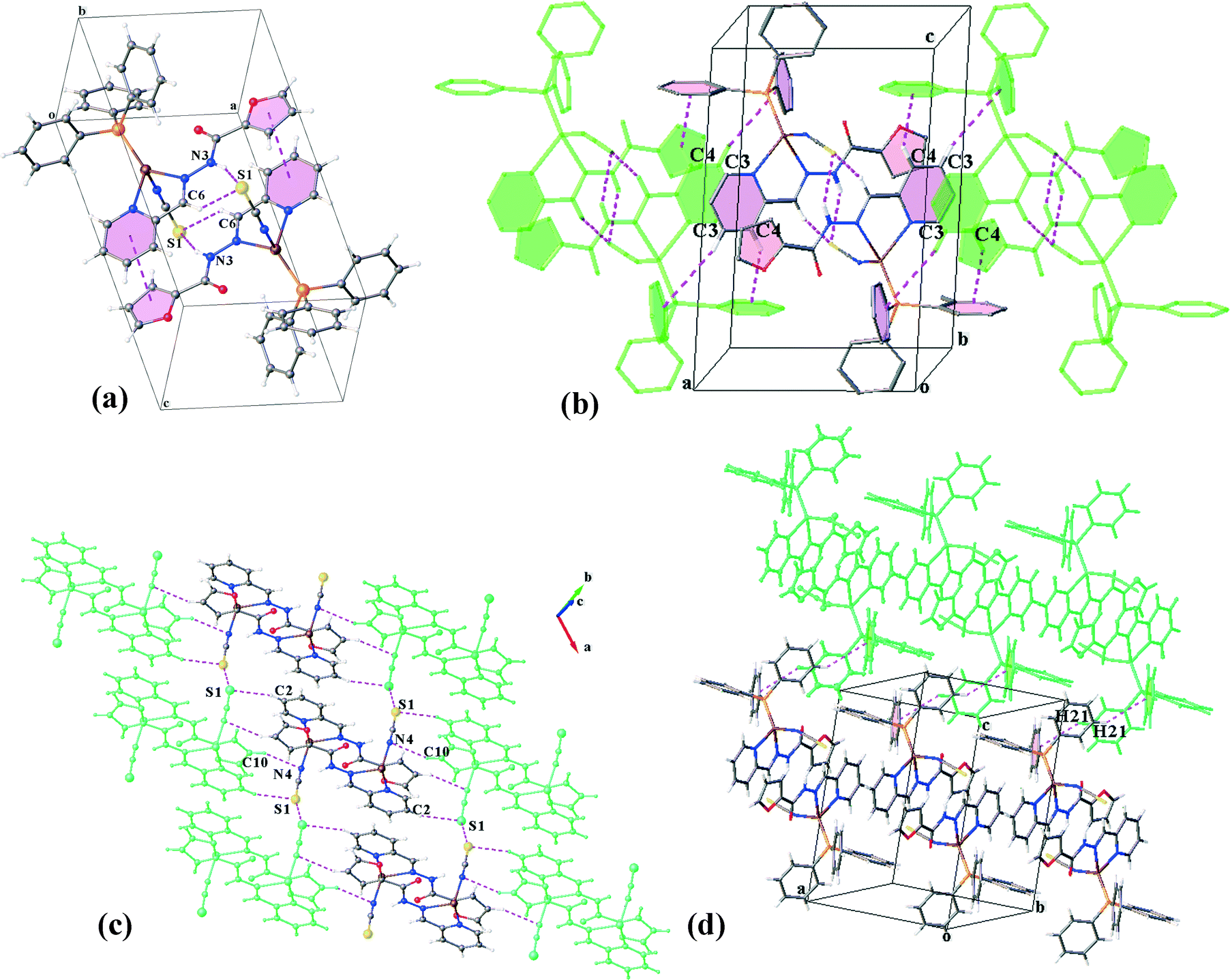 | ||
| Fig. 5 (a) Representation of dimeric units in C2N; (b) association of these units through π–π and C–H⋯π interactions, creating the [100] chain; (c and d) side views of the crystal packing in the ab and ac planes, respectively; which show how the chains are connected in the 3D construction (PPh3 moieties in (c) were omitted for more clarity). | ||
:1 molar ratio of L2 with the mixture of CuSCN and PPh3 results in two kinds of crystals: orange needle crystals, C2N, and dark orange hexagonal ones, C2P. X-ray diffraction analysis confirms that C2P crystallizes in the monoclinic space group P21/c. Unlike the former complexes, PPh3 does not coordinate to copper(I). In return, the trigonal pyramid geometry of the Cu(I) centre (τ4 = 0.80) consists of the chelating ligand (L2), thiocyanate (−SCN) and isothiocyanate (−NCS). Indeed, each thiocyanate anion acts as a bridge between the Cu(I) ions through simultaneous binding from S and N atoms. This coordination pattern leads to the formation of infinite polymeric chains extended along the c-axis (Fig. 6a). The Cu⋯Cu distance within the metal chain is 5.323 Å. The chains are further stabilized via intrachain hydrogen bonds (N3–H3⋯O1, C9–H9⋯O2 and C4–H4⋯S1).
 | ||
| Fig. 6 (a) Intrachain interactions within the coordination polymeric structure of C2P and the side view of the [001] chains to show (b) an overall supramolecular array containing two-fold layers connected to each other; (c) π–π stacking and H-bonding interactions linking a chain to four others and (d) S⋯πpy interactions which connect the chains laterally. | ||
Although the coordination structure of C2P is different from the others, π–π interactions still have an important contribution in the crystal packing. Herein, πfuryl–πfuryl interactions establish two-fold sheets of the neighbouring chains which are also fortified by bifurcated hydrogen bonds. The other side of the chains in the sheets is involved in πpy–πpy stacking and H-bonding interactions, leading to the connection of the (011) layers along the a-direction to complete the overall supramolecular association (Fig. 6b). In other words, each coordination chain is associated with four other chains in a 3D arrangement, from one side through πfuryl–πfuryl, C11–H11⋯N4 and C11–H11⋯O1 linkages and from the other side by πpy–πpy stacking and C2–H2⋯N4 interactions (Fig. 6c). In addition, these chains are laterally linked through interesting S⋯πpy interactions in the b-direction (Fig. 6d).
NCI approach
where ∇ρ is the gradient of ρ. The non-covalent interactions are located in the regions with low RDG and density. Analysis of sign(λ2) of the electron density Hessian can be used to discern different types of interactions. For the strong ones such as H-bonds, sign(λ2)ρ < 0; for the weak van der Waals types, sign(λ2)ρ ≈ 0; and for the non-bonded interactions like steric repulsion, sign(λ2)ρ > 0.
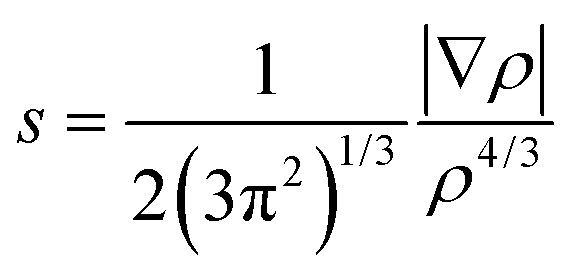
As the sign of λ2 describes the essence of the interaction, 2D plots comprising sign(λ2) × ρ versus RDG s would indicate a non-covalent interaction near-zero area in the horizontal axis.38–44 Close contacts between atoms change the behaviour of the reduced gradient signal more compared to the contacts among the atoms present in the tails, leading to troughs in the 2D NCI plots. These troughs, specially the ρ value at the troughs, are the basis of the NCI approach. The 2D NCI plots are then applied as inputs to construct 3D NCI plots, including isosurfaces of the reduced gradient of the density enabling the spatial visualization of the close contacts.
We applied this method to unravel the nature of supramolecular interactions in the title complexes. NCI analysis has been performed on the structure of complexes including the diverse noncovalent interactions. The considered structures were cut out directly from the CIF data. Since dimerization is the prominent feature of the crystal packing in the monomeric complexes (C1N, C1S and C2N), the main NCIs are related to the interactions involved in the formation of dimers. The 2D and 3D NCI plots of dimers are shown in Fig. 7. Accordingly, we have done calculation once on the dimers including only the carbohydrazone ligands (Cu+ and SCN− ions and PPh3 moieties have been eliminated) and again for the whole dimeric units of complexes. Some of the other interesting intermolecular interactions in the crystal structures have also been investigated by the NCI method. The presence of noncovalent interactions is characterized by spikes at negative to near-zero sign of λ2, whereas the peaks at positive sign indicate the repulsive steric contacts due to the ring formation.45 The spikes at the zero area (sign(λ2)ρ between ±0.015 a.u.) show vdW interactions. Notable points of the NCI calculations have been illustrated in the following:
 | ||
| Fig. 7 Left: The NCI RDG s vs. sign(λ2)ρ plots for dimers of ligands and complexes. Right: Coloured RDG-based NCI isosurfaces for the dimers of complexes. | ||
(i) As shown in Fig. 7, for the ligand dimers, the spikes that appeared at 0.024 a.u. belong to the pyridine ring closure. These spikes shift to lower values (less repulsion) in the whole dimeric units of complexes. It can be explained by the effect of metal ion in the charge redistribution as well as the electrostatic interaction between atoms within the rings.
(ii) In the case of C1N, the thiophene ring closure spike (sign(λ2)ρ) is located between 0.042 and 0.044 a.u. while in the 2D plot and the 3D isosurface of C2N, the furyl ring has a much lesser repulsion of ring closure than the thiophene alternative. It may be caused by the greater charge perturbation due to the presence of an oxygen atom which leads to more electrostatic interactions. Consistent with this, natural bond orbital (NBO) analysis also reveals the stabilizing energy of 54 kcal mol−1 for the electronic delocalization “lone pair (O) → π* (C–C) orbital” which is more than that for the corresponding charge transfer energy in the thiophene ring (LP(S) → π*(C–C): 48 kcal mol−1).
(iii) C1N and C1S compounds are linkage isomers, in a way that SCN− is coordinated from N or S atoms, respectively. Optimization of the isomers, in the gas phase and also acetonitrile and chloroform solutions, indicates that the stability of C1S is approximately 3–4 kcal mol−1 less than that of C1S; however, it has been also formed in the solid state. The formation of C1S can be attributed to stronger intermolecular attractions particularly those involved in dimerization which compensate for the lesser stability of the discrete units of C1S. RDG isosurfaces show stronger interactions in the C1S dimer rather than in C1N. Counterpoise calculations at the M06-2X/6-311G* level indicate that the binding energy of two complexes in a dimer, ΔEdimer, for C1S is 2.8 kcal mol−1 more than for C1N as well.
(iv) It was thought to be of interest to further investigate the sulfur interactions to figure out their nature in the solid state structures. The sulfur atom, due to its large van der Waals radius and high polarizability, is able to establish several interactions with its local environment.46 Morgan and co-workers first proposed the hypothesis that a strong interaction exists between aromatic rings and divalent sulfur atoms.47 The importance of the S⋯π aromatic interaction is revealed in the high degree of its conservation across members in protein folding and stabilization.34,48
Fig. 8 represents the 3D plots of S⋯S and S⋯π interactions in the solid state structures. The compact and small, flat, pill-shaped isosurfaces, concentrated on the NCI critical points indicate that these interactions are significantly attractive and contribute to the crystal packing stability.45
 | ||
| Fig. 8 RDG-based NCI surfaces for S⋯S and S⋯π interactions. | ||
Conclusions
Diverse coordination structures from the reaction of CuSCN with PPh3 and ortho-pyridinyl carbohydrazone containing a thiophene (L1) or furyl ring (L2) were presented. A mixture of thiocyanate linkage isomers, C1N and C1S, was obtained for L1, while the reaction with L2 rendered two monomeric and polymeric compounds, C2N and C2P, respectively. The molecular and supramolecular structures of these systems were elucidated using X-ray diffraction. The structure-directing interactions were also investigated by NCI-RDG calculations.Pyridine, thiophene and furyl rings, the polarized aromatic systems, have an important role in governing the supramolecular assembly of the complexes by establishing π–π interactions. However, the coordination of sulfur to the Cu(I) atom in C1S leads to supernumerary slippage of the neighbouring molecules which prevents the formation of πpy–πthiophene connections. CH–π interactions between PPh3 moieties contribute to further stabilization of the self-association in the monomeric complexes (C1N, C1S and C2N).
A prominent feature of the crystal packing in the monomeric complexes is the formation of the dimeric motifs via hydrogen bonding and π–π stacking interactions. Formation of the less stable S-bound isomer C1S can be attributed to stronger intermolecular attractions particularly those involved in dimerization which compensate for the lesser stability of the discrete units of C1S compared to C1N.
Another interesting feature of the solid state structures is the presence of the lesser known S⋯S and S⋯π interactions. NCI-RDG analysis clearly indicates the significant contribution of these interactions in maintaining favourable packing interactions in the complex.
Experimental
Materials and methods
All chemicals and solvents used in the syntheses were of reagent grade and were used without further purification. 1H and 13C NMR spectra were recorded on a Bruker (Avance DRS) 250 MHz spectrometer. IR spectra were recorded on a Nicolet 510P spectrophotometer using KBr disks. Elemental analysis was performed using a Heraeus CHN-O-RAPID apparatus.Synthesis procedures
The ligands were prepared from the reaction of two equivalent amounts of 2-thiophenecarboxylic acid hydrazide or 2-furoic hydrazide and 2-pyridinecarboxaldehyde in methanol solution.A solution of ligand (0.20 mmol) in CHCl3 (4 mL) was added dropwise to a stirred solution of a mixture of copper(I) thiocyanate (0.20 mmol) and triphenylphosphine (0.20 mmol) in CH3CN (2 ml). The colour of the reaction mixture turned from orange to red. The reaction mixture was filtered; slow diffusion of diethyl ether in the filtered solution afforded suitable single crystals (total yields for the mixture of C1N and C1S: 81%, and for that of C2N and C2P: 76%). The complexes were obtained in good yields. Physical and spectroscopic data of the compounds are presented below:
Crystal structure determination
Single crystal X-ray diffraction data were collected for all compounds on an Agilent Gemini Ultra diffractometer equipped with an Eos CCD area detector and using either Mo-Kα radiation (λ = 0.71073 Å) or Cu-Kα radiation (λ = 1.5418 Å). The data were collected at 173 K using an Oxford Cryosystems Cryostream 600. The data were processed with CrysAlisPro.49 Semi-empirical absorption corrections were carried out using the Multi-Scan50 program. The structures were solved by direct methods using SHELXT51 and refined with full matrix least squares refinement using SHELXL-2013 (ref. 52) within Olex2.53 All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were added at calculated positions and refined using a riding model based on the parent atom. The CIF files have been deposited with the CCDC and have been given the deposition numbers 1401344, 1401345, 1469718 and 1469717 for C1N, C1S, C2N and C2p, respectively.Computational details
The NCI technique was carried out through the analysis of the reduced density gradient (RDG) with low densities32 at the ωB97XD54/6-311+G** level using the Gaussian 09 package55 and Multiwfn program.56 The calculated grid points are plotted for a defined real space function, sign(λ2(r))ρ(r) and reduced density gradient (RDG) and a visualization of the gradient isosurface was depicted using the VMD 1.9.2 software.57 The colour of the isosurfaces was decided using the value of sign(λ2)ρ. Blue, green and red colour codes are commonly used to describe stabilizing H-bonding, van der Waals and steric interaction, respectively. Pictures are provided for an isosurface value of s = 0.5.Natural bond orbital (NBO) analysis58 was performed on the crystal structure of the complexes using the NBO 3.1 module in Gaussian 09 at the B3LYP/6-311+G** level of theory. The binding energy of two complexes in a dimer, ΔEdimer, for C1N, C1S and C2N were calculated at the M062X/6-311G* level based on the energy difference between the dimer and its units. The interaction energies have been corrected for the basis set superposition error (BSSE) using the counterpoise (CP) procedure.59
Acknowledgements
Financial support of this work by Tarbiat Modares University is gratefully acknowledged.Notes and references
- N. Aoyagi, Y. Shinha, A. Ikeda-Ohno, Y. Haga, K. Shimojo, N. R. Brooks, A. Izuoka, H. Naganawa, T. Kimura and K. Binnemans, Cryst. Growth Des., 2015, 15, 1422 CAS.
- E. T. Spielberg, E. Edengeiser, B. Mallick, M. Havenith and A.-V. Mudring, Chem. – Eur. J., 2014, 20, 5338 CrossRef CAS PubMed.
- C. S. Smith, C. W. Branham, B. J. Marquardt and K. R. Mann, J. Am. Chem. Soc., 2010, 132, 14079 CrossRef CAS PubMed.
- L. Li, P. S. Lopes, V. Rosa, C. A. Figueira, M. A. N. D. A. Lemos, M. T. Duarte, T. Avilés and P. T. Gomes, Dalton Trans., 2012, 41, 5144 RSC.
- P. J. Pérez and M. M. Díaz-Requejo, in Comprehensive Organometallic Chemistry III, ed. R. H. Crabtree and D. M. P. Mingos, Elsevier, New York, 2007, vol. 2, pp. 153–195 Search PubMed.
- S. E. Allen, R. R. Walvoord, R. Padilla-Salinas and M. C. Kozlowski, Chem. Rev., 2013, 113, 6234 CrossRef CAS PubMed.
- A. Ouali, J.-F. Spindler, H.-J. Cristau and M. Taillefer, Adv. Synth. Catal., 2006, 348, 499 CrossRef CAS.
- D. G. Cuttell, S.-M. Kuang, P. E. Fanwick, D. R. McMillin and R. A. Walton, J. Am. Chem. Soc., 2002, 124, 6 CrossRef CAS PubMed.
- T. McCormick, W.-L. Jia and S. Wang, Inorg. Chem., 2006, 45, 147 CrossRef CAS PubMed.
- R. Czerwieniec and H. Yersin, Inorg. Chem., 2015, 54, 4322 CrossRef CAS PubMed.
- L. Kang, J. Chen, T. Teng, X.-L. Chen, R. Yu and C.-Z. Lu, Dalton Trans., 2015, 44, 11649 RSC.
- D. Volz, M. Nieger, J. Friedrichs, T. Baumann and S. Bräse, Langmuir, 2013, 29, 3034 CrossRef CAS PubMed.
- G. F. Manbeck, W. W. Brennessel, C. M. Evans and R. Eisenberg, Inorg. Chem., 2010, 49, 2834 CrossRef CAS PubMed.
- J. C. Deaton, S. C. Switalski, D. Y. Kondakov, R. H. Young, T. D. Pawlik, D. J. Giesen, S. B. Harkins, A. J. M. Miller, S. F. Mickenberg and J. C. Peters, J. Am. Chem. Soc., 2010, 132, 9499 CrossRef CAS PubMed.
- Z. Liu, P. I. Djurovich, M. T. Whited and M. E. Thompson, Inorg. Chem., 2012, 51, 230 CrossRef CAS PubMed.
- M. J. Leitl, F.-R. Küchle, H. A. Mayer, L. Wesemann and H. Yersin, J. Phys. Chem. A, 2013, 117, 11823 CrossRef CAS PubMed.
- D. M. Zink, T. Baumann, J. Friedrichs, M. Nieger and S. Bräse, Inorg. Chem., 2013, 52, 13509 CrossRef CAS PubMed.
- D. Volz, T. Baumann, H. Flügge, M. Mydlak, T. Grab, M. Bächle, C. Barner-Kowollik and S. Bräse, J. Mater. Chem., 2012, 22, 20786 RSC.
- D. Volz, M. Wallesch, S. L. Grage, J. Göttlicher, R. Steininger, D. Batchelor, T. Vitova, A. S. Ulrich, C. Heske, L. Weinhardt, T. Baumann and S. Bräse, Inorg. Chem., 2014, 53, 7837 CrossRef CAS PubMed.
- L. Bergmann, J. Friedrichs, M. Mydlak, T. Baumann, M. Nieger and S. Bräse, Chem. Commun., 2013, 49, 6501 RSC.
- X. Liu, H. Nan, W. Sun, Q. Zhang, M. Zhan, L. Zou, Z. Xie, X. Li, C. Lu and Y. Cheng, Dalton Trans., 2012, 41, 10199 RSC.
- D. Volz, D. M. Zink, T. Bocksrocker, J. Friedrichs, M. Nieger, T. Baumann, U. Lemmer and S. Bräse, Chem. Mater., 2013, 25, 3414 CrossRef CAS.
- A. Hazari, L. Kanta Das, A. Bauzá, A. Frontera and A. Ghosh, Dalton Trans., 2014, 43, 8007 RSC.
- R. Loos, S. Kobayashi and H. Mayr, J. Am. Chem. Soc., 2003, 125, 14126 CrossRef CAS PubMed.
- L. Vandenburgh, M. R. Buck and D. A. Freedman, Inorg. Chem., 2008, 47, 9134 CrossRef CAS PubMed.
- M. J. Maroney, E. O. Fey, D. A. Baldwin, R. E. Stenkamp, L. H. Jensen and N. J. Rose, Inorg. Chem., 1986, 25, 1409 CrossRef CAS.
- A. D. Martin, J. Britton, T. L. Easun, A. J. Blake, W. Lewis and M. Schröder, Cryst. Growth Des., 2015, 15, 1697 CAS.
- K. Gholivand, K. Farshadfar, S. M. Roe, A. Gholami and M. D. Esrafili, CrystEngComm, 2016, 18, 2873 RSC.
- P. Qin, S. Tanaka, S. Ito, N. Tetreault, K. Manabe, H. Nishino, M. K. Nazeeruddin and M. Grätzel, Nat. Commun., 2014, 1 Search PubMed.
- T. P. Brewster, W. Ding, N. D. Schley, N. Hazari, V. S. Batista and R. H. Crabtree, Inorg. Chem., 2011, 50, 11938 CrossRef CAS PubMed.
- K. M. Miller, S. M. McCullough, E. A. Lepekhina, I. J. Thibau, R. D. Pike, X. Li, J. P. Killarney and H. H. Patterson, Inorg. Chem., 2011, 50, 7239 CrossRef CAS PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sanchez, J. Contreras-Garcia, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498 CrossRef CAS PubMed.
- (a) L. Wang, Y. Hu, W. Wang, F. Liu and K. Huang, CrystEngComm, 2014, 16, 4142 RSC; (b) K. I. Nättinen and K. Rissanen, Cryst. Growth Des., 2003, 3, 339 CrossRef; (c) B. Piotrkowska, A. Wasilewska, M. Gdaniec and T. Polonski, CrystEngComm, 2008, 10, 1421 RSC; (d) E. Bosch, N. P. Bowling and J. Darko, Cryst. Growth Des., 2015, 15, 1634 CrossRef CAS; (e) C. B. Aakeröy, D. J. Salmon, M. M. Smith and J. Desper, Cryst. Growth Des., 2006, 6, 1033 CrossRef; (f) A. Lemmerer, D. A. Adsmond, C. Esterhuysen and J. Bernstein, Cryst. Growth Des., 2013, 13, 3935 CrossRef CAS.
- B. R. Beno, K.-S. Yeung, M. D. Bartberger, L. D. Pennington and N. A. Meanwell, J. Med. Chem., 2015, 58, 4383 CrossRef CAS PubMed.
- (a) H. Akpinar, J. T. Mague and P. M. Lahti, CrystEngComm, 2013, 15, 831 RSC; (b) M. M. Naseer and S. Hameed, CrystEngComm, 2012, 14, 4247 RSC; (c) B. Sarma, L. S. Reddy and A. Nangia, Cryst. Growth Des., 2008, 8, 4546 CrossRef CAS; (d) C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885 RSC.
- (a) M. Nishio, Y. Umezawa, K. Honda, S. Tsuboyama and H. Suezawa, CrystEngComm, 2009, 11, 1757 RSC; (b) M. Nishio, M. Hirota and Y. Umezawa, The CH/π Interaction. Evidence, Nature, and Consequences, Wiley-VCH, New York, 1998 Search PubMed; (c) M. Nishio, CrystEngComm, 2004, 6, 130 RSC; (d) M. Umezawa, S. Tsuboyama, K. Honda, J. Uzawa and M. Nishio, Bull. Chem. Soc. Jpn., 1998, 71, 1207 CrossRef.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955 RSC.
- S. Pan, S. Mandal and P. K. Chattaraj, J. Phys. Chem. B, 2015, 119, 10962 CrossRef CAS PubMed.
- P. R. Varadwaj, A. Varadwaj and B.-Y. Jin, Phys. Chem. Chem. Phys., 2014, 16, 17238 RSC.
- P. R. Varadwaj, A. Varadwaj and B.-Y. Jin, Phys. Chem. Chem. Phys., 2014, 16, 19573 RSC.
- K. Saadat and H. Tavakol, RSC Adv., 2015, 5, 55227 RSC.
- R. P. Matthews, T. Welton and P. A. Hunt, Phys. Chem. Chem. Phys., 2015, 17, 14437 RSC.
- Y. Chu, X. Sun, Y. Xianfeng, L. Ding, A. Zheng and F. Deng, Catal. Sci. Technol., 2015, 5, 3507 CAS.
- P. R. Varadwaj, A. Varadwaj and B.-Y. Jin, Phys. Chem. Chem. Phys., 2015, 17, 31624 RSC.
- R. Chaudret, B. de Courcy, J. Contreras-Garcia, E. Gloaguen, A. Zehnacker-Rentien, M. Mons and J.-P. Piquemal, Phys. Chem. Chem. Phys., 2014, 16, 9876 RSC.
- G. Saleh, C. Gatti, L. L. Presti and J. Contreras-Garcia, Chem. – Eur. J., 2012, 18, 15523 CrossRef CAS PubMed.
- R. S. Morgan, C. E. Tatsch, R. H. Gushard, J. M. McAdon and P. K. Warme, Int. J. Pept. Protein Res., 1978, 11, 209 CrossRef CAS PubMed.
- R. Bhattacharyya, D. Pal and P. Chakrabarti, Protein Eng., Des. Sel., 2004, 17, 795 CrossRef CAS PubMed.
- CrysAlisPro, Agilent Technologies, Yarnton, England, 2014 Search PubMed.
- R. H. Blessing, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 51, 33 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- J. D. Chai and M. H. Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Cari-cato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- T. Lu and F. J. Chen, Comput. Chem., 2012, 33, 580 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33 CrossRef CAS PubMed.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899 CrossRef CAS.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: IR and NMR spectra of all compounds and crystallographic files in CIF format for structural determination of complexes. CCDC 1401344, 1401345, 1469718 and 1469717. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ce01339b |
| This journal is © The Royal Society of Chemistry 2016 |