
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Supramolecular metallogels with bulk self-healing properties prepared by in situ metal complexation
Marleen
Häring
a and
David Díaz
Díaz
*ab
aInstitute of Organic Chemistry, University of Regensburg, Universitätstr. 31, Regensburg 93053, Germany. E-mail: David.Diaz@chemie.uni-regensburg.de; Fax: +49 (0) 941 943-4121; Tel: +49 (0) 941 943-4373
bIQAC-CSIC, Jordi Girona 18-26, Barcelona 08034, Spain
First published on 30th August 2016
Abstract
In this feature article, we discuss a series of contributions dealing with the in situ fabrication of supramolecular metallogels (i.e. using low molecular weight ligands and metal ions) that show self-healing properties of the bulk gel phase after complete physical segregation. Most of the advances in this area have taken place during the last three years and are mainly represented by organogels, whereas examples of hydrogels and organic-aqueous gels are still a minority. In situ gelation via metal-coordination of low molecular weight compounds is conceptually different from the use of premade (e.g. in solution) coordination polymers and polymeric structures as gelators and ligands, respectively. In the case of in situ gelation, the cooperative effects of all components of the mixture (i.e. ligand, metal ion, counterions and solvent molecules) in an appropriate ratio under well-defined experimental conditions play a crucial role in the gelation phenomenon and self-healing properties of the material.
 Marleen Häring | Marleen Häring obtained both BSc and the MSc degrees from the University of Regensburg (Germany). She completed her Master's thesis on biopolymer-based catalysis under the supervision of Prof. David Díaz Díaz at the University of Regensburg. Since 2015 she has been pursuing her PhD in Chemistry in the same group working on the development and applications of new chemical and physical gels. |
 David Díaz Díaz | David Díaz Díaz received his PhD in Chemistry from the University of La Laguna (Spain). In 2002, he joined Prof. Finn's group as a postdoc at The Scripps Research Institute (USA). Since 2006, he has held various positions in academia and industry (‘Ramón y Cajal’ Researcher, UAM, Spain, 2006; Sr. Chemist, Dow Chemical, Switzerland, 2007; Tenured Scientist, CSIC, Spain, 2009; Alexander von Humboldt Experienced Researcher, University of Regensburg, Germany, 2010). In 2013, he was awarded the DFG Heisenberg Professorship and appointed as Associate Professor at the University of Regensburg. Among other distinctions, he has received the Young Investigator Award from the Polymer Network Group and is the Editor-in-Chief of Gels. His main research interest focuses on the development and understanding of functional soft materials. |
Introduction
Gels prepared in organic solvents (organogels), water (hydrogels) or aqueous organic mixtures represent a very productive research field1–7 due to their large number of applications in many areas including regenerative medicine, drug-delivery, sensors, actuators, cosmetics, foods, environmental remediation, nanoelectronics (‘bottom-up’ approaches), and catalysis, among others.8–11 However, as it has observed with other materials with significant industrial potential, the development of high-tech applications usually takes place much faster than the acquisition of full knowledge of the mechanisms governing the key physical and/or chemical processes that define the properties of the materials.12,13 Gel-based materials are not an exception to the rule because a large number of new gels are still nowadays discovered by serendipity. However, considerable efforts have been made during the last few decades to gain a deeper understanding of the gelation phenomenon, allowing the development of a general set of rules for the rational design of new molecules with the ability to form gels.14Among different criteria, the classification of gels is usually based on the driving forces for molecular aggregation. This allows a fundamental distinction between chemical gels15,16 (i.e. gels based on covalent bonds, usually crosslinked polymers unable to redissolve) and physical gels17–25 (i.e. gels made of either low molecular weight (LMW)) molecules or polymers, which are based on non-covalent bonds, mainly hydrogen-bonding, van der Waals, charge-transfer, dipole–dipole, π–π stacking, and coordination interactions, which typically lead to reversible gel-to-sol phase transitions. Furthermore, systems based on both types of connections have also been described.26,27 The solid-like appearance of a gel results from the entrapment of the liquid (major component in the mixture) in the interstices of a solid 3D matrix with a large surface area (minor component of the mixture), frequently ascribed to surface tension and capillary forces.28 The formation of the solid matrix is a consequence of the entanglement of 1D polymeric (or suprapolymeric) strands of macromolecules (or LMW compounds) – so-called gelators – typically with nanometer scale diameters and micrometer scale lengths through the above described forces. The immobilization of liquid molecules is governed by a metastable dissolution–crystallization equilibrium and usually increases the viscosity of the medium by a factor of 10.10 Overall, these viscoelastic materials may be considered hard or soft based on their rheological characteristics. All these characteristics provide many gels with the potential to respond to a variety of external stimuli29,30 including temperature, light, pH, mechanical action, electro/magnetic fields and/or chemical stimuli, which are manifested as remarkable changes from the molecular (e.g. hierarchical order, conformational state) to the macroscopic level (e.g. surface properties, shape).31–33
On the other hand, one of the most impressive properties of biological materials is their ability to regenerate and heal after the infliction of physical damage. The origin of some natural self-healing processes (e.g., merging of broken bones, cell-wall recovery upon deformation of wood) can be found in the presence of sacrificial bonds between molecules, which by breaking and reforming allow both a better adaptation of biomaterials to external conditions and the repair of damaged materials.34–37 This has inspired many scientists worldwide to develop a variety of materials,38–42 including gels, with self-healing properties through different strategies.43–50 Among different gels that exhibit self-healing properties in bulk,51–60 considerable attention has lately been drawn towards a specific type of gels (made either in organic solvents, water or aqueous organic mixtures) containing metal ions, so-called metallogels,61–67 due to the reversibility nature of metal coordination.68 Besides the tunable coordination binding strength, the redox, electronic, magnetic and optical properties of metal ions are promising features for numerous applications of metallogels in materials science.69–77 The majority of contributions in the field of self-healing metallogels deal with the use of polymeric ligands. During the preparation of this article, a comprehensive article covering the preparation and properties of coordination polymer gels78–88 was published by Sutar and Maji,89 although it contains only two examples of self-healing supramolecular gels. In this context, during the last few years an increasing number of examples describing self-healing metallogels obtained by in situ gelation during metal complexation of LMW ligands have been reported. It is important to realize that in situ gelation using metal–LMW ligand coordination is significantly different from the use of premade (e.g. in solution) coordination polymers or polymeric structures as gelators or ligands, respectively. In the case of in situ gelation, the cooperative effects of all components of the mixture (i.e. ligand, metal and solvent molecules) in an appropriate ratio and under suitable experimental conditions play a crucial role in the gelation phenomenon and self-healing properties of the material.
Herein, we focus on this particular group of in situ formed metallogels, using LMW compounds, showing self-healing properties of the bulk gel phase after physical segregation (the term self-healing is used from now on under this description). The use of polymeric substrates or premade metal coordination polymers to form gels, including self-healing gels,90–104 is out of the scope of this contribution. In addition, those examples in which only rheological thixotropic loop tests are provided but not evidence of self-healing of the bulk material105–108 are not included in this article.
Metallohydrogels
In 2014, the group of A. Banerjee109 described the first supramolecular in situ formed amino acid-based metallohydrogels with self-healing properties. These materials were prepared by combining a series of amphiphilic tyrosine derivatives, bearing alkyl chains of different lengths (1–3, Fig. 1) with NiCl2. In particular, 1–3 (10 mM) were first dissolved in aqueous phosphate buffer (pH 7–8) by heating and subsequently mixed with a NiCl2 solution, which turned the solution into an initial white suspension. Clear and stable metallohydrogels were obtained after sonication of the suspension and resting for a few minutes for 3, 3 h for 2 and 18 h for 1. | ||
| Fig. 1 (a) Chemical structures of various amphiphiles (1–4). (b) Pictures of glass vials containing metallohydrogels obtained from different proportions of 3 and NiCl2. Critical gelation concentrations (CGC) = 10 mM (1), 9 mM (2), and 7 mM (3). Adapted with permission from ref. 109. Copyright©2014 The Royal Society of Chemistry. | ||
The authors found that a gelator![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ni2+ ion ratio between 1:1 and 1:0.3 was necessary in order to ensure complete gelation of the liquid phase and obtain gels with higher thermal stability. As a control experiment regarding the effect of the substitution of the amino acid on gelation, compound 4 was also synthesized and showed no gelation ability under similar conditions. It is also remarkable to note the apparent non-influence of the counterion of the metal salt on the gelation properties as well as the high metal-selectivity showed by 1–3, being unable to form any gel in the presence of other divalent metals such as Zn2+, Cu2+, Co2+, Mn2+, Fe2+, and Hg2+. The results of XRD studies indicated the formation of nickel hydroxide after addition of NiCl2 into the phosphate buffer solution of 1–3 (pH 7.46). Moreover, comparative FT-IR analyses of the non-gel phase (in the absence of Ni2+) and in the gel phase (in the presence of Ni2+) suggested that the phenolic hydroxyl group of 1–3 strongly interacts with the hydrated nickel complex, whereas other functionalities (i.e. carboxylate and amide) of the ligand molecules are likely involved only in hydrogen bonding between the tape-like nanofibrillar assemblies observed by transmission electron microscopy (TEM), which was in accord with the results of small angle X-ray scattering (SAXS) experiments (Fig. 2).
:Ni2+ ion ratio between 1:1 and 1:0.3 was necessary in order to ensure complete gelation of the liquid phase and obtain gels with higher thermal stability. As a control experiment regarding the effect of the substitution of the amino acid on gelation, compound 4 was also synthesized and showed no gelation ability under similar conditions. It is also remarkable to note the apparent non-influence of the counterion of the metal salt on the gelation properties as well as the high metal-selectivity showed by 1–3, being unable to form any gel in the presence of other divalent metals such as Zn2+, Cu2+, Co2+, Mn2+, Fe2+, and Hg2+. The results of XRD studies indicated the formation of nickel hydroxide after addition of NiCl2 into the phosphate buffer solution of 1–3 (pH 7.46). Moreover, comparative FT-IR analyses of the non-gel phase (in the absence of Ni2+) and in the gel phase (in the presence of Ni2+) suggested that the phenolic hydroxyl group of 1–3 strongly interacts with the hydrated nickel complex, whereas other functionalities (i.e. carboxylate and amide) of the ligand molecules are likely involved only in hydrogen bonding between the tape-like nanofibrillar assemblies observed by transmission electron microscopy (TEM), which was in accord with the results of small angle X-ray scattering (SAXS) experiments (Fig. 2).
 | ||
| Fig. 2 Schematic representation illustrating the proposed stepwise assembly of the gelator molecules to form a tape-like network structure. The widths of the nanofibers were in the range 60–80 nm, 60–70 nm and 120–150 nm for gels 1, 2 and 3, respectively. Adapted with permission from ref. 109. Copyright©2014 The Royal Society of Chemistry. | ||
Despite the small differences in the length of the alkyl chains in 1–3, the rigidity of these metallohydrogels increased by increasing the side chain (1 < 2 < 3) as confirmed by oscillatory rheological measurements. Besides responding to multiple stimuli,110i.e. pH, temperature, mechanical stress and metal chelating agents (Fig. 3a), the metallogels exhibited remarkable self-healing properties. Interestingly, the kinetics of the self-healing process was correlated with the stiffness of the gels (i.e. the gel made of 3 (n = 14) recovered immediately, whereas those made of 1 (n = 10) and 2 (n = 12) needed 8 and 5 min, respectively.). However, further investigations are still necessary to explain this apparent relationship. Macroscopic self-healing of all metallogels was demonstrated by cutting the gel blocks into two pieces and subsequently rejoining them with moderate pressing. It was found that after 25–30 min the two pieces merged into a continuous monolithic gel (Fig. 3b). The so-obtained self-healed monoliths displayed the same stability as uncut monoliths and could be stretched by pulling the ends, demonstrating the efficiency of the self-healing phenomenon. The recovery of the gel network phase (storage modulus G′ > loss modulus G′′) and its mechanical properties were also confirmed at the microscale by strain-recovery rheological experiments with alternating low and high strain cycles to ensure rupture of the gel network (G′ < G′′) (Fig. 3c) – note that while bulk self-healing of gels is usually accompanied by a thixotropic behavior at the microscale, the reverse is not necessarily true. The kinetics of the moduli recovery was again confirmed to increase with the increase in the length of the alkyl chain. Thus, the fine control achieved over several properties of these metallohydrogels by small changes in the alkyl chain length encourages us to keep in mind these results when investigating other amphiphilic LMW coordinating gelators with potential self-healing properties.
 | ||
| Fig. 3 (a) Multi-stimuli responsiveness shown by the hydrogel obtained from 3. Above the gel-to-sol transition temperature (Tgel), all the gels turned into solutions and nickel hydroxide was precipitated out. These suspensions were transformed into metallohydrogels upon cooling them to room temperature followed by sonication. These metallohydrogels also showed sensitivity to metal chelating agents like ethylene diamine (EDA) or ethylenediaminetetraacetic acid (EDTA). (b) Illustration of self-healing behavior (i–v) shown by the metallohydrogel obtained from 3. (c) Step strain experimental data obtained from the metallohydrogel made of 1. The concentration was maintained at 10 mM at a 1:0.5 gelator and Ni2+ ratio. The angular frequency was constant at 1 rad s−1 in all steps. Adapted with permission from ref. 109. Copyright©2014 The Royal Society of Chemistry. | ||
Almost at the same time, cooperation between the group of R. Banerjee and our group111 resulted in the publication of the second example of a self-healing amino acid-derived metallohydrogel. Simply mixing two stock aqueous solutions containing a L-valine derivative (i.e.V = L-3-methyl-2-(pyridine-4-yl-methylamino)-butanoic acid), sodium perchlorate (P) and Zn(OAc)2·2H2O (ZA) leads to the instantaneous formation of an opaque hydrogel (ZAVP) with a CGC of 84 g L−1 (Fig. 4a–c). Similarly to the previous example, the ligand system VP showed specific affinity for metal cation (i.e., Zn2+), whereas the effect of counter anions was negligible, with the possibility of obtaining similar gels with zinc perchlorate or zinc nitrate. As observed with other chiral gel systems,112 the enantiomeric purity of the amino acid derivative had a major influence on the gelation ability. Specifically, complete gelation was observed when the enantiomeric excess (ee) was between 85 and 100%, whereas only partial gelation took place for ee between 60 and 80%.
 | ||
| Fig. 4 (a–c) Synthesis of ZAVP at the CGC. Total volume of each solution = 0.5 mL. (d) Scanning electron microscopy (SEM) image of the corresponding xerogel. Adapted with permission from ref. 111. Copyright©2014 The Royal Society of Chemistry. | ||
In terms of the gelation mechanism, mass spectrometry studies of the corresponding xerogels suggested the direct involvement of a reversible octahedral Zn(II)-complex with two carboxylates of one acetate and one ligand molecule forming an octahedral complex bearing two water molecules in the axial positions. This complex may undergo consecutive self-assembly into fibrils (ca. 20–80 nm in diameter) through multiple hydrogen-bonding interactions, which entangle into a 3D fibrillar network as confirmed by scanning electron microscopy (SEM) imaging of the xerogels (Fig. 4d). Furthermore, rheological measurements confirmed the viscoelastic and brittle nature of the metallohydrogel. Strain-recovery experiments with alternating low and high strain cycles of the ZAVP hydrogel revealed full recovery of the gel network within 3 ± 1 min (Fig. 5). The cycles could be repeated at least 15 times without notable reduction of the average magnitude of G′ and G′′, displaying a remarkable and rapid thixotropic behavior, which was in good agreement with the macroscopic thixotropy observed by shaking–resting cycles.
 | ||
| Fig. 5 Oscillatory rheology of the ZAVP hydrogel prepared at the CGC, thixotropy-loop test via continuous step-strain measurements at 0.1 Hz. Reprinted with permission from ref. 111. Copyright©2014 The Royal Society of Chemistry. | ||
Also in this example, and in addition to a multi-responsive behavior towards external stimuli (i.e. temperature, pH, mechanical stress and metal complexing agents),111 the obtained bulk metallohydrogel exhibited remarkable self-sustainability, load-bearing and self-healing properties. The gel was able to support up to 60-fold of its own weight without any visible change in its shape (Fig. 6a–c), which is not often found for LMW gels.113 Furthermore, bulk self-healing was demonstrated by cutting a gel monolith into small pieces and rejoining them by simple physical contact. Complete fusion of the blocks took place within 3 h allowing the reconstruction of long and stable self-healed gel bridges (Fig. 6d–f).
 | ||
| Fig. 6 Pictures of (a) freshly prepared hydrogel cylinder (thickness = 1.4 cm; diameter = 1.6 cm); (b) 2 g mass bearing a load up to 90 g; and (c) maintaining its shape after the load is released. (d) Moldable and shape-persistent hydrogel blocks cut from a larger cylinder. Pink objects correspond to hydrogel pieces obtained upon doping using Rose Bengal dye. (e and f) A bridge constructed by fusing 11 hydrogel blocks together. Light pink coloration of undoped gel blocks is due to the rapid diffusion of the dye through the self-healed hydrogel network. Adapted with permission from ref. 111. Copyright©2014 The Royal Society of Chemistry. | ||
Metalloorganogels
The synthesis of macromolecules containing bipyridine and terpyridine metal complexes has gained considerable attention during the last two decades due to the large number of potential applications showed by these materials.114 Within the context of this structural moiety, Terech and co-workers115 used a tritopic ligand (BTC) (Fig. 7) exhibiting two terpyridine functionalities and a cyclan spacer to form weak (i.e. G′ ca. 250 ± 15 Pa at 1 Hz) and self-healable metalloorganogels in situ when combined with NiCl2·6H2O in DMF at 50 °C for 24 h. The optimal stoichiometry was found to be Ni2+/BTC = 1.8. Above a 2.0 molar ratio, the ligand was found to complex additional Ni2+ ions and showed fragmentation of the aggregates. | ||
| Fig. 7 Formation of Ni2BTC suprapolymers through chelation of nickel ions (purple spheres) by BTC. The situation for stoichiometry s = Ni2+/BTC = 2 is shown. Note that gelation takes place during the reaction without isolation of the proposed Ni2BTC species. Adapted with permission from ref. 115. Copyright©2014 The Royal Society of Chemistry. | ||
Visual inspection of the Ni2BTC gel after macroscopic damage revealed its self-healing ability. The gel was damaged with a spatula and self-healed within 48 h (Fig. 8a), whereas a classical molecular gel made of 12-hydroxy stearic acid (HAS) with a similar elastic modulus (ca. 220 Pa) under the same treatment did not show any visual healing of the wound. Step strain tests were performed with high stresses up to the yield stress in the non-linear regime of deformations. The system was sheared for ca. 30 s and showed instantaneous recovery (albeit not completely) of the elasticity after removal of the applied stress. Moreover, the application of several cycles showed a progressive fatigue of the material, which was related to a decrease of its mean connectivity. Applying different protocols, a small and controlled deformation step in an oscillatory mode within the linear regime of deformations was followed by a shearing step where the gel was disrupted. Again, a significant part of the shear elasticity was recovered but not entirely (Fig. 8b). Moreover, impedance spectroscopy was performed in order to obtain an insight into the healing mechanism. The results confirmed the coexistence of two main dynamical processes: (1) a fast complexation reaction of Ni2+ with BTC (association constant K = 5.0 × 1010 M−1) and (2) a slow repairing step involving optimization of the 3D interconnectivity of the transient gel network.
 | ||
| Fig. 8 (a) Photographs of upside-down vials containing the Ni2BTC gel (s = 1.8, CBTC = 40 mM in DMF) and the HAS gel used as reference (CHSA = 8.0 mM in decane). From top to bottom: at rest; fracture; 24 hours after fracture; 48 hours after fracture, respectively. (b) Cycles of a small deformation (ca. 0.01, frequency 1 Hz) followed by a shearing protocol at dg/dt = 50 s−1 for 300 seconds during which the gel is fractured showing the partial self-healing of the system. Adapted with permission from ref. 115. Copyright©2014 The Royal Society of Chemistry. | ||
Following their previous work on dynamic supramolecular polymers based on host–guest and metal–ligand interactions, Huang and co-workers116 described the use of heteroditopic monomer 5 with a bridging ligand and a metallic cross-linker, [PdCl2(PhCN)2] (0.5 equiv.), for the in situ preparation of a new self-healable metalloorganogel. Heteroditopic monomer 5 contained a 1,2,3-triazole group between the benzo-21-crown-7 host and its complementary guest dialkylammonium salt and the 1,2,3-triazole group acted as a ligand for coordination with Pd(II). With regard to the gel preparation, 5 was first dissolved in acetonitrile and then addition of 0.5 equiv. of PdCl2(PhCN)2 and subsequent heating to 50 °C led to a transparent solution, which spontaneously turned into a shape-persistent, free-standing gel upon cooling at 4 °C (Fig. 9).
 | ||
| Fig. 9 Schematic representation of controlling the topology and rheology of the supramolecular gelator prepared from heteroditopic monomer 5. Adapted with permission from ref. 116. Copyright©2012 Wiley-VCH Verlag GmbH & Co. | ||
Self-aggregation of 5 to form linear or cross-linked supramolecular polymers by addition of Pd(II) was confirmed by 2D diffusion-ordered 1H-NMR spectroscopy (DOSY) which proved an increase in the average aggregation size by addition of the metal salt. A sol-to-gel transition was induced by adding a competitive ligand (PPh3) for Pd(II), which eliminated the cross-linking. SEM imaging of the corresponding xerogels showed an interconnected porous structure of entangled nanofibers (Fig. 10) where the pore size could be modulated by the amount of cross-linker.
 | ||
| Fig. 10 SEM image of the supramolecular gel network with a cross-linking density of (a) 20% and (b) 60%. Adapted with permission from ref. 116. Copyright©2012 Wiley-VCH Verlag GmbH & Co. | ||
At the CGC (30 mM), the gel exhibited reversible gel-to-sol transitions in response to pH, temperature, and addition of cations or metals, which form new complexes with the host. In addition, the gel showed good mechanical properties (i.e. plateau modulus = 101–104 Pa based on rheological analysis), self-sustainability, moldability, elastomeric behavior and shape-persistent properties. Moreover, freshly prepared gel blocks were able to adhere to each other to construct a stable gel bridge made up of up to four gel blocks (Fig. 11a–c). Although the authors did not provide direct data regarding the reversibility of this process (i.e. cutting and rejoining the pieces as usually done to prove self-healing), they found that fresh surfaces of the gel blocks were essential for adhesion. This is a common requisite to most self-healable gels, which suggests that the mechanical stress applied to freshly prepared gels cause significant alteration of the solvated structure due to the solid–liquid interface susceptibility to mechanical stress.117 Thus, we decided to include this interesting example in this feature article because it made us realize that the differences, and likely cooperative effects, between bulk self-adhesion and self-healing processes are still not specifically addressed in most publications.
 | ||
| Fig. 11 (a) Two supramolecular gel blocks. (b) The two gel blocks adhered to each other to form a free-standing object. (c) A bridge constructed by connecting together four gel blocks suspended horizontally. (d) The moving trajectory of the gel blocks after rebounding from the bottom of the beaker (eight pictures overlaid together). Reprinted with permission from ref. 116. Copyright©2012 Wiley-VCH Verlag GmbH & Co. | ||
Interestingly, the gel also showed excellent elastic properties, being able to rebound up to 20 cm after free-falling from a 25 cm height (Fig. 11d). This behavior can be attributed to the dynamically reversible complexation between the host and dialkylammonium salt and the good mechanical properties of the cross-linked 3D supramolecular network. In sharp contrast, the linear polymer (i.e. prepared in the absence of a cross-linker) did not show any elasticity, confirming the necessity of cross-linking to achieve elastic gels.
More recently, other N-containing heterocycles have been also explored as ligands for the preparation of supramolecular moldable and self-healing gels by in situ metal complexation. Thus, Yan and co-workers118 prepared a series of sticky metallogels by mixing two stock solutions of a series of tetrazole-based ligands (Fig. 12) and Pd(OAc)2. Spontaneous gel formation occurred for ligands 6–9 at a 2:1 tetrazolyl unit:Pd2+ molar ratio and a CGC of 3.0, 4.5, 3.6, and 3.9% (w/v), respectively. Methylation of 6 afforded compound 10 lacking H-donor ability, which inhibited the gelation process. TEM, atomic force microscopy (AFM) and dynamic light scattering (DLS) images revealed the initial formation of uniform spherical aggregates with an average diameter of around 30 nm, which further interconnected to form a more dense and entangled network.
 | ||
| Fig. 12 Chemical structures of ligands 6–10. Adapted with permission from ref. 118. Copyright©2014 The Royal Society of Chemistry. | ||
The gel derived from ligand 6 was used as a model gel for further characterization. The viscoelastic and brittle nature of the gel was confirmed by typical rheological measurements. This metallogel displayed a remarkable stability towards a broad range of pH (1–14), heating (ca. 100 °C), shaking or sonication without a visible gel-to-sol transition, suggesting strong intermolecular interactions. The robustness of the gel allowed its molding into self-supporting blocks of any geometrical shape (Fig. 13).
 | ||
| Fig. 13 (a–i) Shape-persistent, free-standing objects moulded from a gel made of 6 (4%). (j) The gel sculpture made from 4% DMF solution of the gel made of 6. Adapted with permission from ref. 118. Copyright©2014 The Royal Society of Chemistry. | ||
Captivatingly, the gel body was able to support up to 30-fold its own weight by unprecedented bottom-up load-bearing properties due to its sticky character (Fig. 14). This deserves attention because usually up–down load-bearing experiments are reported for supramolecular gels.119
 | ||
| Fig. 14 Gel made of 1 (4%) showing bottom-up load-bearing properties. One to three spatulas (ca. 26 g) were inserted into the gel body and suspended just by holding the test tube. Adapted with permission from ref. 118. Copyright©2014 The Royal Society of Chemistry. | ||
Self-healing properties were also confirmed by the construction of a 3 cm self-supporting gel bridge after cutting gel monoliths and placing the pieces into contact for several minutes at room temperature (Fig. 15). The obtained bridge was stable enough to hold when suspended horizontally or vertically from a wire. The same self-healing capability was also found for the gels made from ligands 7–9. In addition, the gels can be restored after any damage by adding DMF or H2O and self-healing can be accelerated by sonication due to a faster permeating speed of the solvent. However, solvent-healed gels lost their viscoelastic response and only 30% can be recovered after aging for several days.
 | ||
| Fig. 15 (a and b) A bridge constructed by connecting freshly prepared blocks of gel 6 can be suspended horizontally and (c) held vertically, (d) a bridge constructed by connecting moulded gel blocks and (e) solvent-healed (DMF) gel blocks. Red objects correspond to metallogel blocks obtained upon doping with an aqueous solution of methyl red (ca. 0.1%). Reprinted with permission from ref. 118. Copyright©2014 The Royal Society of Chemistry. | ||
A year later, the same group120 expanded their investigations using tetrazole-based ligands and reported for the first time a supramolecular self-healing heterometallic gel with synergistic properties. In this case, a DMF solution of simple ligand 11 was mixed with DMF solutions of NiCl2·6H2O and CoCl2·6H2O to spontaneously form homogeneous metallogels at Ni2+:ligand ratios from 1:1 to 3:1. AFM and TEM images confirmed that both monometallic gels (11-Ni and 11-Co) self-assemble into spherical particles, whose interconnection creates a 3D entangled network (Fig. 16). In addition, the necessity of the –NH group in 11 for gelation was confirmed by the methylated derivative 12, which was not capable of evolving into any gel in the presence of Ni2+ or Co2+.
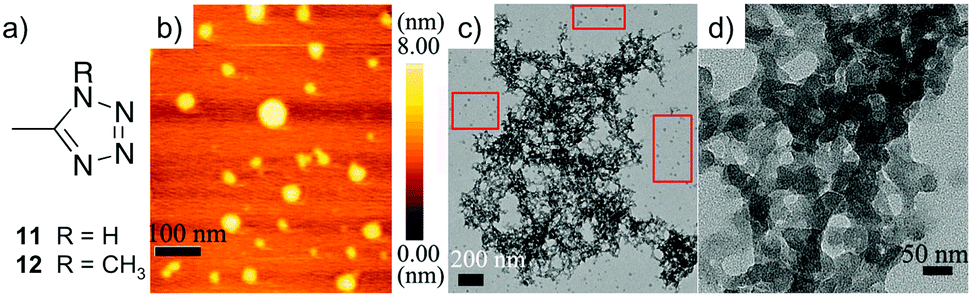 | ||
| Fig. 16 (a) The chemical structures of ligands 11 and 12. (b) Tapping-mode AFM height image of aggregates formed of metallogel 11-Co (0.1 wt%). (c) TEM image of 11-Co on a carbon coated copper grid (unstained, 0.3 wt%). (d) A partial zoomed-in image 11-Co on a carbon coated copper grid (unstained, 0.3 wt%). Adapted with permission from ref. 120. Copyright©2014 The Royal Society of Chemistry. | ||
Gel 11-Ni with a CGC of 2.3 wt% exhibited high stability towards trifluoroacetic acid (TFA), heating (ca. 100 °C), shaking and sonication without a visible gel-to-sol transition. In contrast, Gel 11-Co showed a reversible gel-to-sol transition by heating at 40 °C, which could be regulated by TFA/Et3N. In agreement with rheological measurements 11-Co exhibited an overall weaker stability than 11-Ni (i.e. G′ being ca. 20 times higher for 11-Ni at a three-fold lower concentration than 11-Co). The air stable and stiff 11-Ni gel blocks were not able to adhere to each other, but exhibited an uncommon bouncing behavior, probably due to significant flexibility of the dynamically reversible non-covalent interactions. In sharp contrast, 11-Co collapsed in air accompanied by a visible color change from blue to pink due to a transition from a tetrahedral complex (Td) into an octahedral (Oh) complex induced by water, as supported by UV-vis spectroscopy (Fig. 17b and e). Interestingly, combination of both systems by adding 0.5 equivalents of NiCl2·6H2O into 11-Co to prepare an air stable heterometallic gel 1-Co–Ni(0.5) caused a 5-fold enhancement of the storage modulus compared to the initial value of the pristine gel 1-Co. Strain-recovery tests of heterometallic gels prepared using different Co:Ni ratios showed that gels 11-Co–Ni(0.2) and 11-Co–Ni(0.5) were recovered within ca. 10–15 min after removal of the high oscillatory stress, whereas the gel 11-Co–Ni(0.1) recovered almost immediately (Fig. 17c). Moreover, stable gel blocks allowed the preparation of 5 cm long self-supporting gel bridges by simply rejoining 12 blocks of 11-Co–Ni(0.2) without any external stimuli (Fig. 17a). Due to the synergistic features of the constituent metal ions, visual self-healing was observed after several minutes and for multiple cycles with the wound formed in the same position.
 | ||
| Fig. 17 (a) A self-healing bridge made of heterometallic gel 11-Co–Ni(0.2). (b) The UV-vis spectra of 11-Co with increased Ni2+. (c) A loop test of 11-Co–Ni(0.1) via continuous step-stress measurements at 1 Hz. 11-Co–Ni(0.1) was subjected to 450 Pa stress for 2 min, and then again to 50 Pa for 2 min, and this process was repeated 3 times. (d) The third self-healing cycle of 11-Co–Ni(0.2) (i) after damage, (ii–v) after free standing for 20 min, 40 min, 60 min and 2 h. (e) A schematic illustration of the Ni-supported network of heterometallic gel 11-Co–Ni. Reprinted with permission from ref. 120. Copyright©2014 The Royal Society of Chemistry. | ||
Moving away from alkali and transition metals, Gunnlaugsson and co-workers121 have recently described the use of lanthanide salts (i.e. Ln = Eu3+, Tb3+) to direct the self-assembly of a LMW ligand H2L (i.e.H2L = 4,40-(((pyridine-2,6-dicarbonyl)bis(azanediyl))bis(methylene))-dibenzoic acid (Fig. 18a)) in MeOH affording different luminescent gels with self-healing properties. Although the authors carried out studies with the isolated metal complexes made in solution, they also found the exact conditions to achieve in situ gelation by adjusting the metal:ligand molar ratio, the nature of the counterion and the experimental conditions during the mixing of the reactants. 1H-NMR titrations provided evidence of the involvement of terminal free carboxylic groups during the self-assembly process. The use of a Ln(H2L)3:Ln(CH3COO)3 ratio of 1:0.5 afforded gels with a fibrillar morphology as observed by SEM imaging of the corresponding xerogels (Fig. 18b). However, these gels showed a weak nature as indicated by rheological measurements (i.e. G′ below 1.2 kPa). The self-healing properties of the Ln-based gels were visualized by cutting the gels into two pieces, which reassembled without external stimuli (Fig. 18c–f). An excess number of free carboxylic acid groups from the ligands and a high coordination number of lanthanides were proposed to facilitate the self-healing process.
 | ||
| Fig. 18 (a) Chemical structure of ligand H2L. (b) SEM image of the corresponding Eu(III)-based xerogel dried on a silicon plate (scale bar = 500 nm). Healing experiment of (c) Eu(III) gel in the day light, (d) the same gel under UV light, and (e) gel after being cut in half. (f) Self-healing properties of the gel (scale bars = 1 cm). Reprinted with permission from ref. 121. Copyright©2015 American Chemical Society. | ||
Despite the large number of fascinating supramolecular self-healing gels reported up to 2016, none of them had been proved to impart self-healing properties to a material generated upon combination with other networks. Within this context, our group122 very recently reported the first supramolecular metalloorganogel with self-healing properties and the ability to induce self-healing in other organogel networks lacking this capacity. The metallogel was prepared by mixing two equivolumetric DMF solutions of Cu(OAc)2·H2O (0.36 M) and oxalic acid dihydrate (0.66 M) at room temperature (Fig. 19a–d). The initially formed colloidal suspension was sonicated for 10 min affording a homogeneous gel (CuA-Ox) within 6 h. Previous experimental evidence demonstrated unprecedented proton conductivity for this material with a gelation mechanism involving the spontaneous formation of 1D coordination short oligomers (i.e. n < 7, [OxnCun + 1]), where Cu2+ is surrounded by four oxygen atoms of two oxalic acid molecules in a square-planar geometry.123 Further intermolecular interactions between oxygen and copper atoms as well as hydrogen bonding between oxalic acid units may also play a key role in the overall entanglement trapping the solvent molecules in the interstices of the gel network. No gelation was achieved using commercially available copper oxalate hemihydrate, suggesting that all above-mentioned components must play a synergic role during the gelation phenomenon. The so-obtained gel monoliths displayed self-supporting properties and remarkable load-bearing capacity.123 Moreover, the self-healing ability of the gel was demonstrated after cutting a gel monolith and reconnecting the pieces (Fig. 19e–j). The healing time could be reduced from 5 h to 10 min by applying 2 min sonication after reconnection of the pieces. It is important to emphasize that a self-healed CuA-Ox metalloorganogel was found to have essentially the same entwined nanofibrillar morphology (i.e. fibers width of ca. 20–50 nm), thermal stability (i.e. degradation of the coordination oligomer at ca. 300 °C), rheological properties (i.e. high G′ values of ca. 105 Pa, low frequency dependence G′ ≈ ω0.05; tanδ ≈ 0.12), and proton conductivity (i.e. derived from the Nyquist plot, σ ≈ 1.4 × 10−4 S cm−1 at 27 °C under dehumidified conditions) as the freshly prepared monolithic sample.123
 | ||
| Fig. 19 (a–d) Synthesis of 2 mL of monolithic CuA-Ox gel. (e–g) Self-supporting properties of monoliths. Rhodamine B was used as a dye to stain one monolith and for better visualization of the healing process. (h–j) Self-healing and load-bearing properties of a monolithic gel made by fusion of alternate dye doped and undoped block gels. Reprinted with permission from ref. 122. Copyright©2016 American Chemical Society. | ||
Interestingly, this gel network showed the unprecedented ability to impart self-healing properties to other physical gel networks that inherently lack this capacity. A well-known diaminocyclohexane bis(amide)-based gelator (DACBA) was used to exemplify this behavior. Two strategies were possible, either by fusing alternating gel blocks of CuA-Ox and DACBA affording a stable gel bridge after 12 h or by preparing a (DACBA)–(CuA-Ox) hybrid gel through the simultaneous formation of both gel networks (Fig. 20). The lowest DACBA:CuA-Ox ratio necessary to prepare stable and self-healable hybrid monoliths was found to be 0.8:0.2 (v/v), namely hybrid-0.8. The effective interpenetration of both networks was suggested by comparing both mechanical and thermal properties of each network before and after hybridization. In brief, the average storage moduli G′ of the gels DACBA, CuA-Ox and hybrid-0.8 were 2.1 × 104 Pa, 7.5 × 105 Pa and 4.8 × 104 Pa, respectively. Moreover, the Tgel of DACBA was established at 41 °C, whereas CuA-Ox remained stable until 150 °C and hybrid-0.8 until 120 °C. In terms of morphology, SEM imaging of the xerogel prepared from the hybrid gel revealed morphological features of both networks. It is worth mentioning that not only physical gels but also chemical gels could be hybridized with the CuA-Ox gel network to exhibit self-healing properties to some extent. This was verified using a polyelectrolyte gel based on polyvinylidene difluoride (PVDF) and NaI in DMF instead of DACBA.122
 | ||
| Fig. 20 (a and b) Preparation of a self-supporting monolithic bridge by reconnection of alternate block gels made of CuA-Ox and DACBA. (c and d) Preparation of self-supporting and self-healing hybrid-0.8 (i.e.DACBA/CuA-Ox = 0.8:0.2 v/v) by the simultaneous formation of CuA-Ox and DACBA gel networks. (e) Hybrid-0.9. (f) Hybrid-0.95. (g) Spontaneous collapse of the gel made only of DACBA. Panels (d)–(f) correspond to the material obtained after cutting/reconnecting the gel (four pieces). Total volume in each case = 2 mL. Reprinted with permission from ref. 122. Copyright©2016 American Chemical Society. | ||
Furthermore, conductive composite black gels were also prepared by dispersing different carbon nanomaterials (i.e. graphite = 25 g L−1, graphene oxide = 20 g L−1, MWNT = 12.5 g L−1, SWNT = 5 g L−1) into an oxalic acid stock solution before mixing with a stock solution of copper acetate and sonicated for 2 min. The incorporation of the carbon nanomaterials within the gel network was also confirmed by electron microscopy imaging (Fig. 21a and b). The conductivity of the composite was demonstrated by connecting one monolithic composite to an electrical circuit where a LED was lit with a DC voltage of 3.0 V (Fig. 21c–f). Repeated cutting and reconnection of the composite blocks demonstrated the self-healing ability by restoration of the conductivity.
 | ||
| Fig. 21 (a and b) SEM images of MWNTs embedded in the CuA-Ox gel matrix. (c–f) Demonstration of the restoration of bulk conductivity of the CuA-Ox/MWNTs (10 g L−1) composite gel used to bridge an electrical circuit. Adapted with permission from ref. 122. Copyright©2016 American Chemical Society. | ||
Metallogels in organic-aqueous solvent systems
In some cases, the solvent system used for the preparation of metallogels is neither pure water nor a pure organic solvent, but a combination of both. Thus, strictly speaking, these gels are neither hydrogels nor organogels regardless of the water:organic solvent ratio. In this section, we highlight self-healable metallogels formed in situ in water/organic solvent mixtures.
Alcohols have been used as fuels with high octane rating for a long time. However, the liquid nature and high volatility are serious threats for expanding the use of these fuels. Therefore, strategies to formulate liquid alcohols into gel-like materials will be highly advantageous for easy handling, delivery and safety. Within this context, naturally occurring dipicolinic acid (i.e. it composes 5% to 15% of bacterial spores) is responsible for the heat resistance of bacterial endospores. This inspired Haldar and Bera124 to pursue research aiming to convert liquid alcohols into safe fuel gels aided by alkali metals. In particular, they described the instant formation of a strong self-healable transparent gel by mixing dimethyl dipicolinate (13) with 3 equiv. of KOH in (9:1) MeOH:H2O or EtOH:H2O solvent mixtures at room temperature (Fig. 22). The observed gelation was found to be highly selective towards K+ and OH− ions as well as the above-mentioned solvent systems. Very interestingly, due to its solvent selectivity the gel can be used to separate various miscible organic solvents in a green manner.125 This was exemplified by adding dimethyl dipicolinate and aqueous KOH to a solvent mixture of EtOAc and MeOH. The gel was formed only with MeOH and precipitated out, which facilitated the separation of both solvents by decantation or simple filtration (MeOH can also be collected by distillation of the gel).
 | ||
| Fig. 22 Top: Chemical structure of ligand 13. Bottom: (a) a gel cylinder taken in hand. (b) A gel gate made by fusing three stable gel cylinders. (c) Gel cylinders supporting the weight of several grams. (d) Alternate arrangement of methyl orange-doped and undoped gel cylinders. (e) Self-healing of the gel. (f) A cylinder made from five small gel discs suspended on two glass stoppers. Dye diffusion through the entire gel monolith demonstrated the dynamic exchange of dissolved molecules across the fusion supporting the self-healing process. Adapted with permission from ref. 124. Copyright©2015 The Royal Society of Chemistry. | ||
NMR, mass spectroscopy and powder X-ray diffraction (PXRD) measurements demonstrated the absence of ester hydrolysis of the dipicolinate during gelation and a sheet-like packing arrangement with KOH presumably acting as a crosslinker for the formation of monolayers. This simple and peculiar gel featured a helical tape-like nanofibrillar network as observed by field emission SEM (FE-SEM) imaging of the corresponding xerogel (Fig. 23a). However, in contrast to most chiral gelators, both left and right-handed helices were observed in this case. Moreover, rheological experiments demonstrated the stability and viscoelastic properties of the gel (i.e. the evolution of G′ and G′′ as a function of shear stress and strain at a frequency of 10 rad s−1 showed that G′ was one order of magnitude larger than G′′ with no crossover point). It is remarkable that gels prepared at 1 wt% were stable to handle with tweezers, could be shaped into any self-supporting geometry by using appropriate molds, and could support a weight of several grams (Fig. 22c). The self-healing ability of the gels was also demonstrated by fusing several gel blocks to each other after cutting the monoliths into pieces (Fig. 22d–f). Although a thixotropic behavior was also shown during loop strain-recovery rheological experiments, the storage modulus gradually decreased after each cycle under the tested conditions (Fig. 23b).
 | ||
| Fig. 23 (a) FE-SEM image of the xerogel prepared from the corresponding gel made of KOH responsive compound 13 in 1:9 H2O:MeOH at 0.5% w/v (CGC). (b) Loop step strain experiment of the previous gel at a frequency of 10 rad s−1. Adapted with permission from ref. 124. Copyright©2015 The Royal Society of Chemistry. | ||
Due to high stability at high temperatures and the lack of thermo-reversibility the gel was tested as a potential candidate for a safer fuel. The gel made using EtOH/H2O displayed 1300 kJ mol−1 heat of combustion compared to 726 kJ mol−1 for the gel made using MeOH/H2O. Interestingly, after burning, the gels can be reused up to four times by addition of (9:1) MeOH:H2O. Unfortunately, after that the gelation efficiency decreased with each cycle due to basic hydrolysis of the ester groups at high temperatures (Fig. 24). However, as the gels are not thermoreversible, they remain in a gel phase at the time of burning and take longer times than other standard heating–cooling alcoholic gels to be consumed.
 | ||
| Fig. 24 (a) The burning methanol gel and recharge by 9:1 MeOH:H2O. (b) Efficiency of the gel after various cycles. Adapted with permission from ref. 124. Copyright©2015 The Royal Society of Chemistry. | ||
Very recently, the group of Bhattacharjee116 described self-healing metallogels with excellent selectivity for dye adsorption and separation. Opaque white metallogels were prepared by reacting the disodium salt (Na2HL) of a multidentate ligand (i.e.H3L = carboxymethyl-(3,5-di-tert-butyl-2-hydroxybenzyl)amino acetic acid), as previously described by the same group,127,128 with Cd(II) and Zn(II) halides in an aqueous methanolic solution at room temperature for 1 min. Depending on the metal salt, 4 different metallogels (MOG) were obtained (Fig. 25a). The effective gelation ratio of the gelator and metal to ensure complete gelation was found to be in the range of 1:0.6 to 1:1, with CGCs ranging from 0.8 to 1.27 (w/v). These metallogels showed thixotropic behavior as demonstrated by rheological data (Fig. 25b), and gel-to-sol transitions in response to either temperature, pH outside of the range 6–8 or addition of competitive chelating agents. With regard to the morphological features, FE-SEM imaging of the corresponding xerogels revealed 3D aggregated multisheet arrangements for MOG-1, MOG-2 and MOG-3 and a spongy-like structure for MOG-4 (Fig. 26a and b).
 | ||
| Fig. 25 (a) Synthesis of MOG. CGC (w/v) = 0.89 (MOG-1); 1.02 (MOG-2); 1.27 (MOG-3); 0.8 (MOG-4). (b) Loop step strain experiment of MOG-1 at a frequency of 10 rad s−1 showing complete recovery after each cycle. Adapted with permission from ref. 126. Copyright©2016 American Chemical Society. | ||
 | ||
| Fig. 26 (a) FESEM of MOG-1. (b) TEM of MOG-1. (c) Dye doped and undoped gel blocks. (d–f) A bridge constructed by alternate dye doped and undoped 6 MOG-1 blocks. Diffusion of the dye from doped blocks to the undoped blocks was clearly observed during the formation of the bridge. Adapted with permission from ref. 126. Copyright©2016 American Chemical Society. | ||
Although all metallogels showed good self-sustainability, MOG-1 was found to be the best allowing the construction of free-standing gel blocks in any shape with very good load-bearing capacity (i.e. 18 g cm−2 without compression). Furthermore, the self-healing ability of MOG-1 was demonstrated as usual by cutting the gel blocks into pieces and subsequently reconnection of the pieces by physical contact. In this manner, the reconstruction of a stable 2.5 cm gel bridge was achieved within 10 min (Fig. 26c–f). It is worth mentioning that besides the self-healing behavior of the gels, the corresponding porous xerogels of MOG-1 and MOG-2 were able to absorb N2 gas at 77 K which showed a type-III adsorption isotherm and exhibited a saturated sorption amount of 32.09 and 46.41 cm3 g−1, comprising a total pore volume of 4.96 × 10−2 and 7.17 × 10−2 cm3 g−1, respectively. In addition, these metallogels showed the ability to quickly and selectively remove cationic dyes from mixtures of cationic and anionic dyes (thereby the sodium ions of the ligand are replaced by the cationic dyes).
Conclusions and outlook
In this feature article, we have discussed a series of contributions dealing with the in situ fabrication of supramolecular metallogels, using LMW compounds, which show self-healing properties of the bulk gel phase after physical segregation. Most of the advances in this area within the last three years are represented by organogels, whereas examples of hydrogels and organic-aqueous gels are still a minority. The overview of these contributions should stimulate the scientific community interested in gels to look closer at the possibility of achieving gel materials with multi-stimuli responsiveness and self-healing properties by in situ metal complexation of LMW ligands. Amino acid-based ligands seem to be promising for the preparation of hydrogels, while heterocycle-containing ligands and other non-amino acid ligands with several coordination centers are more favorable for the formation of organogels. The synthetic versatility of LMW ligands may constitute also a major advantage for tuning the moldability, load-bearing and self-healing properties of in situ formed gels. The research carried out with these gels has exposed their potential as safe fuels, conductive materials and materials for environmental remediation. The highly dynamic nature of these gels has also opened an interesting avenue for imparting self-healing properties to other soft gel networks that intrinsically lack the ability to self-repair upon damage. Moreover, the in situ preparation of both monometallic and heterometallic soft gels with self-repair capacity may boost the applications of supramolecular metallogels in areas such as catalysis and drug delivery and overcome some difficulties found in more crystalline structures like metal organic frameworks (MOFs) with lower solubility and processability.129,130We foresee intense work in this emerging area to gain a deeper understanding of both chemical and physical events that govern the self-healing ability of these materials as well as a coherent design of LMW ligands with suitable structures and solubility properties in order to achieve self-healing metallogels under well-defined conditions that are otherwise difficult to prepare. In this sense, a precise description of the experimental conditions in which the gels are formed is crucial for future advances in the field, where researchers should also use or develop new characterization methods to quantify the contribution from both self-adhesion and self-healing processes to the final properties of the bulk material.
Acknowledgements
Financial support from Universität Regensburg and Deutsche Forschungsgemeinschaft (DFG 9209720) is gratefully acknowledged. D. D. D. thanks DFG for the Heisenberg Professorship Award.Notes and references
- P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133–3260 CrossRef CAS PubMed.
- R. G. Weiss and P. Terech, Molecular Gels: Materials with Self-Assembled Fibrillar Networks, Springer, New York, 2006 Search PubMed.
- M. George and R. G. Weiss, Acc. Chem. Res., 2006, 39, 489–497 CrossRef CAS PubMed.
- J. H. van Esch and B. L. Feringa, Angew. Chem., Int. Ed., 2000, 39, 2263–2266 CrossRef CAS.
- O. Gronwald and S. Shinkai, Chem. – Eur. J., 2001, 7, 4328–4334 CrossRef CAS.
- D. J. Abdallah and R. G. Weiss, Adv. Mater., 2000, 12, 1237–1247 CrossRef CAS.
- B.-K. An, J. Gierschner and S. Y. Park, Acc. Chem. Res., 2012, 44, 544–554 CrossRef PubMed.
- N. Fujita, P. Mukhopadhyay and S. Shinkai, Annu. Rev. Nano Res., 2006, 1, 385 CAS.
- A. R. Hirst, B. Escuder, J. F. Miravet and D. K. Smith, Angew. Chem., Int. Ed., 2008, 47, 8002–8018 CrossRef CAS PubMed.
- D. D. Díaz, D. Kühbeck and R. J. Koopmans, Chem. Soc. Rev., 2011, 40, 427–448 RSC.
- Y. Osada and K. Kajiwara, in Gels handbook, ed. T. Fushimi, O. Hirasa, Y. Hirokawa, T. Matsunaga, T. Shimomuraand and L. Wang and H. Ishida, Academic Press, San Diego, 2001, vol. 1–3 Search PubMed.
- J. H. Jung, J. H. Lee, J. R. Silverman and G. John, Chem. Soc. Rev., 2013, 42, 924–936 RSC.
- M. D. Segarra-Maset, V. J. Nebot, J. F. Miravet and B. Escuder, Chem. Soc. Rev., 2013, 42, 7086–7098 RSC.
- J. H. van Esch, Langmuir, 2009, 25, 8392–8394 CrossRef CAS PubMed.
- T. Tanaka, Sci. Am., 1981, 244, 110–116 CrossRef.
- D. Derossi, Y. Kajiwara and Y. Osada, Polymer Gels: Fundamentals and Biomedical Applications, Plenum Press, New York, 1991 Search PubMed.
- L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201–1218 CrossRef CAS PubMed.
- N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821–836 RSC.
- A. Ajayaghosh, V. K. Praveen and C. Vijayakumar, Chem. Soc. Rev., 2008, 37, 109–122 RSC.
- P. Xie and R. Zhang, J. Mater. Chem., 2005, 15, 2529–2550 RSC.
- D. K. Smith, Chem. Commun., 2006, 34–44 RSC.
- M. George, R. Mathew and R. G. Weiss, Mol. Gels, 2006, 449 CAS.
- T. Yi, K. Sada, K. Sugiyasu, T. Hatano and S. Shinkai, Chem. Commun., 2003, 344–345 RSC.
- J. H. Jung, Y. Ono and S. Shinkai, Angew. Chem., Int. Ed., 2000, 39, 1862–1865 CrossRef CAS.
- E. Gazit, Chem. Soc. Rev., 2007, 36, 1263–1269 RSC.
- S. M. Aharoni, in Synthesis, Characterization, and Theory of Polymeric Networks and Gels, ed. S. M. Aharoni, Plenum, New York, 1992 Search PubMed.
- E. R. Zubarev, M. U. Pralle, E. D. Sone and S. I. Stupp, Adv. Mater., 2002, 14, 198–203 CrossRef CAS.
- X. Huang, P. Terech, S. R. Raghavan and R. G. Weiss, J. Am. Chem. Soc., 2005, 127, 4336–4344 CrossRef CAS PubMed.
- F. Ilmain, T. Tanaka and E. Kokufuta, Nature, 1991, 349, 400–401 CrossRef CAS.
- Y. Osada and A. R. Khokhlov, Polymer Gels and Networks, Marcel Dekker, New York, 2002 Search PubMed.
- P. Bawa, V. Pillay, Y. E. Choonara and L. C. du Toit, Biomed. Mater., 2009, 4, 15 Search PubMed.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- M. W. Urban, Handbook of Stimuli-Responsive Materials, Wiley-VCH, Weinheim, 2011 Search PubMed.
- V. Amendola and M. Meneghetti, Nanoscale, 2009, 1, 74–88 RSC.
- P. Fratzl, J. R. Soc., Interface, 2007, 4, 637–642 CrossRef CAS PubMed.
- G. Fantner, E. Oroudjev, G. Schitter, L. Golde, P. Thurner, M. Finch, P. Turner, T. Gutsmann, D. Morse, H. Hansma and P. Hansma, Biophys. J., 2006, 90, 1411–1418 CrossRef CAS PubMed.
- J. Thompson, J. Kindt, B. Drake, H. Hansma, D. Morse and P. Hansma, Nature, 2001, 414, 773–776 CrossRef CAS PubMed.
- F. Herbst, D. Döhler, P. Michael and W. H. Binder, Macromol. Rapid Commun., 2013, 34, 203–220 CrossRef CAS PubMed.
- M. Gragert, M. Schunack and W. H. Binder, Macromol. Rapid Commun., 2011, 32, 419–425 CrossRef CAS PubMed.
- Y. Chen, A. M. Kushner, G. A. Williams and Z. Guan, Nat. Chem., 2012, 4, 467–472 CrossRef CAS PubMed.
- Q. Wei, J. Wang, X. Shen, X. A. Zhang, J. Zhi Sun, A. Qin and B. Z. Tang, Sci. Rep., 2013, 3, 1093, DOI:10.1038/srep01093.
- S. Bode, R. K. Bose, S. Matthes, M. Ehrhardt, A. Seifert, F. H. Schacher, R. M. Paulus, S. Stumpf, B. Sandmann, J. Vitz, A. Winter, S. Hoeppener, S. J. Garcia, S. Spange, S. van der Zwaag, M. D. Hager and U. S. Schubert, Polym. Chem., 2013, 4, 4966–4973 RSC.
- Y. Jin, C. Yu, R. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634–6654 RSC.
- N. Roy, B. Bruchmann and J. Lehn, Chem. Soc. Rev., 2015, 44, 3786–3807 RSC.
- Z. Wei, J. Yang, J. Zhou, F. Xu, M. Zrínyi, P. Dussault, Y. Osada and Y. Chen, Chem. Soc. Rev., 2014, 43, 8114–8131 RSC.
- X. Yu, L. Chen, M. Zhang and T. Yi, Chem. Soc. Rev., 2014, 43, 5346–5371 RSC.
- J. Cui and A. del Campo, Chem. Commun., 2012, 48, 9302–9304 RSC.
- P. Froimowicz, D. Klinger and K. Landfester, Chem. – Eur. J., 2011, 17, 12465–12475 CrossRef CAS PubMed.
- Y. Zhang, L. Tao, S. Li and Y. Wei, Biomacromolecules, 2011, 12, 2894–2901 CrossRef CAS PubMed.
- M. Nakahata, Y. Takashima, H. Yamaguchi and A. Harada, Nat. Commun., 2011, 2, 511, DOI:10.1038/ncomms1521.
- S. Roy, A. Baral and A. Banerjee, Chem. – Eur. J., 2013, 19, 14950–14957 CrossRef CAS PubMed.
- U. K. Das, S. Banerjee and P. Dastidar, Chem. – Asian J., 2014, 9, 2475–2482 CrossRef CAS PubMed.
- A. Vidyasagar, K. Handore and K. M. Sureshan, Angew. Chem., Int. Ed., 2011, 50, 8021–8024 CrossRef CAS PubMed.
- M. Zhang, D. Xu, X. Yan, J. Chen, S. Dong, B. Zheng and F. Huang, Angew. Chem., 2012, 124, 7117–7121 CrossRef.
- D. Liu, D. Wang, M. Wang, Y. Zheng, K. Koynov, G. K. Auernhammer, H.-J. Butt and T. i. Ikeda, Macromolecules, 2013, 46, 4617–4625 CrossRef CAS.
- G. Akay, A. Hassan-Raeisi, D. C. Tuncaboylu, N. Orakdogen, S. Abdurrahmanoglu, W. Oppermann and O. Okay, Soft Matter, 2013, 9, 2254–2261 RSC.
- G. Deng, F. Li, H. Yu, F. Liu, C. Liu, W. Sun, H. Jiang and Y. Chen, ACS Macro Lett., 2012, 1, 275–279 CrossRef CAS.
- J. M. García, G. O. Jones, K. Virwani, B. D. McCloskey, D. J. Boday, G. M. ter Huurne, H. W. Horn, D. J. Coady, A. M. Bintaleb, A. M. S. Alabdulrahman, F. Alsewailem, H. A. A. Almegren and J. L. Hedrick, Science, 2014, 344, 732–735 CrossRef PubMed.
- A. B. South and L. A. Lyon, Angew. Chem., 2010, 122, 779–783 CrossRef.
- T. Kakuta, Y. Takashima, M. Nakahata, M. Otsubo, H. Yamaguchi and A. Harada, Adv. Mater., 2013, 25, 2849–2853 CrossRef CAS PubMed.
- M. H. Filby and J. W. Steed, Coord. Chem. Rev., 2006, 250, 3200–3218 CrossRef CAS.
- J. Zhang and C. Y. Su, Coord. Chem. Rev., 2013, 257, 1373–1408 CrossRef CAS.
- F. Fages, Angew. Chem., Int. Ed., 2006, 45, 1680–1682 CrossRef CAS PubMed.
- M.-O. M. Piepenbrock, G. O. Lloyd, N. Clarke and J. W. Steed, Chem. Rev., 2010, 110, 1960–2004 CrossRef CAS PubMed.
- K. Murata, M. Aoki, T. Nishi, A. Ikeda and S. Shinkai, Chem. Commun., 1991, 1715–1718 RSC.
- A. Y.-Y. Tam and V. W.-W. Yam, Chem. Soc. Rev., 2013, 42, 1540–1567 RSC.
- K. Murata, M. Aoki, T. Suzuki, T. Harada, H. Kawabata, T. Komori, F. Ohseto, K. Ueda and S. Shinkai, J. Am. Chem. Soc., 1994, 116, 6664–6676 CrossRef CAS.
- Q. Lin, T.-T. Lu, X. Zhu, T.-B. Wei, H. Li and Y.-M. Zhang, Chem. Sci., 2016, 7, 5341–5346 RSC.
- Q. Lin, T.-T. Lu, X. Zhu, B. Sun, Q.-P. Yang, T.-B. Wei and Y.-M. Zhang, Chem. Commun., 2015, 51, 1635–1638 RSC.
- Q. Lin, B. Sun, Q.-P. Yang, Y.-P. Fu, X. Zhu, T.-B. Wei and Y.-M. Zhang, Chem. – Eur. J., 2014, 20, 11457–11462 CrossRef CAS PubMed.
- A. Kishimura, T. Yamashita and T. Aida, J. Am. Chem. Soc., 2005, 127, 179–183 CrossRef CAS PubMed.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed.
- J. H. Lee, S. Kang, J. Y. Lee, J. Jaworski and J. H. Jung, Chem. – Eur. J., 2013, 19, 16665–16671 CrossRef CAS PubMed.
- R. Afrasiabi and H. B. Kraatz, Chem. – Eur. J., 2013, 19, 17296–17300 CrossRef CAS PubMed.
- A. Y.-Y. Tam, K. M.-C. Wong and V. W.-W. Yam, J. Am. Chem. Soc., 2009, 131, 6253–6260 CrossRef CAS PubMed.
- S. Bhowmik, B. N. Ghosh, V. Marjomki and K. Rissanen, J. Am. Chem. Soc., 2014, 136, 5543–5546 CrossRef CAS PubMed.
- W. Bai and D. A. Spivak, Angew. Chem., Int. Ed., 2014, 53, 2095–2098 CrossRef CAS PubMed.
- H. B. Aiyappa, S. Saha, P. Wadge, R. Banerjee and S. Kurungot, Chem. Sci., 2015, 6, 603–607 RSC.
- S. Saha, G. Das, J. Thote and R. Banerjee, J. Am. Chem. Soc., 2014, 136, 14845–14851 CrossRef CAS PubMed.
- R. Banerjee, Cryst. Growth Des., 2014, 14, 3434–3437 Search PubMed.
- S. Sarkar, S. Dutta, S. Chakrabarti, P. Bairi and T. Pal, ACS Appl. Mater. Interfaces, 2014, 6, 6308–6316 CAS.
- A. E. Rowan, M. C. Feiters and R. J. M. Nolte, Chem. Commun., 1997, 545–546 Search PubMed.
- Q. Wei and S. L. James, Chem. Commun., 2005, 1555–1556 RSC.
- M. Shirakawa, S. Kawano, N. Fujita, K. Sada and S. Shinkai, J. Org. Chem., 2003, 68, 5037–5044 CrossRef CAS PubMed.
- M. George, G. P. Funkhouser, P. Terech and R. G. Weiss, Langmuir, 2006, 22, 7885–7893 CrossRef CAS PubMed.
- B. Xing, M.-F. Choi and B. Xu, Chem. – Eur. J., 2002, 8, 5028–5032 CrossRef CAS.
- P. Terech, G. Gebel and R. Ramasseul, Langmuir, 1996, 12, 4321–4323 CrossRef CAS.
- N. Fujita, Y. Sakamoto, M. Shirakawa, M. Ojima, A. Fujii, M. Ozaki and S. Shinkai, J. Am. Chem. Soc., 2007, 129, 4134–4135 CrossRef CAS PubMed.
- P. Sutar and T. K. Maji, Chem. Commun., 2016, 52, 8055–8074 RSC.
- A. Phadke, C. Zhang, B. Arman, C.-C. Hsu, R. A. Mashelkar, A. K. Lele, M. J. Tauber, G. Arya and S. Varghese, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4383–4388 CrossRef CAS PubMed.
- Y. Shi, M. Wang, C. Ma, Y. Wang, X. Li and G. Yu, Nano Lett., 2015, 15, 6276–6281 CrossRef CAS PubMed.
- Z. Shafiq, J. Cui, L. Pastor-Pérez, V. San Miguel, R. A. Gropeanu, C. Serrano and A. del Campo, Angew. Chem., Int. Ed., 2012, 51, 4332–4335 CrossRef CAS PubMed.
- M. Krogsgaard, M. A. Behrens, J. S. Pedersen and H. Birkedal, Biomacromolecules, 2013, 14, 297–301 CrossRef CAS PubMed.
- M. Guvendiren, P. B. Messersmith and K. R. Shull, Biomacromolecules, 2008, 9, 122–128 CrossRef CAS PubMed.
- N. Holten-Andersena, M. J. Harrington, H. Birkedal, B. P. Lee, P. B. Messersmith, K. Y. C. Lee and J. H. Waite, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2651–2655 CrossRef PubMed.
- T.-A. Asoh, H. Yoshitake, Y. Takano and A. Kikuchi, Macromol. Chem. Phys., 2013, 214, 2534–2539 CrossRef CAS.
- F. R. Kersey, D. M. Loveless and S. L. Craig, J. R. Soc., Interface, 2007, 4, 373–380 CrossRef CAS PubMed.
- J. Yuan, X. Fang, L. Zhang, G. Hong, Y. Lin, Q. Zheng, Y. Xu, Y. Ruan, W. Weng, H. Xia and G. Chen, J. Mater. Chem., 2012, 22, 11515–11522 RSC.
- B. Yang, H. Zhang, H. Peng, Y. Xu, B. Wu, W. Weng and L. Lia, Polym. Chem., 2014, 5, 1945–1953 RSC.
- Y. Zhang, B. Yang, X. Zhang, L. Xu, L. Tao, S. Li and Y. Wei, Chem. Commun., 2012, 48, 9305–9307 RSC.
- J. Yuan, X. Fang, L. Zhang, G. Hong, Y. Lin, Q. Zheng, Y. Xu, Y. Ruan, W. Weng, H. Xia and G. Chen, J. Mater. Chem., 2012, 22, 11515–11522 RSC.
- M. Zhang, D. Xu, X. Yan, J. Chen, S. Dong, B. Zheng and F. Huang, Angew. Chem., Int. Ed., 2012, 124, 7117–7121 CrossRef.
- S. Varguese, A. Lele and R. Mahelkar, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 666–670 CrossRef.
- T. Sato, M. Ebara, S. Tanaka, T.-A. Asoh, A. Kikuchi and T. Aoyagi, Phys. Chem. Chem. Phys., 2013, 15, 10628–10635 RSC.
- W. Fang, X. Liu, Z. Lu and T. Tu, Chem. Commun., 2014, 50, 3313–3316 RSC.
- W. Fang, Z. Sun and T. Tu, J. Phys. Chem. C, 2013, 117, 25185–25194 CAS.
- A. Dey, S. K. Mandal and K. Biradha, CrystEngComm, 2013, 15, 9769–9778 RSC.
- T. D. Hamilton, D.-K. Bučar, J. Baltrusaitis, D. R. Flanagan, Y. Li, S. Ghorai, A. V. Tivanski and L. R. MacGillivray, J. Am. Chem. Soc., 2011, 133, 3365–3371 CrossRef CAS PubMed.
- S. Basak, J. Nanda and A. Banerjee, Chem. Commun., 2014, 50, 2356–2359 RSC.
- Z. Sun, Q. Huang, T. He, Z. Li, Y. Zhang and L. Yi, ChemPhysChem, 2014, 15, 2421–2430 CrossRef CAS PubMed.
- S. Saha, J. Bachl, T. Kundu, D. D. Díaz and R. Banerjee, Chem. Commun., 2014, 50, 3004–3006 RSC.
- J.-S. Shen, G.-J. Mao, Y.-H. Zhou, Y.-B. Jiang and H.-W. Zhang, Dalton Trans., 2010, 39, 7054–7058 RSC.
- S. Samai and K. Biradha, Chem. Mater., 2012, 24, 1165–1173 CrossRef CAS.
- U. S. Schubert and C. Eschbaumer, Angew. Chem., Int. Ed., 2002, 41, 2892–2926 CrossRef CAS.
- P. Terech, M. Yan, M. Maréchal, G. Royal, J. Galvez and S. K. P. Velu, Phys. Chem. Chem. Phys., 2013, 15, 7338–7344 RSC.
- X. Yan, D. Xu, X. Chi, J. Chen, S. Dong, X. Ding, Y. Yu and F. Huang, Adv. Mater., 2012, 24, 362–369 CrossRef CAS PubMed.
- H. Ibach, Surf. Sci. Rep., 1997, 29, 195–263 CrossRef.
- L. Yan, S. Gou, Z. Ye, S. Zhang and L. Ma, Chem. Commun., 2014, 50, 12847–12850 RSC.
- P. Sahoo, R. Sankolli, H.-Y. Lee, S. R. Raghavan and P. Dastidar, Chem. – Eur. J., 2012, 18, 8057–8063 CrossRef CAS PubMed.
- L. Yan, L. Shen, M. Lv, W. Yu, J. Chen, S. Wang, X. Fu and Z. Ye, Chem. Commun., 2015, 51, 17627–17629 RSC.
- M. Martínez-Calvo, O. Kotova, M. E. Möbius, A. P. Bell, T. McCabe, J. J. Boland and T. Gunnlaugsson, J. Am. Chem. Soc., 2015, 137, 1983–1992 CrossRef PubMed.
- T. Feldner, M. Haring, S. Saha, J. Esquena, R. Banerjee and D. D. Díaz, Chem. Mater., 2016, 28, 3210–3217 CrossRef CAS.
- S. Saha, E.-M. Schön, C. Cativiela, D. D. Díaz and R. Banerjee, Chem. – Eur. J., 2013, 19, 9562–9568 CrossRef CAS PubMed.
- S. Bera and D. Haldar, J. Mater. Chem. A, 2016, 4, 6933–6939 CAS.
- S. Song, A. Song, L. Feng, G. Wei, S. Dong and J. Hao, ACS Appl. Mater. Interfaces, 2014, 6, 18319–18328 CAS.
- C. K. Karan and M. Bhattacharjee, ACS Appl. Mater. Interfaces, 2016, 8, 5526–5535 CAS.
- S. Khatua, H. Stoeckli-Evans, T. Harada, R. Kuroda and M. Bhattacharjee, Inorg. Chem., 2006, 45, 9619–9621 CrossRef CAS PubMed.
- S. Khatua, T. Harada, R. Kuroda and M. Bhattacharjee, Chem. Commun., 2007, 3927–3929 RSC.
- Themed Issue: Metal–organic Frameworks, Chem. Rev., 2012, 112, 673–674.
- Themed Issue: Metal–organic Frameworks, Chem. Soc Rev., 2014, 43, 5415–5418.
| This journal is © The Royal Society of Chemistry 2016 |