
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dioxazoles, a new mild nitrene transfer reagent in gold catalysis: highly efficient synthesis of functionalized oxazoles†
Ming
Chen‡
,
Ning
Sun‡
,
Haoyi
Chen
and
Yuanhong
Liu
*
State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: yhliu@sioc.ac.cn
First published on 4th April 2016
Abstract
A gold-catalyzed regioselective [3+2] cycloaddition of ynamides with 1,4,2-dioxazoles was developed and offers a novel approach to obtain highly functionalized oxazoles under mild reaction conditions. 1,4,2-Dioxazole was found to act as an efficient N-acyl nitrene equivalent to trigger a facile generation of α-imino gold–carbene intermediate through the elimination of a ketone.
In recent years, gold–carbene-mediated reactions have attracted considerable attention since they serve as promising intermediates in the synthesis of various types of carbo- or heterocycles.1 Compared with α-carbonyl gold carbenes,2 the generation and reactions of α-imino gold carbenes have been less explored.3 These highly reactive gold-species are mainly accessed through gold-catalyzed nitrene transfer to alkynes using azides as the nitrene equivalent, as reported by Toste,4a Gagosz,4b Zhang4c–e and others.4 Recently, 2H-azirines,5N-iminopyridium ylides,6 isoxazoles,7 benzoisoxazoles8 and triazapentalene9 have also been used as nitrene equivalents. Despite the impressive progress made so far, the development of new methods for the generation of α-imino gold carbenes involving the utilization of less reactive/sensitive nitrene transfer reagents with high chemo- and regioselectivities under milder reaction conditions is still highly desired. 1,4,2-Dioxazol-5-one a, a cyclic carbonate of hydroxamic acids, and its derivative 1,4,2-dioxazol-5-thione b, were found in 1968 to undergo thermal or photo-induced decomposition leading to highly reactive N-acyl nitrene intermediates via the elimination of CO2 or SO2.10 1,4,2-Dioxazole c decomposed similarly at elevated temperatures (above 150 °C) into isocyanates and ketones.11 These attractive and easily accessible heterocyclic compounds are potentially useful as N-acyl nitrene precursors in place of hazardous acyl azides, and could produce the N-acyl nitrene or N-acyl nitrenoid intermediates under mild reaction conditions, such as in the presence of a metal catalyst. Recently, Bolm et al. described an elegant light-induced ruthenium-catalyzed synthesis of N-acyl sulfoximines and sulfimides at room temperature via a ruthenium N-acyl nitrene intermediate using dioxazolone a as the nitrene precursor.12 More recently, Chang and others13 revealed that the substrates a–c could also be used as amidating reagents in metal-catalyzed C–H amidation reactions, in which a metal–nitrene complex is proposed to be involved (Scheme 1). During our continuous work on gold-catalyzed oxidative reactions, we hypothesized that these five-membered heterocycles could be employed as a nucleophilic nitrene equivalent to trigger an efficient generation of α-imine gold–carbene species through nucleophilic attack of the gold-activated alkyne followed by expulsion of a leaving group. In this design, no metal–nitrene complex is formed, which is different from the other metal-catalyzed reactions shown above. Herein, we describe a novel reaction of dioxazole derivatives, which act as a new type of nitrene transfer reagents and undergo gold-catalyzed [3+2] cycloaddition with ynamides, leading to a facile synthesis of highly functionalized oxazoles.6b–d
 | ||
| Scheme 1 Metal-catalyzed reactions involving nitrene equivalents of 1,4-2-dioxazolone derivatives and the design of gold-catalyzed nitrene transfer reactions. | ||
To test our hypothesis, we initially investigated the reactions of mesylamide-derived ynamide 1a with three different types of dioxazole derivatives 2a–2c in the presence of 5 mol% Johnphos(MeCN)AuSbF6 (catalyst A) in DCE at room temperature. However, in the case of dioxazolone 2a, a non-clean reaction mixture resulted with significant remaining 2a, possibly as the rapid self-reaction of ynamide had occurred under gold-catalyzed conditions.14 No desired cyclization product was observed also in the case of dioxathiazole 2b (Table 1, entries 1 and 2). Considering the lower nucleophilicity of 2a and 2b, we reasoned that employing more nucleophilic dioxazole might be feasible for the successful transformation. Gratifyingly, employing dioxazole 2c led to the desired 4-amino-oxazole 3a in a 92% yield within 2 h (entry 3). The results implied that an efficient [3+2] cycloaddition of ynamide with dioxazole had taken place, and that the self-reaction of ynamide was mostly suppressed. A similar reaction outcome was found when N-heterocyclic carbene gold(I) complex B (IPrAu(MeCN)SbF6) or C (IPrAuNTf2)15 was used as the catalyst (IPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene) (entries 4 and 5). Various commonly used gold catalysts also catalyze the desired cyclization efficiently, furnishing 3a in lower yields of 72–87% (entries 6–8). The reaction could also be performed smoothly in the DCM, THF, toluene or CH3CN solvents (entries 9–12). No reaction was observed catalyzed by IPrAuCl or AgNTf2 alone or in the absence of any catalyst (entries 13–15).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Catalyst | Solvent | Time (h) | Yielda (%) |
| a Isolated yields. Ms = methanesulfonyl. The yields of recovered 1a are shown in parentheses. | |||||
| 1 | 2a | A | DCE | 3 | — |
| 2 | 2b | A | DCE | 3 | — |
| 3 | 2c | A | DCE | 2 | 92 |
| 4 | 2c | B | DCE | 2 | 90 |
| 5 | 2c | C | DCE | 2 | 95 |
| 6 | 2c | PPh3AuNTf2 | DCE | 2 | 85 |
| 7 | 2c | PPh3AuSbF6 | DCE | 3 | 87 |
| 8 | 2c | PPh3AuOTf | DCE | 3 | 72 |
| 9 | 2c | C | DCM | 3 | 91 |
| 10 | 2c | C | THF | 3 | 86 |
| 11 | 2c | C | Toluene | 3 | 88 |
| 12 | 2c | C | MeCN | 3 | 92 |
| 13 | 2c | IPrAuCl | DCE | 3 | −(99) |
| 14 | 2c | AgNTf2 | DCE | 3 | −(98) |
| 15 | 2c | None | DCE | 12 | −(99) |

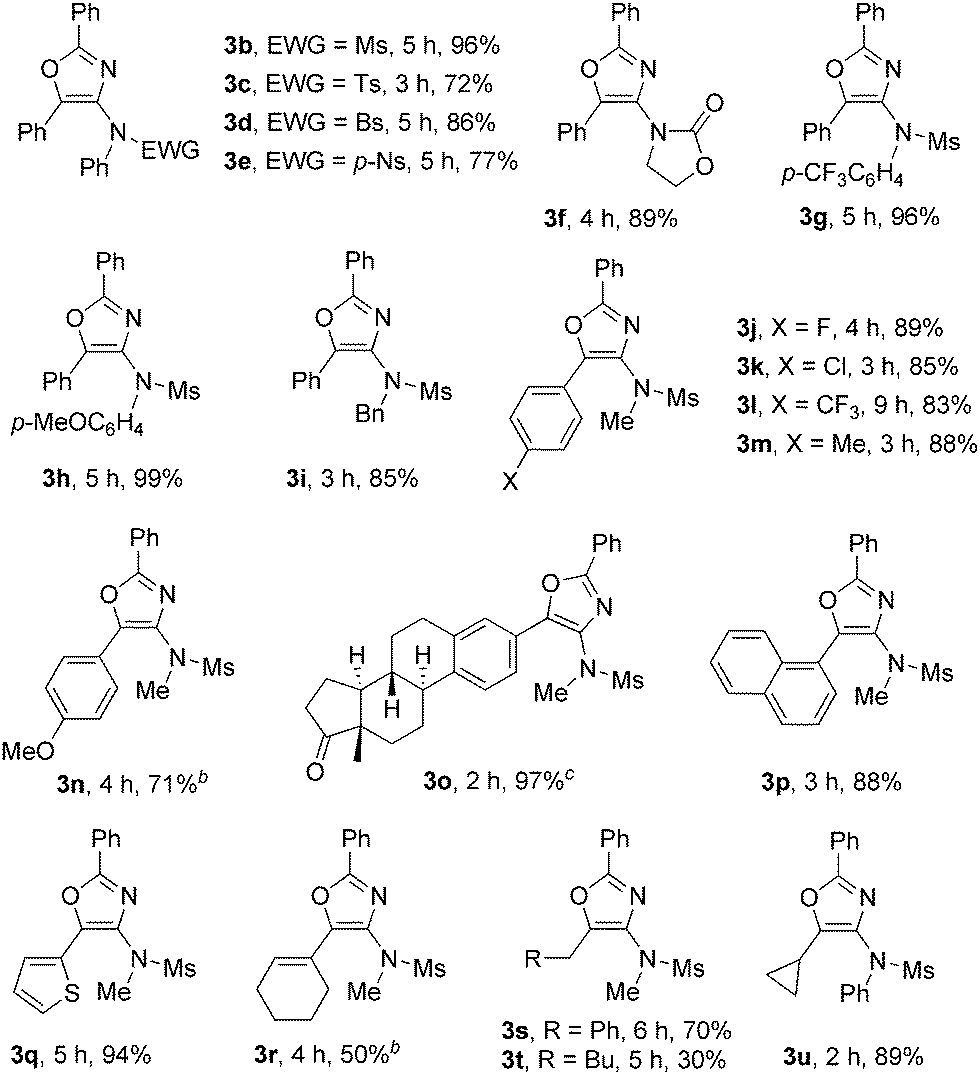
Encouraged by these results, we next investigated the substrate scope of the reaction. The scope of ynamides was first investigated using dioxazole 2c as the reaction partner under the reaction conditions given in Table 1, entry 5. The results are shown in Table 2. The effects of the electron-withdrawing groups on nitrogen were first examined. The reactions proceeded very well with tosyl, para-bromobenzenesulfonyl (Bs) and a stronger electron-withdrawing para-nitrobenzenesulfonyl (p-Ns) moiety, furnishing 3c–3e in 72–86% yields. The more electron-rich ynamide with an oxazolidine group also afforded the corresponding oxazole 3f in an 89% yield. N-Aryl mesylamide, whenever bearing an electron-neutral, electron-deficient CF3, or electron-rich MeO substituent on its aromatic ring, was tolerated well in this reaction, leading to 3b and 3g–3h in 96–99% yields. N-Benzyl mesylamide was also suitable, providing 3i in an 85% yield. Next, the effect of the R1 group on the alkyne terminus was examined. For aryl substituted alkynes, a wide range of functionalities, such as F, Cl, CF3, Me and MeO, on aromatic rings were compatible, furnishing 3j–3n in good to high yields. It was noted that when p-MeO-substituted aryl alkyne was used, part of the product precipitated during the reaction process at room temperature, which appeared to interfere with the reaction process. A higher reaction temperature (50 °C) was then required to achieve a better conversion. Ynamide with an 1,3,5(10)-estratrien-3-ol-17-one derivative also reacted efficiently to produce the oxazole 3o in a 97% yield. 1-Naphthyl and 2-thienyl-substituted alkynes converted into the corresponding 3p and 3q in excellent yields. Cyclohexenyl-substituted alkyne transformed to the corresponding 3r in a moderate yield. Alkyl-substituted alkynes, such as benzyl- or cyclopropyl-substituted alkynes, underwent the reaction smoothly to obtain 3s and 3u in 70% and 89% yields, respectively. However, a pentyl-substituted ynamide afforded 3t only in a 30% yield. No alkene product derived from 1,2-C–H insertion of the gold–carbene intermediate was observed in these cases. The results indicated that intramolecular nucleophilic attack of the N-acyl group to gold–carbene is much faster than 1,2-C–H insertion due to the ease of aromatization.

The scope of dioxazoles was also investigated using ynamide 1a as the reaction partner (Table 3). Due to the lower solubility of the products in DCE, all the reactions were carried out at 80 °C. Under this reaction condition, we were pleased to see that the reactions were quite general with the substituted dioxazoles, since aryl, heteroaryl and alkenyl as well as an alkyl-substituted one were all suitable for this reaction, leading to the highly functionalized oxazoles in good to excellent yields. The reaction efficiency was affected by the nature of aryl substituents: p-FC6H4 (3v, 90%), p-ClC6H4 (3w, 93%), p-CF3C6H4 (3x, 77%), p-MeOC6H4 (3y, 92%). Sterically encumbered o-Me-substituted aryl dioxazole reacted efficiently to afford 3z in a 90% yield, suggesting that the steric hindrance had little effect on the reaction course. Heteroaryl-substituted dioxazoles, such as pyridyl, furanyl and a thienyl-substituted dioxazoles, transformed to 3za–3zc successfully in 75–95% yields. High product yields were also observed in 2-naphthyl or alkenyl-substituted dioxazoles.
|
|
|---|
| a Isolated yields. |

|

Alkyl-substituted dioxazoles, such as methyl, cyclohexyl or even bulky adamantyl, turned out to be also perfect substrates to afford 3zf–3zh in 71–96% yields. It was noted that in the case of 3zf, dioxazole 2n was used instead of 3,5,5-trimethyl-1,4,2-dioxazole since it is not convenient to prepare the latter with a lower boiling point. Oxazoles constitute important classes of natural products, drugs and biologically active substances. These compounds are commonly prepared by cyclization of an acyclic precursor or ring derivatization. However, the construction of oxazoles through convergent and one-pot methods from readily available substrates is still limited.16 Our method provided a mild and efficient route to these compounds.
To demonstrate the practicality of our method, a gram scale reaction was performed. It was found that by using only 2 mol% of IPrAuNTf2, the reaction of 1a with dioxazole 2c at 5 mmol scale delivered oxazole 3a in a high yield of 89% (Scheme 2).
 | ||
| Scheme 2 Gram scale synthesis of 3a. | ||
The reaction can be extended to other activated alkynes. As shown in Scheme 3, gold-catalyzed reactions of alkynyl ester 4 or alkynyl ketone 5 with 2c afforded the functionalized oxazoles 6 or 7 in 51% and 50% yields, respectively. However, when a terminal alkyne, such as phenylacetylene, was used, no clean reaction was observed.
 | ||
| Scheme 3 Reactions of dioxazole with activated alkynes. | ||
We propose the following reaction mechanism for this novel transformation (Scheme 4). Initially, dioxazole 2 attacks the gold-coordinated ynamide 8 or 8′ regioselectively at the carbon adjacent to the nitrogen due to the polarity of the ynamide to afford the iminium ion intermediate 10. Subsequently, the ring fragmentation of 10 generates α-imino gold–carbene 11 with the concomitant elimination of acetone. In fact, acetone was formed quantitatively and could be detected in the crude reaction mixture.17 Intermediate 11 may prefer an E-form of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond due to the steric repulsion with the R3 substituent on dioxazole with the amino moiety,6a resulting in a cis orientation of an N-acyl group with gold–carbene. Nucleophilic attack of the acyl oxygen in 11 to gold–carbene18 is followed by elimination of the gold catalyst, leading to the oxazole products 3. The reaction pathway involving the formation of N-acylaziridine via gold–nitrene followed by cyclization is unlikely, since an oxazole with a different regioselectivity would possibly have resulted.6b,19
N bond due to the steric repulsion with the R3 substituent on dioxazole with the amino moiety,6a resulting in a cis orientation of an N-acyl group with gold–carbene. Nucleophilic attack of the acyl oxygen in 11 to gold–carbene18 is followed by elimination of the gold catalyst, leading to the oxazole products 3. The reaction pathway involving the formation of N-acylaziridine via gold–nitrene followed by cyclization is unlikely, since an oxazole with a different regioselectivity would possibly have resulted.6b,19
 | ||
| Scheme 4 Possible reaction mechanism. | ||
To understand the reaction mechanism, we also tried to trap the α-imino gold–carbene intermediate via an intramolecular cyclization of dioxazole-ynamide 13, since the C–O bond formation can be avoided in such a case. To our delight, 13 cyclized efficiently to give the fused indole derivative 1515 in a 70% yield (Scheme 5). The results indicated that the α-imino gold–carbene 14 was likely generated in the process, and could be trapped by the N-aryl ring, followed by deauration to furnish the cyclized product.
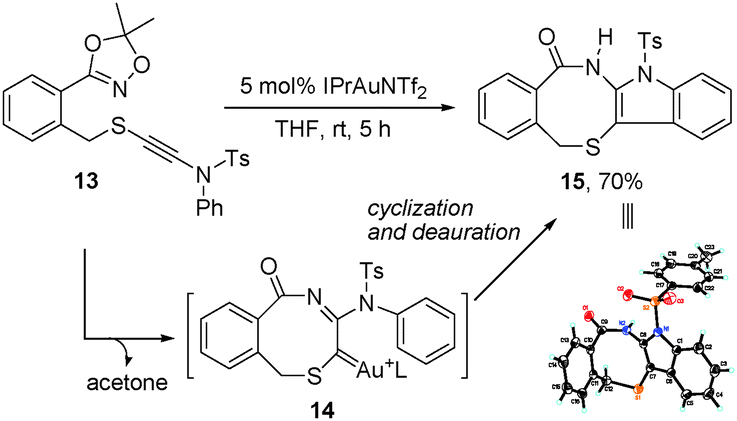 | ||
| Scheme 5 Trapping of the α-imino gold–carbene intermediate. | ||
In summary, we disclosed that 1,4,2-dioxazole can be used as an efficient nitrene equivalent in gold-catalyzed nitrene transfer reactions to ynamides. The reaction proceeds under mild reaction conditions to afford highly functionalized oxazoles in good to excellent yields likely via the formation of an α-imino gold–carbene intermediate followed by cyclization. This method offers several advantages, such as easily accessible starting materials, high regioselectivity, wide functional group compatibility and high efficiency. Further investigations on the detailed reaction mechanism and application of this chemistry are in progress.
We thank the National Natural Science Foundation of China (Grant No. 21421091, 21372244, 21572256) for financial support.
References
- For reviews, see: (a) Gold Carbenes: L. Zhang, in Contemporary Carbene Chemistry, ed. R. A. Moss and M. P. Doyle, Wiley, Hoboken, 2013, p. 526 Search PubMed; (b) Y. Wang, M. E. Muratore and A. M. Echavarren, Chem. – Eur. J., 2015, 21, 7332 CrossRef CAS PubMed.
- For reviews, see: (a) J. Xiao and X. Li, Angew. Chem., Int. Ed., 2011, 50, 7226 CrossRef CAS PubMed; (b) L. Zhang, Acc. Chem. Res., 2014, 47, 877 CrossRef CAS PubMed.
- For a review, see: P. W. Davies and M. Garzón, Asian J. Org. Chem., 2015, 4, 694 CrossRef CAS.
- (a) D. J. Gorin, N. R. Davis and F. D. Toste, J. Am. Chem. Soc., 2005, 127, 11260 CrossRef CAS PubMed; (b) A. Wetzel and F. Gagosz, Angew. Chem., Int. Ed., 2011, 50, 7354 CrossRef CAS PubMed; (c) B. Lu, Y. Luo, L. Liu, L. Ye, Y. Wang and L. Zhang, Angew. Chem., Int. Ed., 2011, 50, 8358 CrossRef CAS PubMed; (d) Y. Xiao and L. Zhang, Org. Lett., 2012, 14, 4662 CrossRef CAS PubMed; (e) Z.-Y. Yan, Y. Xiao and L. Zhang, Angew. Chem., Int. Ed., 2012, 51, 8624 CrossRef CAS PubMed; (f) Y. Tokimizu, S. Oishi, N. Fujii and H. Ohno, Org. Lett., 2014, 16, 3138 CrossRef CAS PubMed; (g) Y. Wu, L. Zhu, Y. Yu, X. Luo and X. Huang, J. Org. Chem., 2015, 80, 11407 CrossRef CAS PubMed.
- (a) A. Prechter, G. Henrion, P. Faudot dit Bel and F. Gagosz, Angew. Chem., Int. Ed., 2014, 53, 4959 CrossRef CAS PubMed; (b) L. Zhu, Y. Yu, Z. Mao and X. Huang, Org. Lett., 2015, 17, 30 CrossRef CAS PubMed.
- (a) C. Li and L. Zhang, Org. Lett., 2011, 13, 1738 CrossRef CAS PubMed; (b) P. W. Davies, A. Cremonesi and L. Dumitrescu, Angew. Chem., Int. Ed., 2011, 50, 8931 CrossRef CAS PubMed; (c) E. Chatzopoulou and P. W. Davies, Chem. Commun., 2013, 49, 8617 RSC; (d) A. D. Gillie, R. J. Redd and P. W. Davies, Adv. Synth. Catal., 2016, 358, 226 CrossRef CAS . In the reactions of ref. 6b–d, oxazoles were formed via gold-catalyzed [3+2] cycloaddition of pyridine-N-aminides with ynamides or alkynyl indoles; (e) H.-H. Hung, Y.-C. Liao and R.-S. Liu, J. Org. Chem., 2013, 78, 7970 CrossRef CAS PubMed.
- A. Zhou, Q. He, C. Shu, Y. Yu, S. Liu, T. Zhao, W. Zhang, X. Lu and L. Ye, Chem. Sci., 2015, 6, 1265 RSC.
- H. Jin, L. Huang, J. Xie, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2016, 55, 794 CrossRef CAS PubMed.
- J. González, J. Santamaría, Á. L. Suárez-Sobrino and A. Ballesterosa, Adv. Synth. Catal., 2016 DOI:10.1002/adsc.201600022.
- J. Sauer and K. K. Mayer, Tetrahedron Lett., 1968, 9, 319 CrossRef.
- H. Nouira, K. Inoue, H. Hattori, T. Okawa and T. Mukaiyama, Bull. Chem. Soc. Jpn., 1967, 40, 664 CrossRef.
- V. Bizet, L. Buglioni and C. Bolm, Angew. Chem., Int. Ed., 2014, 53, 5639 CrossRef CAS PubMed.
- (a) Y. Park, K. T. Park, J. G. Kim and S. Chang, J. Am. Chem. Soc., 2015, 137, 4534 CrossRef CAS PubMed; (b) J. Park and S. Chang, Angew. Chem., Int. Ed., 2015, 54, 14103 CrossRef CAS PubMed; (c) Y. Liang, Y. Liang, C. Tang, Y. Yuan and N. Jiao, Chem. – Eur. J., 2015, 21, 16395 CrossRef CAS PubMed.
- S. Kramer, Y. Odabachian, J. Overgaard, M. Rottländer, F. Gagosz and T. Skrydstrup, Angew. Chem., Int. Ed., 2011, 50, 5090 CrossRef CAS PubMed.
- CCDC 1455247 (IPrAuNTf2), 1450076 (3b) and 1455246 (15).
- For selected papers, see: (a) C. Wan, J. Zhang, S. Wang, J. Fan and Z. Wang, Org. Lett., 2010, 12, 2338 CrossRef CAS PubMed; (b) C. Lalli, J. M. Bouma, D. Bonne, G. Masson and J. Zhu, Chem. – Eur. J., 2011, 17, 880 CrossRef CAS PubMed; (c) W. He, C. Li and L. Zhang, J. Am. Chem. Soc., 2011, 133, 8482 CrossRef CAS PubMed; (d) Y. Luo, K. Ji, Y. Li and L. Zhang, J. Am. Chem. Soc., 2012, 134, 17412 CrossRef CAS PubMed; (e) Ref. 6b–d. For Ru/Cu-catalyzed reaction of 1,4,2-dioxazol-5-ones with terminal alkynes to oxazoles, see: ; (f) C. Zhong, B. Tang, P. Yin, Y. Chen and L. He, J. Org. Chem., 2012, 77, 4271 CrossRef CAS PubMed.
- See ESI†.
- (a) S. Kramer and T. Skrydstrup, Angew. Chem., Int. Ed., 2012, 51, 4681 CrossRef CAS PubMed; (b) X. Huang, B. Peng, M. Luparia, L. F. R. Gomes, L. F. Veiros and N. Maulide, Angew. Chem., Int. Ed., 2012, 51, 8886 CrossRef CAS PubMed.
- H. Li and R. P. Hsung, Org. Lett., 2009, 11, 4462 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1455247, 1450076 and 1455246. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc02776h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |