
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On the behaviour of biradicaloid [P(μ-NTer)]2 towards Lewis acids and bases†
Alexander
Hinz
a,
Axel
Schulz
*bc and
Alexander
Villinger
b
aDepartment of Chemistry, University of Oxford, 12 Mansfield Road, Oxford OX1 3TA, UK
bInstitut für Chemie, Universität Rostock, Albert-E-Str. 3a, 18059 Rostock, Germany. E-mail: axel.schulz@uni-rostock.de
cAbteilung Materialdesign, Leibniz-Institut für Katalyse e.V. an der Universität Rostock, Albert-Einstein-Str. 29a, 18059 Rostock, Germany
First published on 7th April 2016
Abstract
The well-known diphosphadiazane-1,3-diyl [P(μ-NTer)]2 (Ter = 2,6-bis(2,4,6-trimethyl-phenyl)-phenyl) was treated with Lewis bases such as N-heterocyclic carbenes and Lewis acids e.g. gold(I) chloride complexes. In the reaction with the Lewis base, fragmentation of the P2N2 framework was observed, yielding a salt of the type [(NHC)2P]+[(TerN)2P]− in a clean reaction. The reaction of [P(μ-NTer)]2 with gold(I) chloride afforded 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1:2 complexes. The dinuclear complex [(ClAu)2P(μ-NTer)2P] displays a bridging P atom between both gold centers, as has been observed for P based zwitterions.
:1 and 1:2 complexes. The dinuclear complex [(ClAu)2P(μ-NTer)2P] displays a bridging P atom between both gold centers, as has been observed for P based zwitterions.
In the mid-1980s Dewar et al. investigated the mechanisms of Cope1 and Claisen rearrangements,2 as well as Diels–Alder3 reactions. For all three examples, the available experimental data, regarding both kinetic effects and regioselectivity as well as computational investigations, corroborated the existence of transient biradicaloids in the course of these chemical transformations. Dewar stated that “Biradicals are very polarizable species” as illustrated in Scheme 1.3
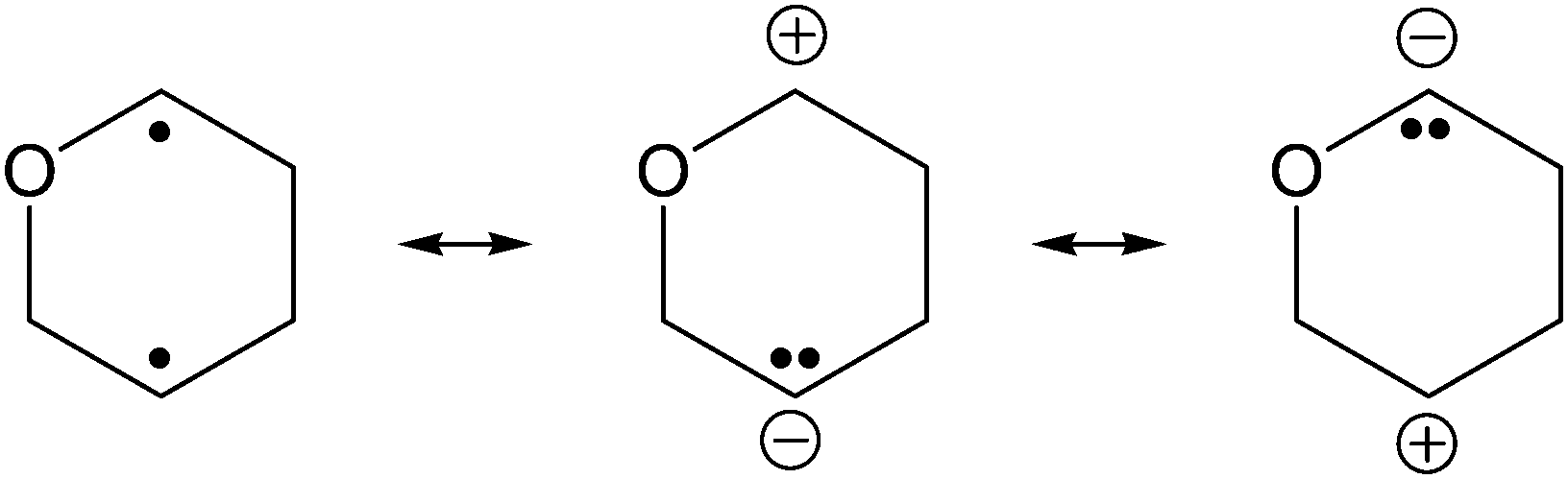 | ||
| Scheme 1 Intermediate in the Claisen rearrangement of vinyl allyl ether. | ||
To date, several examples of stable cyclic singlet biradicaloids have been known, which recently were parts of excellent reviews by F. Breher, M. Abe, and Rivard et al.4–6 The chemistry of singlet biradicaloids is rich and was investigated mainly in the examples of [ClC(μ-PMes*)]2 (Mes* = 2,4,6-tri(tertbutyl)phenyl) by the groups of Niecke7–10 and Yoshifuji11–13 and [iPr2P(μ-BtBu)]2 by Bertrand et al.14–16 More recently, the activation of small molecules17–21 and the one-electron oxidation22 of group 15 singlet biradicaloids of the type [E(μ-NTer)2E′] (E, E′ = P, As) were studied by our group. Another instance of well-investigated, but non-cyclic singlet biradicaloids are digermynes. Digermynes were studied by Power et al. who could demonstrate the high reactivity of this class of compounds in the activation of dihydrogen23 and several other small molecules like alkynes, alkenes, diazenes, azides, diazomethanes, isonitriles, nitriles, or nitrosotoluene.24–27
It is noteworthy to mention that in a recent computational study by Streubel and Frontera et al.,28 several isomers of complexes of P2N2 singlet biradicaloids were investigated, based on complexes which were experimentally obtained29 by the group of Paine in the early 1980s as a result of the first attempt of the reduction of dichlorodiphosphadiazanes [ClP(μ-NR)]2 (R = e.g.tBu).30,31 However, even though the computational study predicted several intriguing properties of the metal complexes, e.g. conservation or even an increase of the biradical character, our experimental results differ significantly. This is foremost attributed to steric congestions, as the previously investigated systems bear small substituents on the N atom (tBu as the bulkiest), and bulkier metal fragments, e.g. W(CO)5 or Mo(CO)2Cp*. In contrast, our species contain sterically very demanding terphenyl groups on the N atoms, but the metal fragment was chosen to be as small as possible. But still, the coordination of the first AuCl moiety causes the “pocket” formed by the two terphenyls to be distorted in such a way that only the P atom already bearing the AuCl fragment is available for further coordination. Thus, dinuclear complexes were obtained with a structure as observed by Ragogna et al. for zwitterionic bisphosphinoborate ligands in the complex Ph2B(μ-{CH2PiPr2}2)P(AuCl)2.32
Dewar's abovementioned statement stimulated our investigations to tackle the question if the singlet biradicaloid [P(μ-NTer)]2 (1, Ter = 2,6-bis(2,4,6-trimethylphenyl)-phenyl) could exhibit the reactivity of a Lewis acid and/or base (Scheme 2).
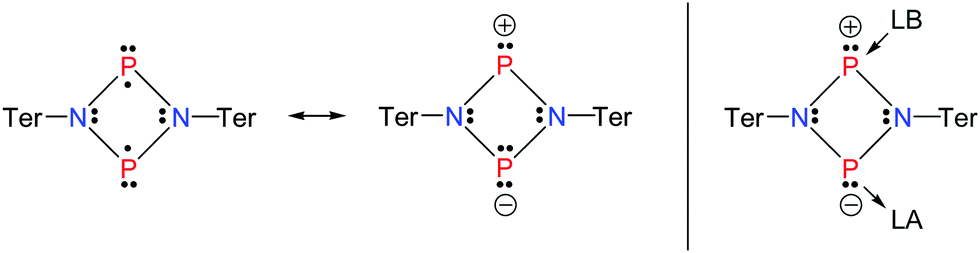 | ||
| Scheme 2 Targeted reactivity (right, LA = Lewis acid, LB = Lewis base) and resonance structures of [P(μ-NTer)]2 (1). | ||
For this purpose, a suitable Lewis base was found with the N-heterocyclic carbene (NHC), 1,3,4,5-tetramethylimidazol-2-ylidene (Scheme 3).33,34 Furthermore, for reasons of steric congestion, a linear gold metal fragment was utilized as Lewis acid, which was introduced by the addition of a dimethylsulfide gold(I) chloride complex.
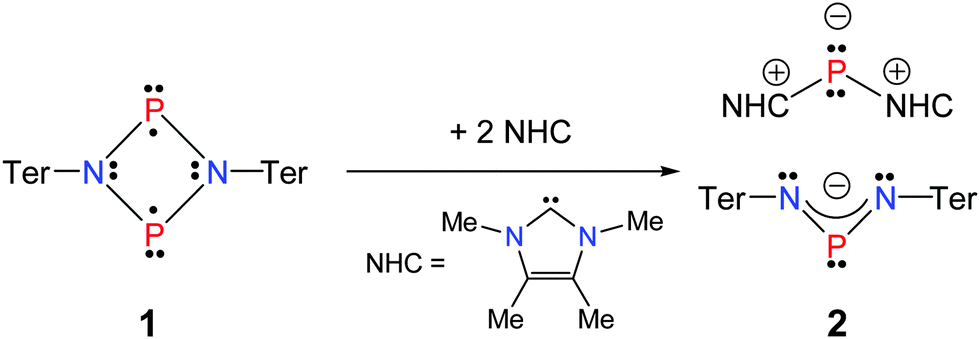 | ||
| Scheme 3 Reaction of [P(μ-NTer)]2 with a small NHC. | ||
As previously reported, the reactivity of biradicaloid 1 towards small molecules such as chalcogens, alkenes and alkynes is very high.20 As expected, similar reactivity was also observed in the reactions with the NHC 1,3,4,5-tetramethylimidazol-2-ylidene as illustrated in Scheme 3. Immediately after the addition of carbene, the orange solution turned yellow. Interestingly, the formation of the product did not depend on the stoichiometry. Initial attempts were carried out in a 1:1 stoichiometric ratio between the two starting materials. However, in these experiments according to 31P NMR data 50% of the biradicaloid remained in solution and two new species formed, exhibiting singlet resonances at +308.0 and −113.2 ppm. Changing the stoichiometric ratio to 1:2 rendered this reaction quantitative according to 31P NMR data. Single crystals of the yellow product could be obtained by concentration of the reaction mixture and undisturbed storage overnight, allowing the elucidation of the molecular structure. Therefore, the formation of a salt [(NHC)2P]+[(TerN)2P]− (2) bearing a phosphorus centred cation as well as a phosphorus centred anion became evident in accord with the 31P NMR data (Fig. 1). This type of reaction can be regarded as a redox disproportionation of 1 triggered by the Lewis base. For 1 both P atoms are in the oxidation state +II, which changes to +I in the cation and +III in the anion for 2.
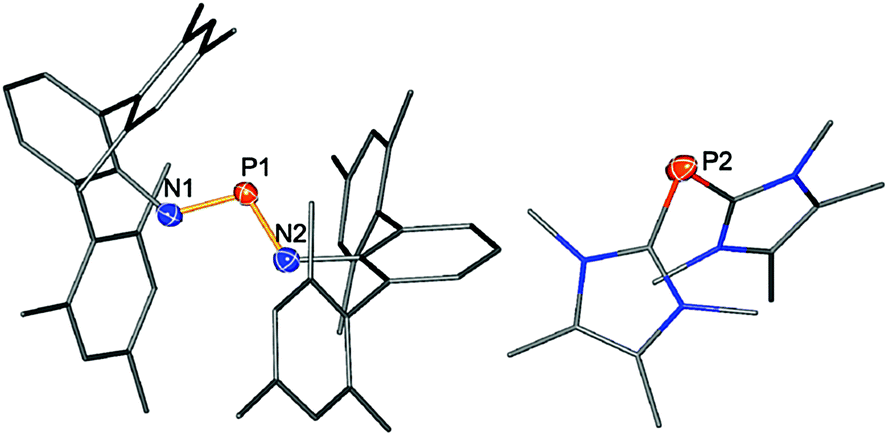 | ||
| Fig. 1 Molecular structure of 2 (left: anion, right: cation). Thermal ellipsoids are drawn at 50% probability (123 K). Selected bond lengths [Å]: P1–N1 1.586(1), P1–N2 1.593(1), P2–C49 1.790(2), P2–C56 1.801(2), N3–C49 1.365(2), N4–C49 1.354(2), N1–P1–N2 106.31(7), C49–P2–C56 96.96(7), N4–C49–N3 105.2(1), N4–C49–P2 125.4(1), N3–C49–P2 129.3(1), C49–P2–C56–N6 −143.4(1). | ||
Cations of the type [(NHC)2P]+ have been described before by the groups of Schmidpeter, Macdonald, and Bertrand.35–38 It is interesting to note that treatment of dichlorodiphosphadiazane [ClP(μ-NTer)]2 with two equivalents of the NHC resulted in a mixture of compounds, among them is the starting material as well as the PI cation [(NHC)2P]+.
The chemical shift for both components of compound 2 compares well with literature known values (cation: −93 to −127 ppm;38,39 anion: K[(TerN)2P] +322, Li[(TerN)2P] +351 ppm).19,40 The 31P NMR resonance of the [(TerN)2P]− anion with +308.0 ppm is less downfield shifted compared to those of K[(TerN)2P] and Li[(TerN)2P], which can be attributed to the absence of direct anion–cation interactions via the N atoms. Hence the shielding of the P nucleus is stronger. In accord with this, in the “free” [(TerN)2P]− anion of 2 the N–P distances are slightly shorter (averaged 1.589 in 2, cf. K[(TerN)2P] 1.595 Å−1) and the N–P–N angle with 106.31(7)° is less acute than in K[(TerN)2P] (104.88(6)°). The structure of the cation in 2 is similar to the known [(carbene)2P]+ compounds reported by Macdonald et al. and Bertrand et al.35–38
After establishing the reactivity of the singlet biradicaloid 1 towards a Lewis base, the second part of our study focused on the reaction with (Me2S)AuCl and the in situ generated Lewis acidic AuCl.
When 1 was treated with (Me2S)AuCl, depending on the stoichiometry, either the monoadduct [ClAuP(μ-NTer)2P] (3) or the diadduct [(ClAu)2P(μ-NTer)2P] (4) were isolated (Scheme 4). Obviously, both lone pairs at the phosphorus atom in the zwitterionic Lewis representation of Scheme 2 can be utilized to form either monoadduct 3 or diadduct 4 depending on the stoichiometry. These reactions have to be carried out in very dilute solutions, since 3 and 4 are only sparingly soluble in organic solvents. Therefrom a problem arises for the 1:1 reaction due to the fact that a local excess of (Me2S)AuCl causes a mixture of 3 and 4 to precipitate, leaving some of the starting material 1 in solution. Furthermore, an excess of (Me2S)AuCl has to be avoided as well, because otherwise under decomposition of complex 4, the well-known dichlorodiphosphadiazane [ClP(μ-NTer)]2 and elemental gold are formed.
 | ||
| Scheme 4 Reaction of [P(μ-NTer)]2 with (Me2S)AuCl. | ||
Both complexes 3 and 4 are characterized by a downfield-shifted AB spin system in the 31P NMR spectrum [3: 290.6 (d, JPP = 151 Hz, PAuCl), 307.3 (d, JPP = 151 Hz, NPN); 4: 286.0 (d, JPP = 26 Hz, PAuCl), 330.7 (d, JPP = 26 Hz, NPN)]. While in 3, the shifts are quite similar (Δδ = 16 ppm), the difference increases upon coordination of the second equivalent of AuCl (Δδ = 44 ppm). It is interesting to note that the coupling decreases considerably from 151 to 26 Hz upon diadduct formation.
Single crystal structure elucidation unequivocally allowed the identification of 3 and 4. In 3, the trigonal phosphorus atom is pyramidalized; however, the four-membered P2N2 ring remains almost planar (deviation from planarity <5°). A similar situation is found for the tetrahedrally coordinated P atom in 4 (deviation from planarity <4°). Upon adduct formation the planar ring skeleton distorts affording two shorter P2-coordinated–N (1.600(3), 1.645(3) Å) and two longer P3-coordinated–N bond lengths (1.776(3), 1.781(3) Å). Again, for diadduct 4 the same situation was observed with two sets of different P–N distances (Fig. 2). The Au–Cl distance amounts to 2.2361(6) Å (cf. Σrcov(Au–P) = 2.35 Å)41 which even shortens upon diadduct formation (2.228(1), 2.124(1) Å).
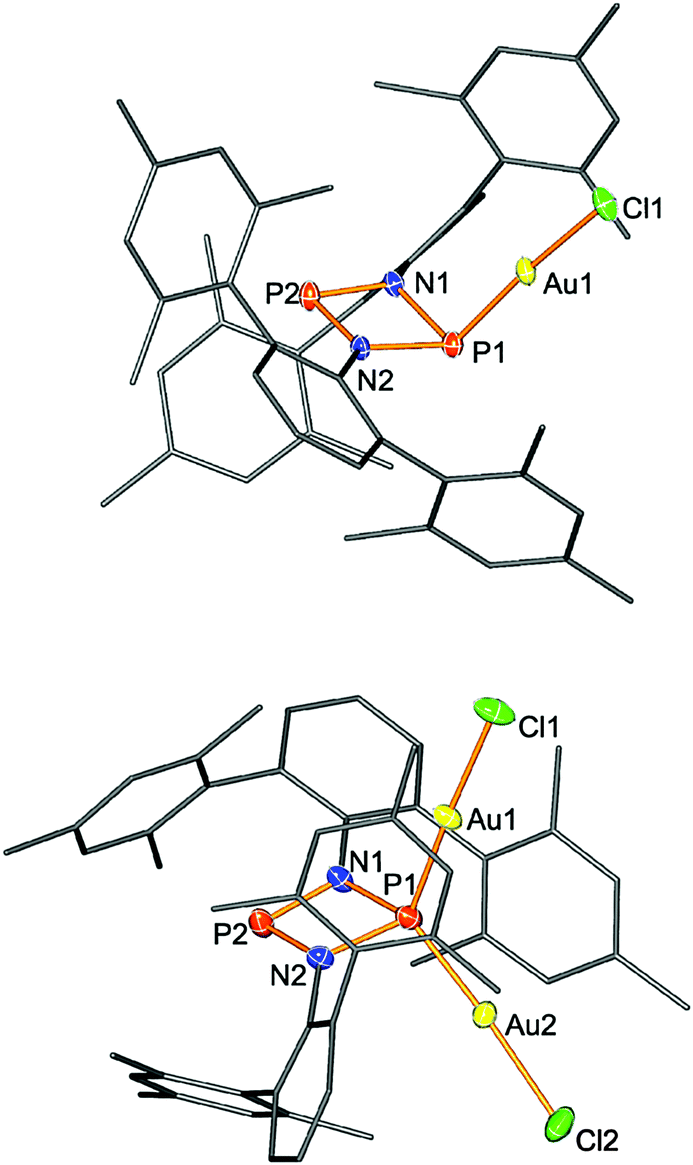 | ||
| Fig. 2 Molecular structure of 3. Thermal ellipsoids are drawn at 50% probability (123 K). Selected bond lengths [Å]: 3: Au1–P1 2.2361(6), Au1–Cl1 2.3140(6), P1–N1 1.785(2), P1–N2 1.790(2), P2–N1 1.654(2), P2–N2 1.663(2), P1–P2 2.6117(9), N1–P1–N2 77.65(9), Au1–P1–P2 114.63(3), N1–P2–N2 85.02(9). 4: P1–N1 1.776(3), P1–N2 1.781(3), P2–N2 1.645(3), P2–N1 1.660(3), P1–Au2 2.124(1), P1–Au1 2.228(1), P1–P2 2.595(2), P2–N2 1.645(3), N1–P1–N2 78.1(2), Au2–P1–Au1 118.50(5). | ||
Computations at the PBE1PBE level of theory revealed a closed shell singlet ground state for 3 and 4, respectively, in contrast to biradicaloid 1. NBO (Natural bond orbital) analysis data feature the existence of a zwitterionic resonance structure as depicted in Scheme 4 for compounds 3 and 4 with a formal oxidation state of +III for the two-coordinated phosphenium center and +I for the metal-coordinated P species.
Singlet biradicaloid [P(μ-NTer)]2 (1) was found to react with a strong Lewis base, 1,3,4,5-tetramethylimidazol-2-ylidene, to afford the salt [(NHC)2P]+[(TerN)2P]− in a clean reaction under redox disproportionation. Similarly, upon addition of a gold(I) chloride complex redox disproportionation occurred to give the zwitterionic complexes [ClAuP(μ-NTer)2P] (3) and [(ClAu)2P(μ-NTer)2P] (4). From these results it can be deduced that singlet biradicaloids such as 1 are highly polarizable species (with a considerable zwitterionic character), which can act either as Lewis base or Lewis acid and might open new vistas for all known singlet biradicaloids.
The authors thank the DFG (SCHU 1170/11-1) for financial support. Dr Christian Hering-Junghans is acknowledged for a gift of (Me2S)AuCl. MSc Jonas Bresien is gratefully acknowledged for setting up and maintaining Gaussian and NBO software on the cluster computer. A. H. thanks the GDCh for financial support.
Notes and references
- M. J. S. Dewar and E. F. Healy, Chem. Phys. Lett., 1987, 141, 521–524 CrossRef CAS.
- M. J. S. Dewar and E. F. Healy, J. Am. Chem. Soc., 1984, 106, 7127–7131 CrossRef CAS.
- M. J. S. Dewar, S. Olivella and J. J. Stewart, J. Am. Chem. Soc., 1986, 108, 5771–5779 CrossRef CAS PubMed.
- F. Breher, Coord. Chem. Rev., 2007, 251, 1007–1043 CrossRef CAS.
- M. Abe, Chem. Rev., 2013, 113, 7011–7088 CrossRef CAS PubMed.
- G. He, O. Shynkaruk, M. W. Lui and E. Rivard, Chem. Rev., 2014, 114, 7815–7880 CrossRef CAS PubMed.
- E. Niecke, A. Fuchs, F. Baumeister, M. Nieger and W. W. Schoeller, Angew. Chem., 1995, 107, 640–642 CrossRef.
- O. Schmidt, A. Fuchs, D. Gudat, M. Nieger, W. Hoffbauer, E. Niecke and W. W. Schoeller, Angew. Chem., 1998, 110, 995–998 CrossRef.
- E. Niecke, A. Fuchs, M. Nieger, O. Schmidt and W. W. Schoeller, Angew. Chem., 1999, 111, 3216–3219 CrossRef.
- M. Sebastian, M. Nieger, D. Szieberth, L. Nyulászi and E. Niecke, Angew. Chem., 2004, 116, 647–651 CrossRef.
- H. Sugiyama, S. Ito and M. Yoshifuji, Angew. Chem., 2003, 115, 3932–3934 CrossRef.
- S. Ito, M. Kikuchi, M. Yoshifuji, A. J. Arduengo, T. A. Konovalova and L. D. Kispert, Angew. Chem., Int. Ed., 2006, 45, 4341–4345 CrossRef CAS PubMed.
- H. Sugiyama, S. Ito and M. Yoshifuji, Chem. – Eur. J., 2004, 10, 2700–2706 CrossRef CAS PubMed.
- D. Scheschkewitz, H. Amii, H. Gornitzka, W. W. Schoeller, D. Bourissou and G. Bertrand, Science, 2002, 295, 1880–1882 CrossRef CAS PubMed.
- H. Amii, L. Vranicar, H. Gornitzka, D. Bourissou and G. Bertrand, J. Am. Chem. Soc., 2004, 126, 1344–1345 CrossRef CAS PubMed.
- G. Fuks, N. Saffon, L. Maron, G. Bertrand and D. Bourissou, J. Am. Chem. Soc., 2009, 131, 13681–13689 CrossRef CAS PubMed.
- T. Beweries, R. Kuzora, U. Rosenthal, A. Schulz and A. Villinger, Angew. Chem., Int. Ed., 2011, 50, 8974–8978 CrossRef CAS PubMed.
- S. Demeshko, C. Godemann, R. Kuzora, A. Schulz and A. Villinger, Angew. Chem., 2013, 125, 2159–2162 CrossRef.
- A. Hinz, A. Schulz and A. Villinger, Angew. Chem., Int. Ed., 2014, 54, 668–672 Search PubMed.
- A. Hinz, R. Kuzora, U. Rosenthal, A. Schulz and A. Villinger, Chem. – Eur. J., 2014, 20, 14659–14673 CrossRef CAS PubMed.
- A. Hinz, A. Schulz, W. W. Seidel and A. Villinger, Inorg. Chem., 2014, 53, 11682–11690 CrossRef CAS PubMed.
- A. Brückner, A. Hinz, J. B. Priebe, A. Schulz and A. Villinger, Angew. Chem., 2015, 127, 7534–7538 CrossRef.
- G. H. Spikes, J. C. Fettinger and P. P. Power, J. Am. Chem. Soc., 2005, 127, 12232–12233 CrossRef CAS PubMed.
- P. P. Power, Organometallics, 2007, 26, 4362–4372 CrossRef CAS.
- Z. D. Brown and P. P. Power, Inorg. Chem., 2013, 52, 6248–6259 CrossRef CAS PubMed.
- X. Wang, Y. Peng, M. M. Olmstead, J. C. Fettinger and P. P. Power, J. Am. Chem. Soc., 2009, 131, 14164–14165 CrossRef CAS PubMed.
- X. Wang, Y. Peng, Z. Zhu, J. C. Fettinger, P. P. Power, J. Guo and S. Nagase, Angew. Chem., Int. Ed., 2010, 49, 4593–4597 CrossRef CAS PubMed.
- A. Bauzá, D. Escudero, A. Frontera and R. Streubel, Organometallics, 2015, 34, 355–360 CrossRef.
- D. A. DuBois, E. N. Duesler and R. T. Paine, Organometallics, 1983, 2, 1903–1905 CrossRef CAS.
- D. A. DuBois, E. N. Duesler and R. T. Paine, Organometallics, 1984, 3, 1913–1915 CrossRef CAS.
- D. A. DuBois, E. N. Duesler and R. T. Paine, Inorg. Chem., 1985, 24, 3–5 CrossRef CAS.
- J. W. Dube, C. L. B. Macdonald and P. J. Ragogna, Angew. Chem., Int. Ed., 2012, 51, 13026–13030 CrossRef CAS PubMed.
- A. J. Arduengo, H. V. R. Dias, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1992, 114, 5530–5534 CrossRef CAS.
- N. Kuhn and T. Kratz, Synthesis, 1993, 561–562 CrossRef CAS.
- B. D. Ellis, C. A. Dyker, A. Decken and C. L. B. Macdonald, Chem. Commun., 2005, 1965–1967 RSC.
- B. D. Ellis and C. L. B. Macdonald, Coord. Chem. Rev., 2007, 251, 936–973 CrossRef CAS.
- O. Back, G. Kuchenbeiser, B. Donnadieu and G. Bertrand, Angew. Chem., 2009, 121, 5638–5641 CrossRef.
- J. F. Binder, A. Swidan, M. Tang, J. H. Nguyen and C. L. B. Macdonald, Chem. Commun., 2015, 51, 7741–7744 RSC.
- A. Schmidpeter, S. Lochschmidt and A. Willhalm, Angew. Chem., Int. Ed., 1983, 22, 545–546 CrossRef.
- A. Hinz, J. Rothe, A. Schulz and A. Villinger, Dalton Trans., 2016, 45, 6044–6052 RSC.
- P. Pyykkö and M. Atsumi, Chem. – Eur. J., 2009, 15, 186–197 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational details and information on X-ray structure elucidation. CCDC 1457705–1457708. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc01935h |
| This journal is © The Royal Society of Chemistry 2016 |