DOI:
10.1039/C5CE01893E
(Paper)
CrystEngComm, 2016,
18, 1156-1163
Hydrogen bonding-induced conformational change in a crystalline sugar derivative†
Received
24th September 2015
, Accepted 7th January 2016
First published on 7th January 2016
Abstract
We report crystallographic evidence of the change of a regular chair conformation to a skew boat conformation in a partially protected sugar derivative. Crystallographic correlation with two more analogous derivatives indicates that a particular O–H⋯O hydrogen bonding network is responsible for the change in conformation.
Hydrogen bonding plays a pivotal role in regulating various biological pathways significant to life processes, e.g. it is responsible for DNA base pairing1 and several protein–protein2 or protein–ligand3 interactions. Apart from recognition in the biological domain, it has a direct role in regulating the physicochemical properties of non-living matter like polymers.4 Hydrogen bonding networks (HBNs) greatly influence the supramolecular architecture5 created by a molecule and therefore, they have an immense impact on the crystal packing and the properties of the material.6
Sugars are interesting molecules in supramolecular chemistry7 on account of the fact that their OH groups can offer both hydrogen-bond donor and acceptor sites. Moreover, it is relevant to have an insight into the HBNs in the supramolecular architecture of sugars as they are involved in vital biological processes through sugar–sugar or sugar–protein8 interactions. It is challenging to develop an understanding of the HBNs and their individual impact on the ultimate crystal packing when multiple supramolecular functionalities are present.9 This can be preferably carried out by either systematically changing the position of the functional groups (positional isomers) of interest in the desired compound or a systematic replacement of the functional group with others.
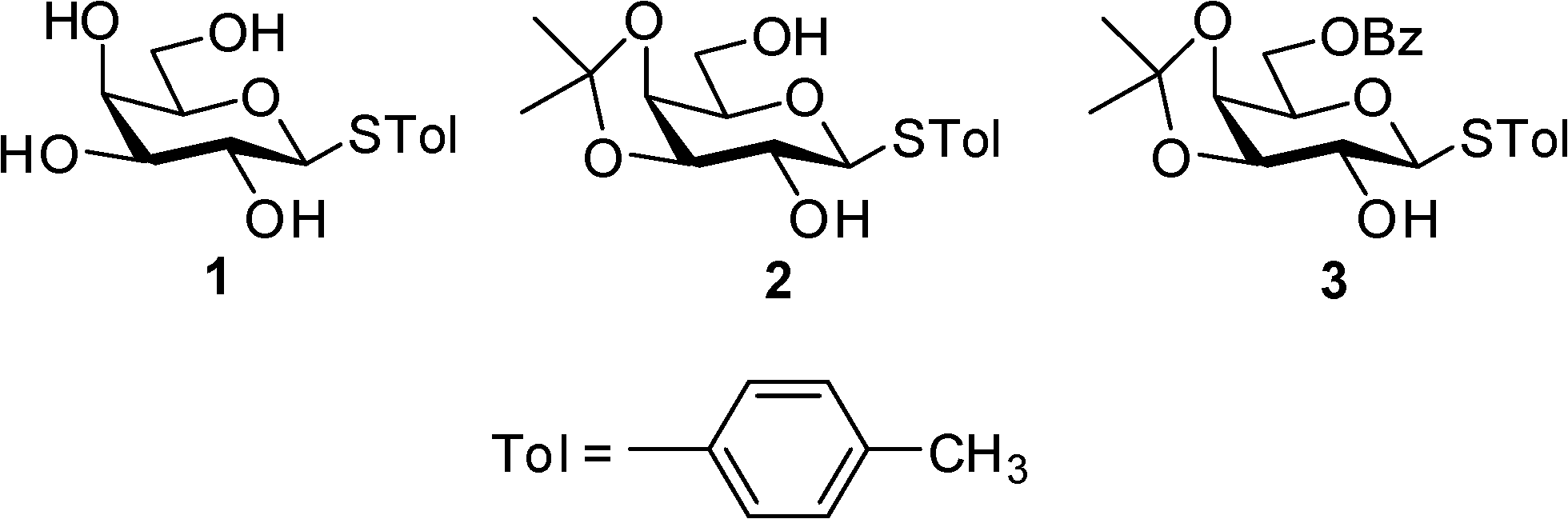
In continuation of our effort in elucidating the driving forces governing the supramolecular architecture of sugar derivatives, we report the crystal structures of three partially protected galactose derivatives (1–3, Fig. 1) and insights into the HBNs involved. The set of molecules reported in this communication is essentially the same except for the fact that these are p-thiotolyl glycosides instead of p-methoxyphenyl glycosides reported in our previous article.10 As expected, compound 1 gave a gel and we were able to obtain single crystals of all three compounds. However, critical insights into the crystal structures revealed a significant difference in the HBNs involved and more interestingly in the case of compound 3, the pyranose ring has taken a skew boat conformation instead of a regular 4C1 chair conformation.
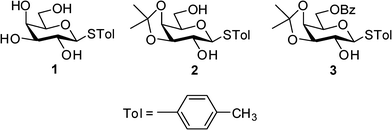 |
| | Fig. 1 Structures of the derivatives in this study. | |
2. Experimental
2.1. Materials
D-Galactose and other reagents were purchased from SigmaAldrich. Commercially available solvents were used as received without further purification.
2.2 Chemical synthesis
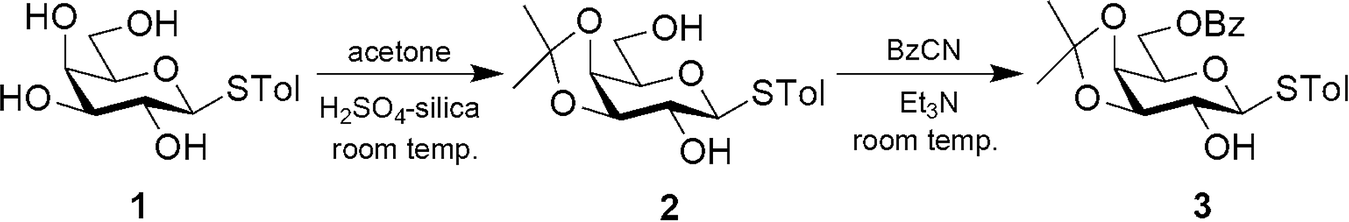
Tolyl-1-thio-β-D-galactopyranoside (1) was synthesized by following the literature procedure.11 It was reacted with dry acetone in the presence of H2SO4-silica to furnish the corresponding 3,4-O-isopropylidene derivative 2 in 85% yield. Selective protection of the primary OH group using benzoyl cyanide and Et3N gave the mono-benzoate derivative 3 in 89% yield (Scheme 1). All three derivatives were purified by flash chromatography using an n-hexane–EtOAc mixture in a suitable ratio and characterized by 1H and 13C NMR and mass spectrometry.
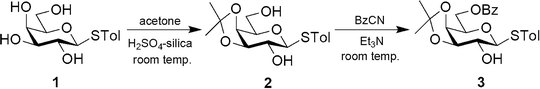 |
| | Scheme 1 | |
2.3. NMR
1H and 13C nuclear magnetic resonance (NMR) analyses of compounds 1, 2 and 3 were performed using a Bruker 500 MHz NMR at 298 K. CD3OD was used as NMR solvent for compound 1, whereas CDCl3 was used for 2 and 3.
2.4. Mass spectrometry
The mass spectra were recorded on a Q-TOF Micro YA263 high resolution (Waters Corporation) mass spectrometer by positive-mode electrospray ionization.
2.5. Gelation and gel characterization
A required amount of the compound and a measured volume of the desired pure solvent were placed in a screw-capped vial with an internal diameter (i.d.) of 10 mm and were slowly heated till the solid was completely dissolved. The clear solution thus obtained was cooled to room temperature in air to form the gel. The vials were inverted to confirm the formation of the gel. The minimum gelation concentration (MGC) was determined by finding the minimum amount of compound required for gel formation at room temperature. The gels were found to be thermally reversible. Upon heating above their gel dissociation temperature, they transformed to solution state and returned to their original gel state upon cooling.
2.6. Determination of gel–sol transition temperature (Tgel)
The gel-to-sol transition temperature (Tgel) was determined by the dropping ball method. A small glass ball (230 mg) was placed on 1 mL of gel in a standard 15 mm vial. The vial was heated slowly in a thermostated oil bath while observing the rising temperature carefully. The temperature at which the ball dropped to the bottom of the vial was recorded as Tgel.
2.7. FT-IR spectroscopy
The FT-IR spectra of compounds 1, 2 and 3 in their crystalline state and compound 1 in its xerogel state were obtained using a Fourier-transform infrared spectrometer (PerkinElmer 502). KBr samples (2 mg in 20 mg of KBr) were prepared and 10 scans were collected at 4 cm−1 resolution for each sample. The spectra were measured at room temperature over the range of 4000–500 cm−1.
2.8. Field emission scanning electron microscopy (FE-SEM)
The morphologies of the reported gels were investigated using field emission-scanning electron microscopy (FE-SEM). A small amount of gel/solution was placed on a clean microscope cover glass and then dried by slow evaporation. The material was then allowed to dry under vacuum at 30 °C for two days. The materials were gold-coated, and the micrographs were taken using a FE-SEM apparatus (Jeol Scanning Microscope JSM-6700F).
2.9. Rheological study
To understand the mechanical strengths of the swollen hydrogels (swollen in DI water overnight), we performed rheological measurements on a AR-G2 rheometer (TA Instrument) using a steel parallel plate geometry with a 40 mm diameter at 25 °C. The rheometer is attached to a Peltier circulator thermo cube that helps with accurate control of temperature during the experiment. The storage modulus (G′) and loss modulus (G′′) of the polymer gels have been recorded in the linear viscoelastic regime at a strain of γ = 2% of the angular frequency (0.1–100 rad s−1).
2.10. Single crystal preparation
Crystals of the compounds 1, 2 and 3 were obtained for single crystal X-ray diffraction. Crystals of compound 1 were obtained by slow evaporation of a methanolic solution. Block-shaped colourless crystals appeared in seven days. In the case of galactoside 2, colourless needles were obtained after twenty five days from a dilute solution in 1,2-dichlorobenzene. Compound 3 gave colourless needles after eight days upon slow evaporation of a solution of the compound in an n-hexane–ethyl acetate (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture.
:1) mixture.
2.11. Crystallography
The single-crystal X-ray diffraction data of the crystals were collected on a SuperNova, Dual, Mo at zero, Eos diffractometer at 292 K for compound 1 and 100 K for compounds 2 and 3 using graphite-monochromatic Mo Kα radiation (λ = 0.71073 Å). Atomic coordinates, and isotropic and anisotropic displacement parameters of all the non-hydrogen atoms of the two compounds were refined using Olex2,12 and the structure was solved with the Superflip13 structure solution program using Charge Flipping and refined with the ShelXL14 refinement package using least squares minimization. The structure graphics shown in the figures were created using the X-Seed software package version 2.0. and mercury software package version 3.5.
2.12. Melting point
The melting points of the three crystals were measured using a digital melting point apparatus, SECOR INDIA. The melting points for the crystals of compounds 1, 2 and 3 were found to be 115 °C, 54 °C and 113 °C, respectively.
3. Results and discussion
Compounds 1, 2 and 3 were tested for their ability to form gels in different solvents. The results of those experiments are summarized in Table 1. Only compound 1 showed significant gelation ability in bromobenzene, chlorobenzene, 1,2-dichlorobenzene and 1,3-dichlorobenzene (Fig. 2) with a minimum gelation concentration (MGC) range between 0.35 and 1.2% w/v. 1,3-Dichlorobenzene was found to be the best solvent when the MGC is concerned. The gels are thermo-reversible in nature and no significant change in the gelation behaviour was observed upon several heating and cooling cycles. The gels were stable at room temperature and no noticeable change was observed when kept in a closed container for a couple of months. It shows the temporal stability of the gel. The effect of the concentration of the gelator on the gel-to-sol transition temperature (Tgel) is shown in Fig. S1 (ESI†). The Tgel value increased up to 381 K with an increase in the gelator concentration of 6% w/v and remained almost constant till 9% w/v showing no further concentration dependence.
Table 1 Gelation abilities of compound 1, 2 and 3 in various solventsa
| Solvents |
Compound 1 |
Compound 2 |
Compound 3 |
| MGC (% w/v) |
MGC (% w/v) |
MGC (% w/v) |
|
OG = opaque gel; S = soluble; I = insoluble, P = precipitate, PS = partially soluble, C = crystalline
|
| 1,2-Dichlorobenzene |
1.2 (OG) |
PS |
PS |
| 1,3-Dichlorobenzene |
0.35 (OG) |
PS |
PS |
| Toluene |
C |
S |
C |
| Xylene |
P |
PS |
S |
| Mesitylene |
P |
PS |
S |
| Benzene |
P |
P |
P |
| Nitrobenzene |
C |
PS |
C |
| Bromobenzene |
0.5 (OG) |
PS |
PS |
| Acetone |
C |
S |
S |
| Butanol |
C |
S |
PS |
| Methanol |
C |
S |
S |
| Water |
I |
I |
I |
| CCl4 |
PS |
S |
S |
| Fluorobenzene |
P |
I |
P |
| Chlorobenzene |
0.4 (OG) |
PS |
PS |
 |
| | Fig. 2 Photograph showing the gels of compound 1 in different solvents: (a) chlorobenzene, (b) 1,3-dichlorobenzene, (c) 1,2-dichlorobenzene, (d) bromobenzene. | |
3.1. FT-IR study
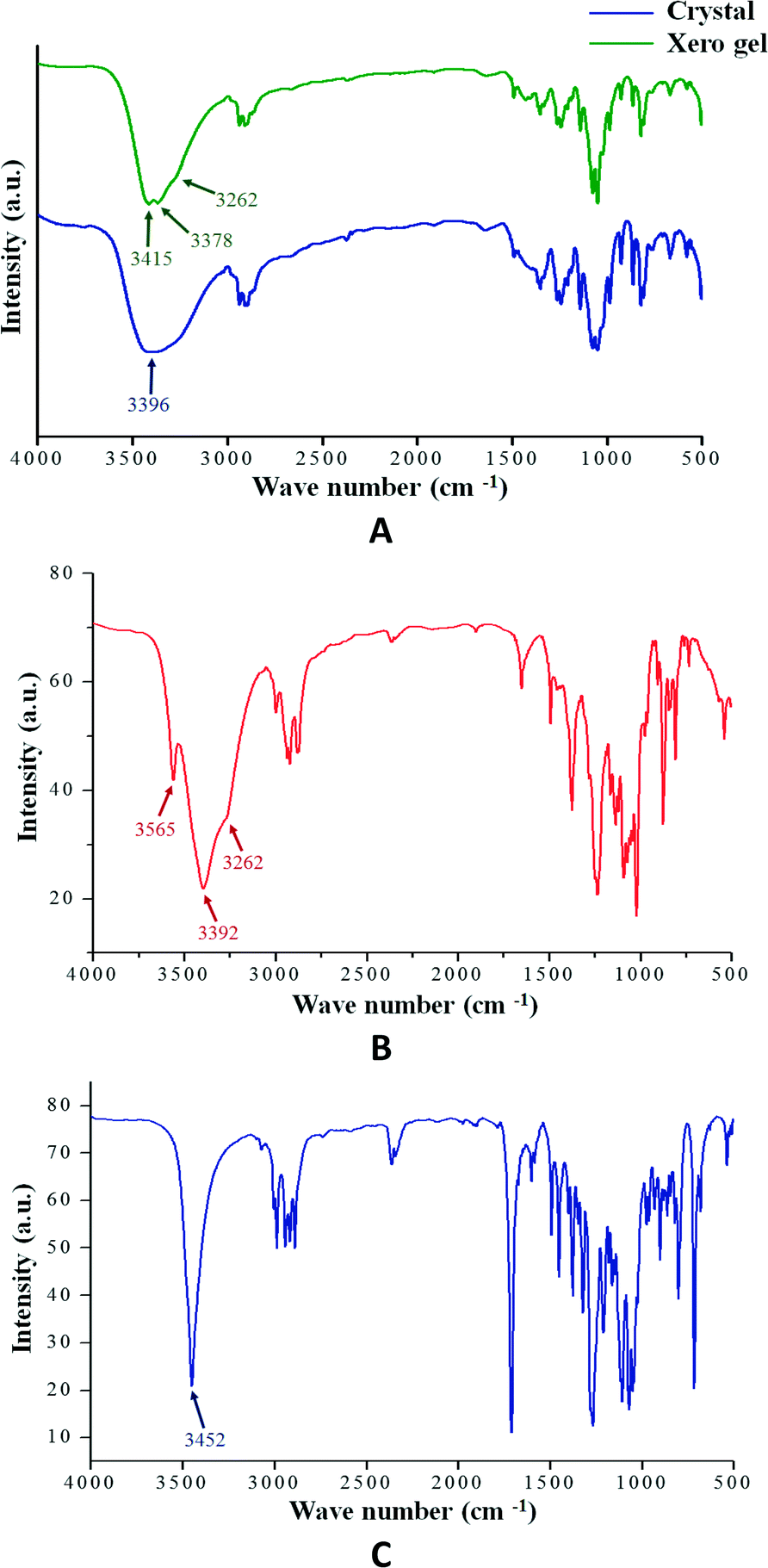
Hydrogen bonding is one of most important interactions among the non-covalent interactions responsible for the formation of the supramolecular architecture in gel or crystalline state. The FT-IR study is particularly useful for the detection and characterization of hydrogen bonding in the self-assembled state. Fig. 3 shows the FT-IR spectra of the crystalline state of compounds 1, 2 and 3 and the xerogel state of 1. In all cases, the absence of any band at 3600 cm−1 (free OH stretching vibration) clearly indicates that all the O–H groups are hydrogen bonded. Moreover, it is observed that the IR bands for the hydrogen bonded O–H groups are well resolved in the case of the crystalline states compared to the gel state. This suggests the involvement of the hydrogen bonding network towards the formation of the supramolecular architecture.
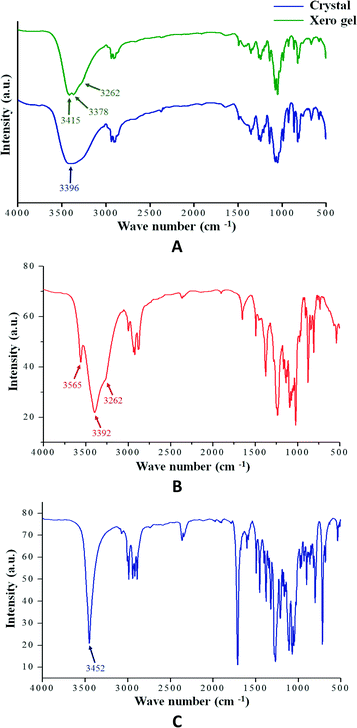 |
| | Fig. 3 (A) FT-IR spectra of compound 1 in crystalline and xerogel state (0.25% w/v in 1,3-dichlorobenzene); (B) compound 2 in crystalline state and (C) compound 3 in crystalline state. | |
3.2 Microscopic studies
The microstructures of the gels formed by compound 1 in different solvents were inspected by FE-SEM to get the visual insight into their morphology. Fig. 4a–d show the FE-SEM images of the xerogels of compound 1 in 1,2-dichlorobenzene, 1,3-dichlorobenzene, bromobenzene and chlorobenzene, respectively. All of them show three-dimensional networks formed by cross-linking of self-assembled fibrillar networks (SAFiNs),15 which are approximately 70–100 nm in diameter and several micrometers in length, indicating the entrapment of the solvent molecules into the spaces of the 3D network.
 |
| | Fig. 4 FE-SEM images of the xerogels of compound 1 in (a) 1,2-dichlorobenzene, (b) 1,3-dichlorobenzene, (c) bromobenzene and (d) chlorobenzene. | |
3.3. Rheological study (frequency sweep) of the gel
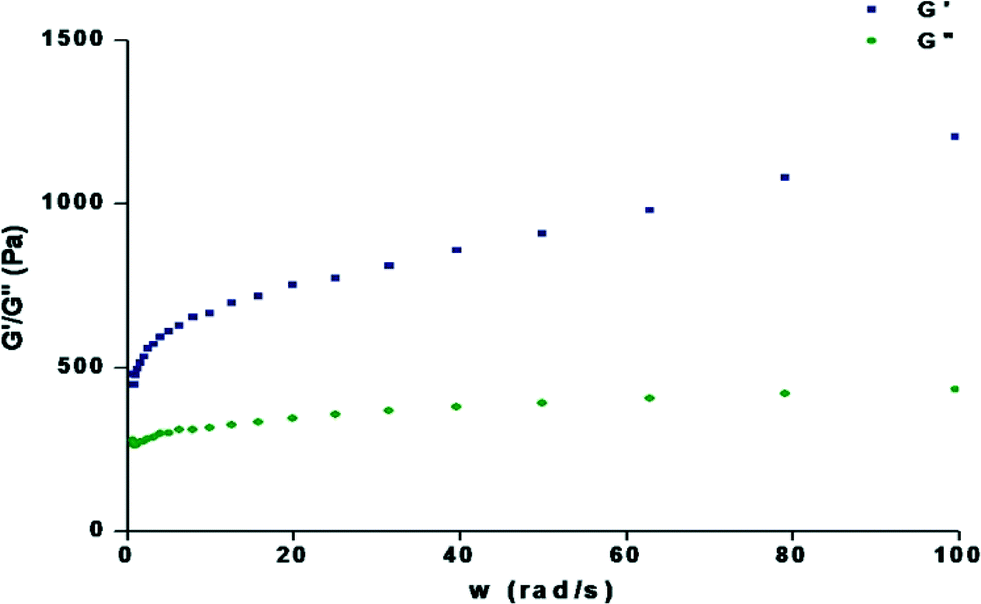
To judge the tolerance power of the gel, rheological measurements were done to determine the frequency sweep. Frequency sweep rheometric measurements were carried out using the gels (1.0%, wt/v) with 1,2-dichlorobenzene, 1,3-dichlorobenzene, bromobenzene and chlorobenzene at a low shear stress. In all cases (Fig. 5 and S2†), the storage modulus (G′) was higher than the loss modulus (G′′) and no crossover is observed over the whole frequency range, showing a typical gel behaviour with a good tolerance performance against external forces.
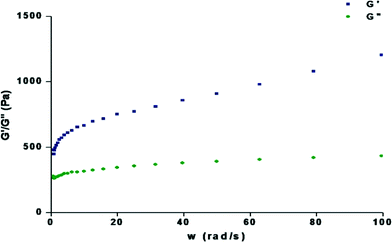 |
| | Fig. 5 Dynamic rheology of the organogel containing 1.2% w/v compound 1 in 1,3-dichlorobenzene as a function of angular frequency (rad s−1) at 15 °C. | |
3.4. Single crystal X-ray diffraction studies
Crystals of the three galactose derivatives (1, 2 and 3) were subjected to single crystal X-ray diffraction. The experimental data and structure refinement parameters are given in Table S1 (ESI†). Critical analysis of the crystal structures reveals that the pyranose ring remains in the typical 4C1 conformation for compounds 1 and 2. However, it has taken a skew boat conformation in the case of compound 3 which is a significant and interesting deviation from our observed results with the p-methoxyphenyl galactosides having similar functionalities reported earlier.10
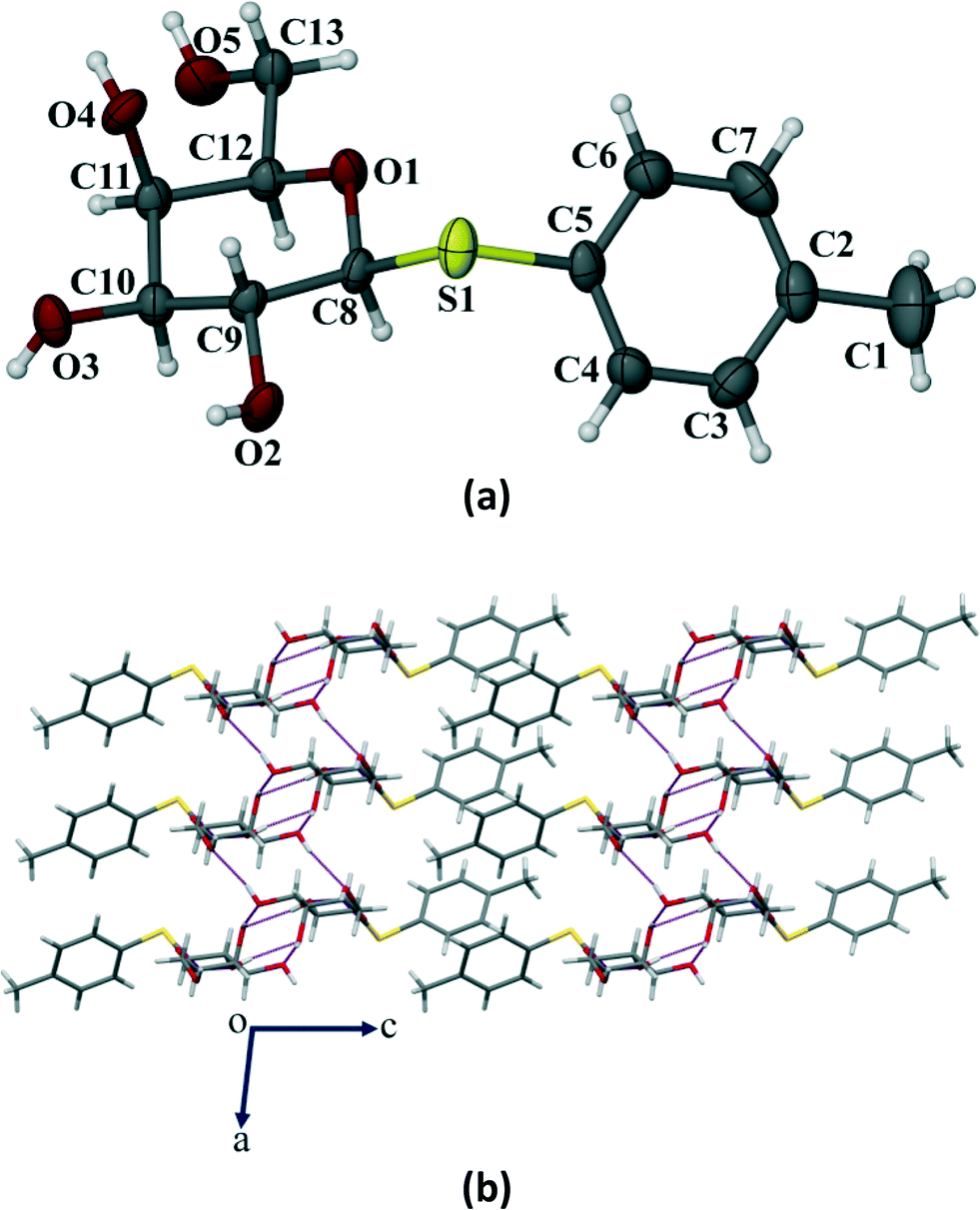
Compound 1 crystallized in the monoclinic P21 space group with 2 molecules (Z = 2) in the unit cell (Fig. 6a). The p-thiotolyl ring of the molecule in the asymmetric unit and the sugar moiety are not co-planar. The torsional angles of the p-thiotolyl ring and the sugar moieties are 158.5(2)° (C5–S1–C8–C9) and −82.7(3)° (C8–S1–C5–C4), respectively, indicating that there is no planarity in the molecule. The four free hydroxyl groups in the molecule led to the formation of multiple intermolecular hydrogen bonds (selected bond lengths and angles are given in Table S2, ESI†). The sugar moieties of adjacent molecules are connected via multiple O–H⋯O hydrogen bonding interactions in the crystal structure and extended in 2D HBN as shown in Fig. 6b. As a result, the molecules form thick two-sided comb-like sheets running along the c-axis. In addition, the adjacent sheets are interlocked by the close packing of p-thiotolyl groups in a space-filling manner and therefore form a 1D layer structure with intra-layer π-stacking interactions (Fig. 6b).
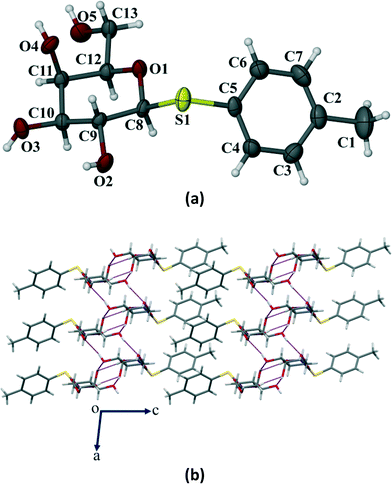 |
| | Fig. 6 (a) ORTEP representation of the crystal of compound 1 with thermal ellipsoids drawn at the 50% probability level. (b) Molecular packing showing a 2D HBN in the crystal structure. Notice the formation of double-sided comb-like sheets via strong intermolecular O–H⋯O hydrogen bonds, interlocked by p-thiotolyl groups. | |
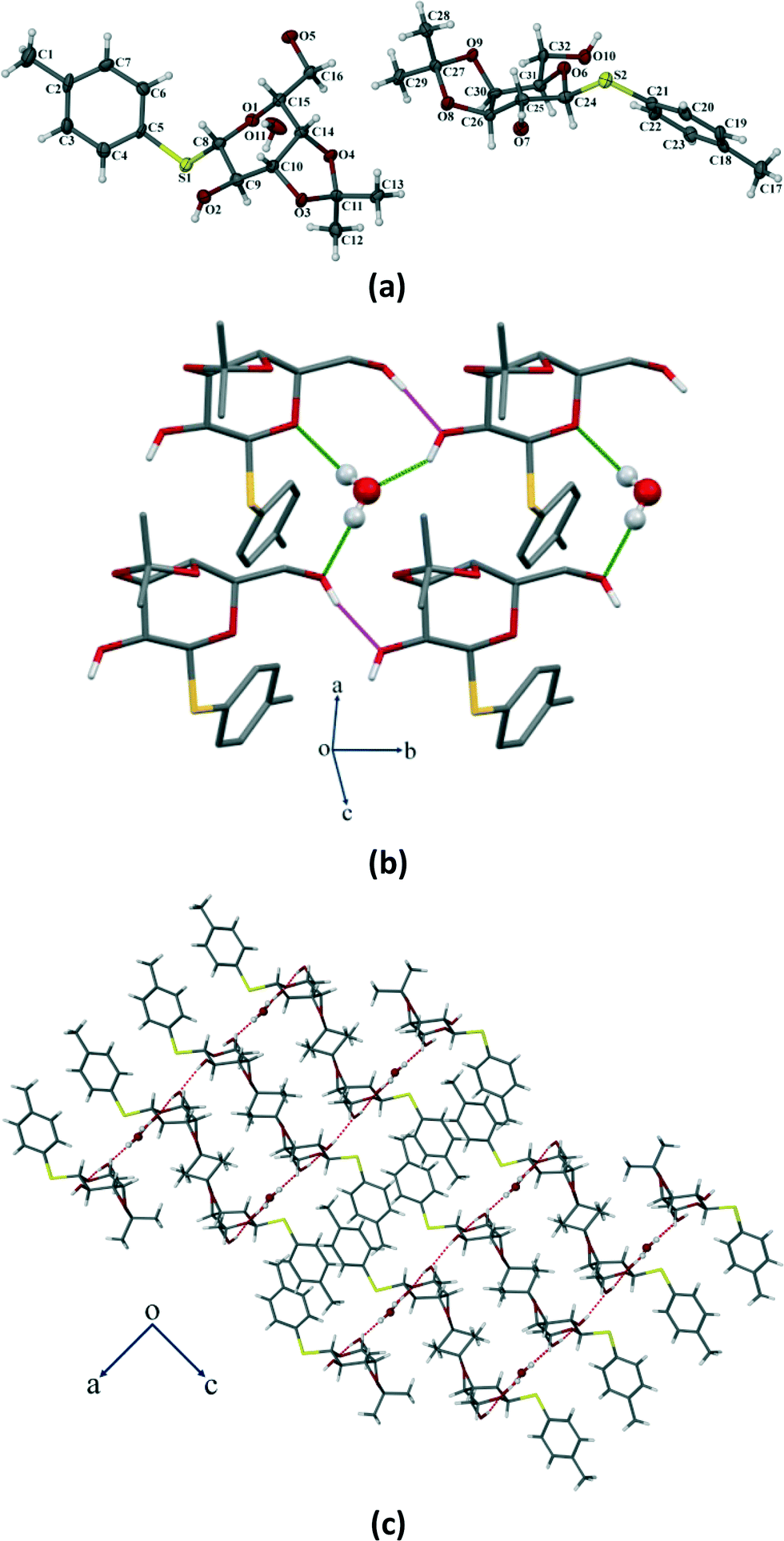
Compound 2 crystallizes in the monoclinic P21 space group with two molecules of compound 2 and one water molecule in the asymmetric unit (Fig. 7a). The molecule contains two free hydroxyl functional groups along with a water of crystallization which form several O–H⋯O hydrogen bonds (Table S2, ESI†) and leads to the formation of a 2D HBN in the single crystal structure (Fig. 7b). The molecules form thick two-sided comb-like sheets interlocked by the close packing of p-thiotolyl groups of the adjacent sheets in a space-filling manner. As a result, the 2D HBN between water molecules and sugar molecules leads to a ladder motif along the b-axis (Fig. 7c). These layers are stacked along the b-axis to further stabilize the structure by multiple weak C–H⋯O interactions (selected bond lengths and angles are given in Table S2, ESI†).
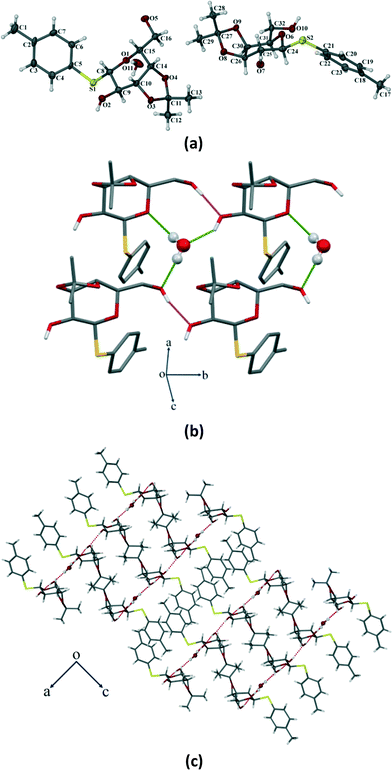 |
| | Fig. 7 (a) ORTEP representation of the crystal of compound 2 with thermal ellipsoids drawn at the 50% probability level. (b) Molecular packing showing a 2D HBN involving sugar–sugar and sugar–water interactions in the crystal structure (except the hydroxyl H, all other H have been omitted for clarity). (c) The ladder motif along the b-axis. Notice the formation of double-sided comb-like sheets via strong intermolecular O–H⋯O hydrogen bonds, interlocked by p-thiotolyl groups. | |
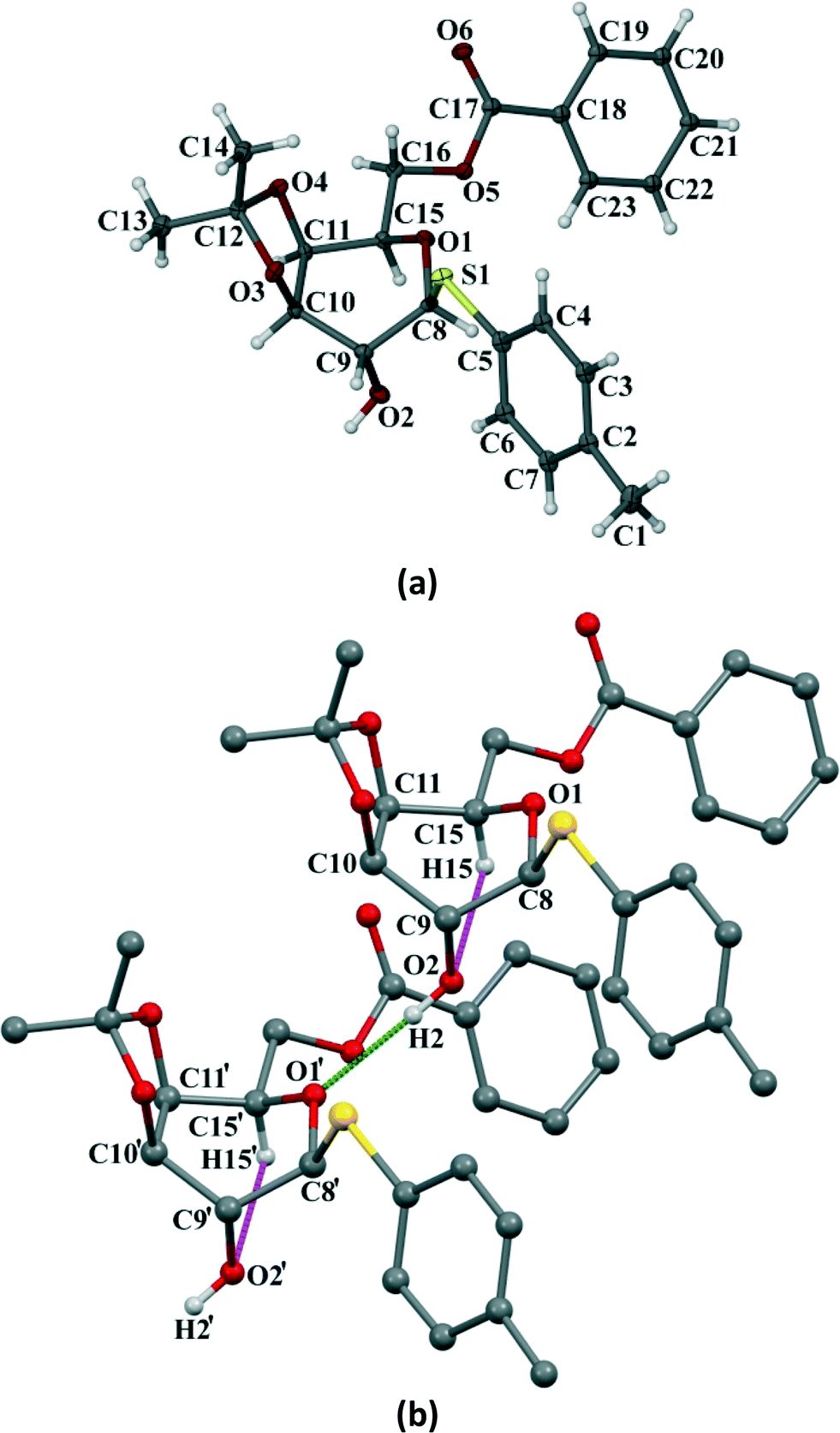
Compound 3 crystallizes in the orthorhombic P212121 space group with four molecules in the unit cell (Z = 4) (Fig. 8a). The p-thiotolyl ring of the molecule in the asymmetric unit is not co-planar with a galactoside moiety. The torsional angles of the p-thiotolyl ring and the sugar moieties are 94.2(3)° (C5–S1–C8–C9) and 156.5(3)° (C8–S1–C5–C4), respectively, indicating that there is no planarity in the molecule. The molecule contains only one free hydroxyl group that forms a O–H⋯O hydrogen bond (O2–H2⋯O1; 2.858(3) Å, 165°) with the O-atom of the next pyranose ring. This leads to the conformational perturbation in the molecule by dragging the C9 of the sugar ring containing the free hydroxyl group downwards and the ring oxygen (O1) of the adjacent molecule upwards. In addition to the strong O–H⋯O HBN, an intramolecular C–H⋯O hydrogen bond (C15–H15⋯O2; 2.937(4) Å, 110°) is responsible for dragging C15 downwards. On the other hand, the isopropylidene ring gives the rigidity at C10 and C11 carbons and due to the required cis-geometry of the isopropylidene ring oxygen atoms, C10 and C11 remain in the same plane. The cumulative effects of these interactions bring O1, C8, C10 and C11 upward and they remain in a plane, whereas C9 and C15 go downwards resulting in a skew boat conformation of the pyranose ring (Fig. 8b). As far as our knowledge is concerned, this is the first report of such a conformational change in a sugar derivative driven by a hydrogen bonding network.
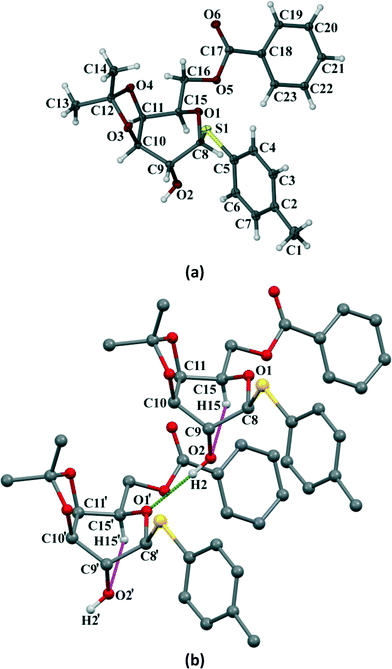 |
| | Fig. 8 (a) ORTEP representation of the crystal of compound 3 with thermal ellipsoids drawn at the 50% probability level. (b) HBN involved for the change of conformation in the pyranose ring (except the hydroxyl H, all other H have been omitted for clarity). Two discrete molecules are shown in a ball and stick model to illustrate the HBN responsible for conformational change. | |
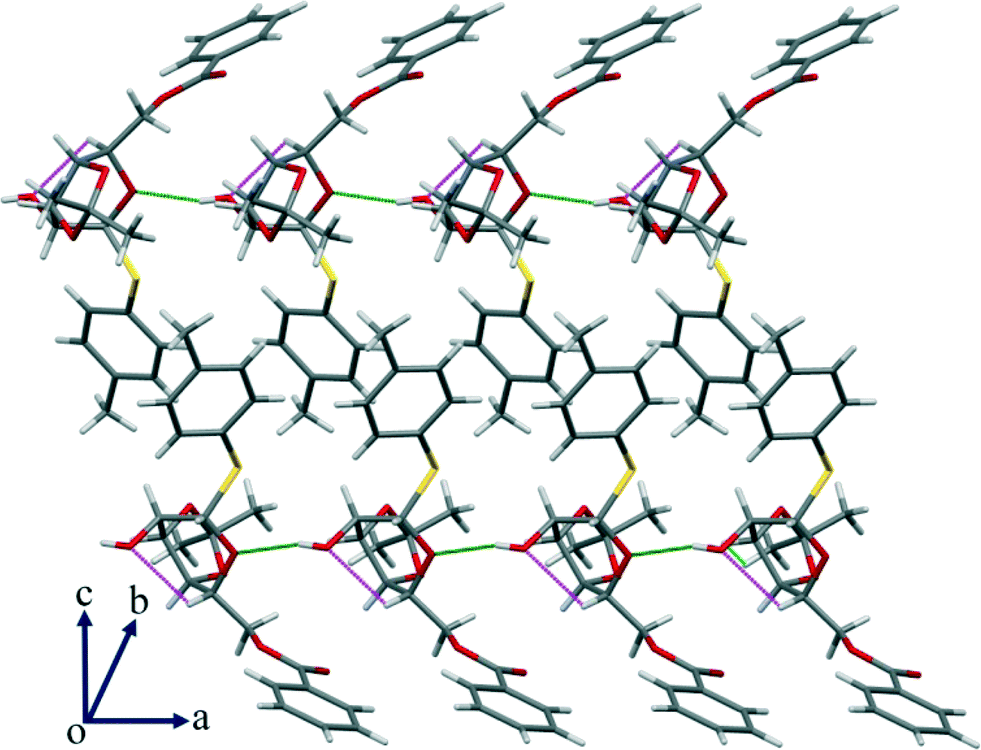
In addition to the interactions that seem relevant for the conformational change, there are other significant C–H⋯O hydrogen bonds that are present (Table S2, ESI†) in the crystal structure that led to a wave-type packing along the given axis (Fig. 9).
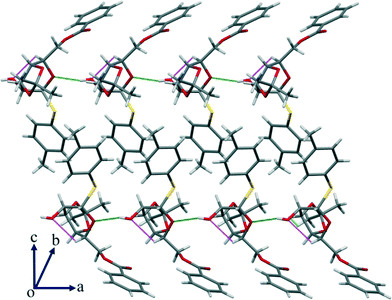 |
| | Fig. 9 In addition to the strong O–H⋯O interaction, there is a significant intramolecular C–H⋯O HBN shown in the packing of compound 3 along the given axis. | |
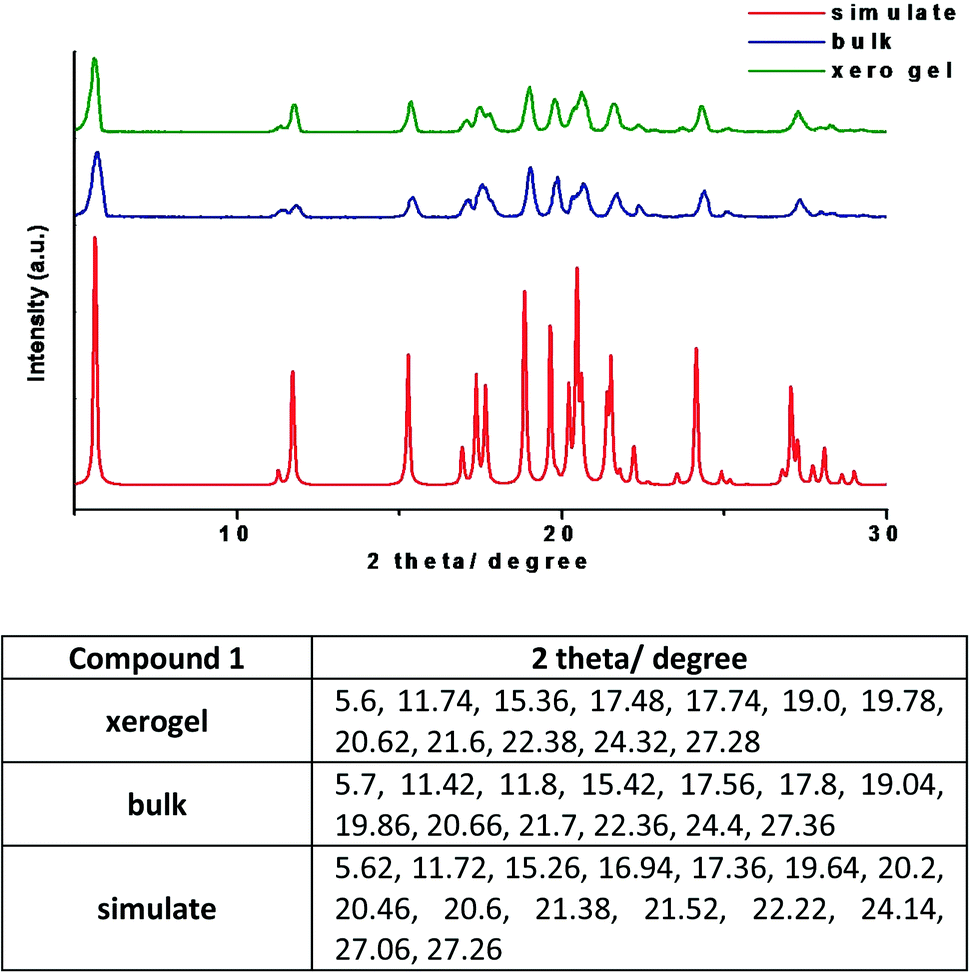
3.5. Powder X-ray diffraction (PXRD)
The PXRD patterns of compound 1 in the xerogel state was compared with the patterns obtained from samples in bulk state and the simulated PXRD pattern extracted from single crystal X-ray. The patterns are in good agreement with each other confirming that the molecular packing in different states is very much the same (Fig. 10). Normally, it is difficult to have clear PXRD patterns from gel samples due to their poor diffraction. However, the correlation between the PXRD patterns in different states,16 is a useful tool to get an insight into the molecular packing. It is worth noting that xerogel samples can only be regarded as logical representative as the presence of other morphs or solvates cannot be ruled out.
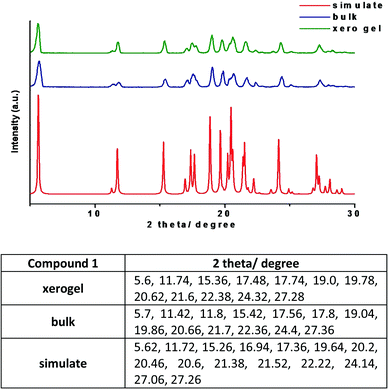 |
| | Fig. 10 PXRD patterns of compound 1 from various states with the list of the major peaks (the xerogel of 1 was prepared from 1,3-dichlorobenzene). | |
3.6. Significant deviation from the reported p-methoxyphenyl galactosides with same substituent
Correlation with our previously reported series of galactose derivatives10 revealed that there are significant differences in the supramolecular architecture of the thioglycoside derivatives reported here with that of the p-methoxyphenyl galactosides. Since the protecting groups present in both series are the same except the reducing end glycoside, we expected to see a close similarity in their supramolecular behaviour. Indeed, compound 1 gave a strong gel and all compounds (1–3) gave quality crystals. Compound 1 and 2 have shown a 2D HBN, whereas compound 3 have a 1D HBN as revealed by their respective crystal packing. Therefore, in this series too, compounds 1 and 3 violate Shinkai's hypothesis17 and the views reflected in the review by Dastidar18 on the gelling and non-gelling behaviour and HBN. Compound 2 behaves in line with the hypothesis. Although the p-methoxyphenyl galactoside and p-thiotolyl glycoside series matched nicely, in-depth evaluation of the HBN showed significant difference between the two series. It is clear that an apparently minor difference in functionality contributes distinctly to the supramolecular arrangement in the crystal structures. In fact, the presence of sulphur at the reducing end glycoside plays a crucial role and influences the HBN significantly. Particularly in the case of the derivative 3, it results in the change of the regular chair conformation of the galactose unit to a skew boat conformation.
Conclusions
In summary, we have analyzed the role of HBN in the single crystal X-ray structures and gelation abilities of three p-thiotolyl galactosides and compared the data with the previously reported series of p-methoxyphenyl galactosides having similar protecting groups. There are similarities between the two series of derivatives with respect to their gelation and crystallization abilities. However, striking differences have been observed between the series considering the HBN involved. The presence of sulphur in the reducing end glycoside influences the difference in the HBN considerably and most interestingly, the altered HBN results in a change in the conformation of the galactose moiety from a regular chair form to a skew boat form in the case of compound 3 as evident from its single crystal X-ray structure. It is important to note that a minimal change in the protecting group decoration on the sugar molecule can have such an impact on the HBN in their molecular packing.
Acknowledgements
K. B. P. is thankful to UGC, New Delhi for Senior Research Fellowship. V. S. is thankful to IISER Kolkata for Senior Research Fellowship. This work is funded by SERB, DST, New Delhi through grant SB/S1/OC-48/2013. We sincerely thank Mr. Ramesh Devarapalli, IISER Kolkata for his help in crystallographic analysis.
Notes and references
-
(a) K. S. Gates, Nat. Chem. Biol., 2013, 9, 412–414 CrossRef CAS PubMed;
(b) K. M. Bradley and S. A. Benner, Beilstein J. Org. Chem., 2014, 10, 1826–1833 CrossRef PubMed;
(c) K. Kristen, K. K. Merritt, K. M. Bradley, D. Hutter, M. F. Matsuura, D. J. Rowold and S. A. Benner, Beilstein J. Org. Chem., 2014, 10, 2348–2360 CrossRef PubMed;
(d) O. Khakshoor, S. E. Wheeler, K. N. Houk and E. T. Kool, J. Am. Chem. Soc., 2012, 134, 3154–3163 CrossRef CAS PubMed;
(e) C. F. Guerra, F. M. Bickelhaupt, J. G. Snijders and E. J. Baerends, Chem. – Eur. J., 1999, 5, 3581–3594 CrossRef CAS.
-
(a) B. Dalhus, A. S. Arvai, I. Rosnes, Ø. E. Olsen, P. H. Backe, I. Alseth, H. Gao, W. Cao, J. A. Tainer and M. Bjørås, Nat. Struct. Mol. Biol., 2009, 16, 138–143 CrossRef CAS PubMed;
(b) R. D. Makde, J. R. England, H. P. Yennawar and S. Tan, Nature, 2010, 467, 562–566 CrossRef CAS PubMed;
(c) G. Lebon, T. Warne, P. C. Edwards, K. Bennett, C. J. Langmead, A. G. W. Leslieand and C. G. Tate, Nature, 2011, 474, 521–526 CrossRef CAS PubMed;
(d) A. K. W. Leung, K. Nagai and J. Li, Nature, 2011, 473, 536–539 CrossRef CAS PubMed;
(e) K. Welke, H. C. Watanabe, T. Wolter, M. Gausaband and M. Elstner, Phys. Chem. Chem. Phys., 2013, 15, 6651–6659 RSC.
-
(a) J. Ju, M. Park, J.-M. Suk, M. S. Lah and K.-S. Jeong, Chem. Commun., 2008, 3546–3548 RSC;
(b) G. A. Kochetov, Biochemistry (Moscow), 2001, 66, 1077–1085 CrossRef CAS;
(c) M. Murár, G. Addová and A. A. Boháč, Beilstein J. Org. Chem., 2013, 9, 173–179 CrossRef PubMed;
(d) Z. Zhang and P. R. Schreiner, Chem. Soc. Rev., 2009, 38, 1187–1198 RSC;
(e) K. Dobi, I. Hajdú, B. Flachner, G. Fabó, M. Szaszkó, M. Bognár, C. Magyar, I. Simon, D. Szisz, Z. Lőrincz, S. Cseh and G. Dormán, Molecules, 2014, 19, 7008–7039 CrossRef PubMed;
(f) N. Srimongkolpithak, S. S. Sundriyal, F. Li, M. Vedadic and M. J. Fuchter, Med. Chem. Commun., 2014, 5, 1821–1828 RSC.
-
(a) W. B. Stockton and M. F. Rubner, Macromolecules, 1997, 30, 2717–2725 CrossRef CAS;
(b) D. A. Dikin, S. Stankovich, E. J. Zimney, R. D. Piner, G. H. B. Dommett, G. Evmenenko, S. T. Nguyen and R. S. Ruoff, Nature, 2007, 448, 457–460 CrossRef CAS PubMed;
(c) V. Berl, M. Schmutz, M. J. Krische, R. G. Khoury and J.-M. Lehn, Chem. – Eur. J., 2002, 8, 1227–1244 CrossRef CAS PubMed;
(d) J. Y. Lee, P. C. Painter and M. M. Coleman, Macromolecules, 1988, 21, 954–960 CrossRef CAS;
(e) B. J. B. Folmer, R. P. Sijbesma, R. M. Versteegen, J. A. J. van der Rijt and E. J. Meijer, Adv. Mater., 2000, 12, 874–878 CrossRef CAS;
(f) C. Liang and S. Dai, J. Am. Chem. Soc., 2006, 128, 5316–5317 CrossRef CAS PubMed;
(g) S. Y. Yang and M. F. Rubner, J. Am. Chem. Soc., 2001, 124, 2100–2101 CrossRef.
-
(a) Y. Li and F. Jian, Molecules, 2015, 20, 14435–14450 CrossRef CAS PubMed;
(b) P. Bhyrappa, S. R. Wilson and K. S. Suslick, J. Am. Chem. Soc., 1997, 119, 8492–8502 CrossRef CAS;
(c) B. Moulton and M. J. Zaworotko, Chem. Rev., 2001, 101, 1629–1658 CrossRef CAS PubMed;
(d) S. Griessl, M. Lackinger, M. Edelwirth, M. Hietschold and W. M. Heckl, Single Mol., 2002, 3, 25–31 CrossRef CAS;
(e) A. Y. Robin and K. M. Fromm, Coord. Chem. Rev., 2006, 250, 2127–2157 CrossRef CAS;
(f) I. A. Baburin, V. A. Blatov, L. Carlucci, G. Cianib and D. M. Proserpio, CrystEngComm, 2008, 10, 1822–1838 RSC;
(g) H. Kihara, T. Kato, T. Uryu and J. M. Fréchet, J. Mater. Chem., 1996, 8, 961–968 CrossRef CAS;
(h) X.-L. Zhang and X.-M. Chen, Cryst. Growth Des., 2005, 5, 617–622 CrossRef CAS;
(i) J. J. V. Gorp, J. A. J. M. Vekemans and E. W. Meijer, J. Am. Chem. Soc., 2002, 124, 14759–14769 CrossRef PubMed;
(j) V. A. Russell, C. C. Evans, W. Li and M. D. Ward, Science, 1997, 276, 575–579 CrossRef CAS PubMed.
-
(a) E. D. Gtwacki, M. Irimia-Vladu, S. Bauerband and N. S. Sariciftcia, J. Mater. Chem. B, 2013, 1, 3742–3753 RSC;
(b) P. Dauber and A. T. Hagler, Acc. Chem. Res., 1980, 13, 105–112 CrossRef CAS;
(c) B. Sarmaand and B. Saikia, CrystEngComm, 2014, 16, 4753–4765 RSC;
(d) S. M. Martin, J. Yonezawa, M. J. Horner, C. W. Macoskoand and M. D. Ward, Chem. Mater., 2004, 16, 3045–3055 CrossRef CAS;
(e) H. Zhang, Z. Zhang, K. Ye, J. Zhang and Y. Wang, Adv. Mater., 2006, 18, 2369–2372 CrossRef CAS;
(f) G. E. Delgado, A. J. Mora, M. Guillén-Guillén, J. W. Ramírez and J. E. Contreras, Cryst. Struct. Theory Appl., 2012, 1, 30–34 Search PubMed;
(g) A. Arunkumar and P. Ramasamy, Mater. Lett., 2014, 123, 246–249 CrossRef CAS;
(h) J. Fielden, P. T. Gunning, D. L. Long, M. Nutley, A. Ellern, P. Kögerler and L. Cronin, Polyhedron, 2006, 25, 3474–3480 CrossRef CAS;
(i) S. Dasgupta, W. B. Hammond and W. A. Goddard, J. Am. Chem. Soc., 1996, 118, 12291–12301 CrossRef CAS.
-
(a) K. Sakurai, Y. Jeong, K. Koumoto, A. Friggeri, O. Gronwald, S. Sakurai, S. Okamoto, K. Inoue and S. Shinkai, Langmuir, 2003, 19, 8211–8217 CrossRef CAS;
(b) A. V. Eliseevand and H.-J. Schneider, J. Am. Chem. Soc., 1994, 116, 6081–6088 CrossRef;
(c) N. Kimizukaand and T. Nakashima, Langmuir, 2001, 17, 6759–6761 CrossRef;
(d) X. Sun and T. D. James, Chem. Rev., 2015, 115, 8001–8037 CrossRef CAS PubMed;
(e) O. Gronwald, E. Snip and S. Shinkai, Curr. Opin. Colloid Interface Sci., 2002, 7, 148–156 CrossRef CAS;
(f) S.-I. Tamaru, M. Nakamura, M. Takeuchi and S. Shinkai, Org. Lett., 2001, 3, 3631–3634 CrossRef CAS PubMed;
(g) T. Shimizu and M. Masuda, J. Am. Chem. Soc., 1997, 119, 2812–2818 CrossRef CAS.
-
(a) F. A. Quiocho, Pure Appl. Chem., 1989, 61, 1293–1306 CrossRef CAS;
(b) T. G. Setty, C. Cho, S. Govindappa, M. A. Apicellab and S. Ramaswamy, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2014, 70, 1801–1811 Search PubMed;
(c) M. Ambrosi, N. R. Cameron and B. G. Davis, Org. Biomol. Chem., 2005, 3, 1593–1608 RSC.
-
(a) S. Chris, C. S. Hawes, C. Chen, A. Tranand and D. R. Turner, Crystals, 2014, 4, 53–63 CrossRef;
(b) G. R. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 31, 2311–2321 CrossRef;
(c) P. Bhyrappa, S. R. Wilson and K. S. Suslick, J. Am. Chem. Soc., 1997, 119, 8492–8502 CrossRef CAS;
(d) S. K. Nayak, K. N. Venugopala, D. Chopra and T. N. Guru Row, CrystEngComm, 2011, 13, 591–605 RSC;
(e) J. Lewiński, J. Zacharaa, I. Justyniak and M. Dranka, Coord. Chem. Rev., 2005, 249, 1185–1199 CrossRef;
(f) J. V. Barth, J. Weckesser, G. Trimarchi, M. Vladimirova, A. De Vita, C. Cai, H. Brune, P. Günter and K. Kern, J. Am. Chem. Soc., 2002, 124, 7991–8000 CrossRef CAS PubMed.
- S. Mukherjee, G. M. Krishna, B. Mukhopadhyay and C. M. Reddy, CrystEngComm, 2015, 17, 3345–3353 RSC.
-
(a) B. Yang, K. Yoshida, Z. Yin, H. Dai, H. Kavunja, M. H. El-Dakdouki, S. Sungsuwan, S. B. Dulaney and X. Huang, Angew. Chem., Int. Ed., 2012, 51, 10185–10189 CrossRef CAS PubMed;
(b) K.-K. T. Mong and C.-H. Wong, Angew. Chem., Int. Ed., 2002, 41, 4087–4090 CrossRef CAS;
(c) M. Wilstermann, J. Balogh and G. Magnusson, J. Org. Chem., 1997, 62, 3659–3665 CrossRef CAS;
(d) C.-M. Huang, R.-S. Liu, T.-S. Wuc and W.-C. Cheng, Tetrahedron Lett., 2008, 49, 2895–2898 CrossRef CAS;
(e) C. Wang, Q. Li, H. Wang, L.-H. Zhang and X.-S. Ye, Tetrahedron, 2006, 62, 11657–11662 CrossRef CAS;
(f) M.-Y. Chen, L. N. Patkar, K.-C. Lu, A. S.-Y. Lee and C.-C. Lin, Tetrahedron, 2004, 60, 11465–11475 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2: a complete structure solution, refinement and analysis program, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- L. Palatinus and G. Chapuis, SUPERFLIP, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS.
- G. M. Sheldrick, SHELXL, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
-
(a) T. Sagawa, S. Fukugawa, T. Yamada and H. Ihara, Langmuir, 2002, 18, 7223–7228 CrossRef CAS;
(b) M. A. Rogers, A. J. Wright and A. G. Marangoni, Soft Matter, 2008, 4, 1483–1490 RSC;
(c) M. Hamedi, A. Herland, R. H. Karlsson and O. Inganäs, Nano Lett., 2008, 8, 1736–1740 CrossRef CAS PubMed;
(d) R. Lam, L. Quaroni, T. Pederson and M. A. Rogers, Soft Matter, 2010, 6, 404–408 RSC.
- E. Ostuni, P. Kamaras and R. G. Weiss, Angew. Chem., Int. Ed. Engl., 1996, 35, 1324–1326 CrossRef CAS.
- R. Luboradzki, O. Gronwald, M. Ikeda, S. Shinkai and D. N. Reinhoudt, Tetrahedron, 2000, 56, 9595–9599 CrossRef CAS.
- P. Dastidar, Chem. Soc. Rev., 2008, 37, 2699–2715 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Details of synthesis, analytical data, crystal data, refinement parameters and hydrogen bonding parameters for all three compounds. Gel to solution temperature (Tgel) graph for compound 1. CCDC 1052160 (1), 1411666 (2), 1052154 (3). For ESI and crystallographic data in CIF or other electronic format See DOI: 10.1039/c5ce01893e |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a