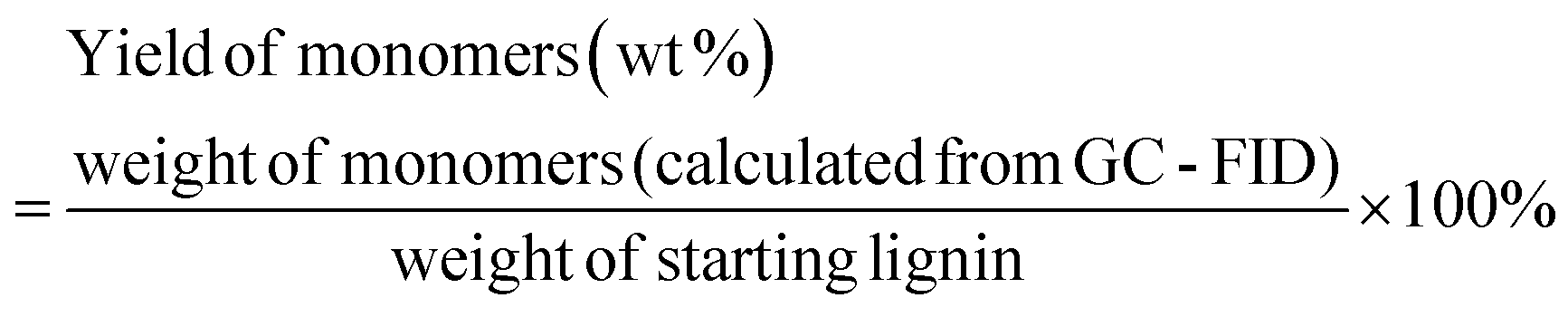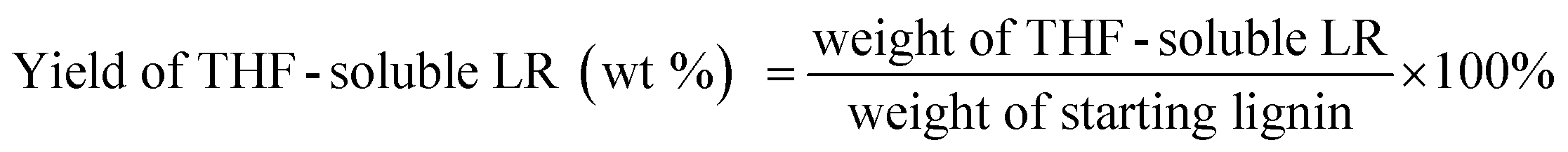DOI:
10.1039/C5GC01120E
(Paper)
Green Chem., 2015,
17, 4941-4950
Ethanol as capping agent and formaldehyde scavenger for efficient depolymerization of lignin to aromatics†
Received
25th May 2015
, Accepted 22nd June 2015
First published on 23rd June 2015
Abstract
Obtaining renewable fuels and chemicals from lignin presents an important challenge to the use of lignocellulosic biomass to meet sustainability and energy goals. We report on a thermocatalytic process for the depolymerization of lignin in supercritical ethanol over a CuMgAlOx catalyst. Ethanol as solvent results in much higher monomer yields than methanol. In contrast to methanol, ethanol acts as a scavenger of formaldehyde derived from lignin decomposition. Studies with phenol and alkylated phenols evidence the critical role of the phenolic –OH groups and formaldehyde in undesired repolymerization reactions. O-alkylation and C-alkylation capping reactions with ethanol hinder repolymerization of the phenolic monomers formed during lignin disassembly. After reaction in ethanol at 380 °C for 8 h, this process delivers high yields of mainly alkylated mono-aromatics (60–86 wt%, depending on the lignin used) with a significant degree of deoxygenation. The oxygen-free aromatics can be used to replace reformate or can serve as base aromatic chemicals; the oxygenated aromatics can be used as low-sooting diesel fuel additives and as building blocks for polymers.
Introduction
The utilization of biomass as a renewable source of energy and chemicals requires significant technological breakthroughs.1 With cellulosic ethanol production approaching commercial practice,2 it becomes necessary to economically process the lignin co-product obtained from lignocellulosic feedstock. The amount of lignin will exceed both the internal energy needs of biorefineries and the world market for lignin-derived specialty products by a large margin.1 If lignin could be efficiently depolymerized, it could serve as a renewable feedstock for aromatic compounds. Such a process would not only help to meet sustainability goals, but also to secure an aromatics supply for the chemical industry that increasingly makes use of natural gas.3
The depolymerization of lignin into value-added chemicals such as aromatics and fuels has already been explored by approaches such as pyrolysis, hydrocracking, hydrogenolysis, oxidation and hydrolysis.4,5 Hydrogenolysis in the presence of hydrogen or hydrogen-donating solvents is promising, because higher monomer yields can be obtained in this way and less char is formed.4 Solvents such as sub- and supercritical water,6–8 methanol,9–11 ethanol,11–13 iso-propanol,11,14 ethanol/water15–17 and methanol/water18 have been investigated for the solvolysis and hydrogenolysis of lignin. The yield and product distribution strongly depends on the solvent used. For example, catalytic single-step deconstruction of lignin into monomeric cyclohexyl derivatives in supercritical methanol at 300 °C was reported by Ford and co-workers.9,10 At lower temperatures (140–220 °C), mainly aromatics were formed in the presence of H2.19 Switch grass lignin was converted into phenolic products in ethanol at 350 °C over a Pt/C catalyst with formic acid as the hydrogen source, resulting in significant reduction in molecular weight and oxygen content.20 Wang and Rinaldi14 compared various alcohols as solvent for the catalytic depolymerization of lignin model compounds and organosolv lignin at 300 °C over a RANEY® Ni catalyst. They found that iso-propanol is the preferred alcohol solvent because of its good transfer hydrogenation properties in the catalytic depolymerization of organosolv lignin. The Weckhuysen group15,21 developed an aqueous phase reforming process of lignin in ethanol/water solvent using a Pt/Al2O3 catalyst. At 220 °C, a combined yield of monomeric aromatic oxygenates such as guaiacol and substituted guaiacols of 17% was obtained without char formation. It was observed that ethanol hinders lignin repolymerization; without ethanol, highly recalcitrant solids are formed. Recent work by Ma et al.13 reported on the catalytic conversion of Kraft lignin in supercritical ethanol at 280 °C over an α-MoC1−x/activated carbon catalyst without the addition of gaseous hydrogen. Ethanol was found as a much more effective solvent than pure water, methanol or iso-propanol. Song et al.11 tested the different alcohols including methanol, ethanol, iso-propanol, ethylene glycol, etc. at 200 °C using Ni/carbon catalyst in the presence of molecular H2. In this case, methanol was preferred over ethanol. In our earlier study, we reported that the use of ethanol is much more effective than that of methanol in the presence of a mixed non-noble-metal oxides (CuMgAlOx) catalyst.12 Despite the apparent promise of alcohol for lignin depolymerization, there is a lack of detailed knowledge about its role in obtaining high product yield. Understanding the influence of the solvent effect on the hydrogenolysis is highly desirable to rationally design catalyst/solvent systems for the valorization of lignin. Herein, we report on a novel catalytic process that can convert a variety of lignins into aromatics with high yield in supercritical ethanol. A non-noble-metal oxide catalyst protects the monomers and larger fragments from repolymerization by alkylation with the solvent. Another important aspect of the use of ethanol in this process is that it scavenges formaldehyde formed during lignin decomposition.
Results and discussion
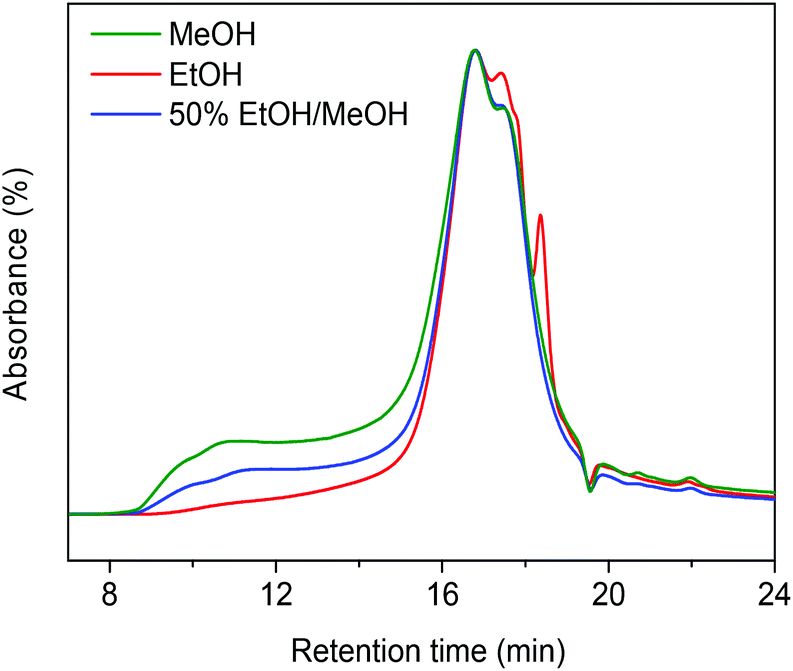
Table 1 compares typical data for soda lignin conversion in supercritical methanol and ethanol. A workup procedure (Scheme 2) was developed to distinguish smaller, tetrahydrofuran (THF)-soluble and larger (THF-insoluble) lignin fragments and char.12 In the presence of the CuMgAlOx catalyst, the monomers yield in the ethanol solvent was 17 wt% and no char was formed under these conditions (entry 1). The effect of alkylation on hindering repolymerization and char formation has been discussed in our previous work.12 In this respect, one would expect that methanol is preferred over ethanol, because alkylation of aromatics with methanol proceeds at a higher rate.22 However, the monomers yield was much lower when lignin was catalytically depolymerized in methanol; the THF-insoluble LR was also higher in this case (entry 2). Gel gel permeation chromatography (GPC) analysis of this residue revealed that much more repolymerized products formed in methanol than in ethanol (Fig. 1). These findings indicate that catalytic depolymerization of lignin is more effective in ethanol than in methanol.6,13,23 The better performance of ethanol compared with methanol as a solvent for lignin depolymerization is also evident from literature.6,13,23 For example, Miller reported that, in the base-catalyzed depolymerization of lignin at 290 °C, supercritical ethanol resulted in much less ether-insoluble residue compared to supercritical methanol.6 Ma et al. also observed that Kraft lignin can be more effectively converted in ethanol than in methanol.13 These reports and our own findings prompted us to investigate in detail the reason for this substantial difference between methanol and ethanol.
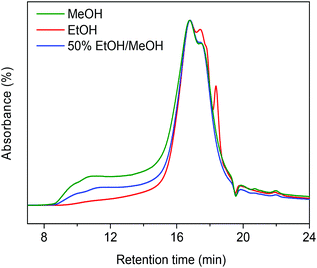 |
| | Fig. 1 GPC chromatograms of the THF-insoluble fraction of the lignin residue following reaction in methanol, ethanol and 50%/50% (v/v) methanol/ethanol mixture at 300 °C for 4 h over the CuMgAlOx catalyst (chromatograms normalized to the total peak area). | |
Table 1 Yields of monomers, lignin residues (LR), and char and the mass balance following lignin depolymerization under varying conditions over the CuMgAlOx catalyst
| Entry |
Lignin |
Solvent |
Temp. (°C) |
Time (h) |
Yield of products (wt%) |
Mass balance (wt%) |
| Monomers |
THF-soluble LR |
THF-insoluble LR |
Char |
|
50 ml autoclave conditions: 1 g of lignin, 0.5 g of catalyst, and 20 ml of solvent.
100 ml autoclave conditions: 1 g of lignin, 0.5 g of catalyst, and 40 ml of solvent.
1.07 g of THF-soluble LR was obtained from a reaction of 2 g of lignin, 1 g of catalyst, and 40 ml of ethanol at 380 °C for 8 h.
|
|
|
Reactions in 50 ml autoclavea |
| 1 |
P1000 |
EtOH |
300 |
4 |
17 |
73 |
18 |
0 |
108 |
| 2 |
P1000 |
MeOH |
300 |
4 |
6 |
57 |
39 |
1 |
103 |
| 3 |
P1000 |
50% MeOH/EtOH |
300 |
4 |
9 |
77 |
18 |
0 |
104 |
|
|
Reactions in 100 ml autoclaveb |
| 4 |
P1000 |
EtOH |
380 |
8 |
60 |
52 |
1 |
10 |
123 |
| 5 |
THF-soluble LRc |
EtOH |
380 |
8 |
47 |
72 |
0 |
3 |
122 |
| 6 |
Alcell |
EtOH |
380 |
8 |
62 |
47 |
1 |
6 |
116 |
| 7 |
Kraft |
EtOH |
380 |
8 |
86 |
26 |
3 |
31 |
146 |
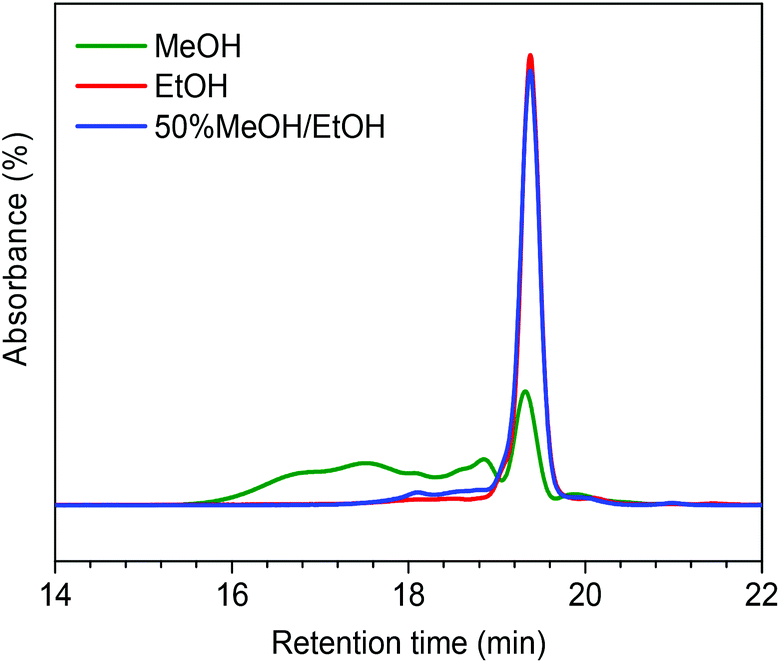
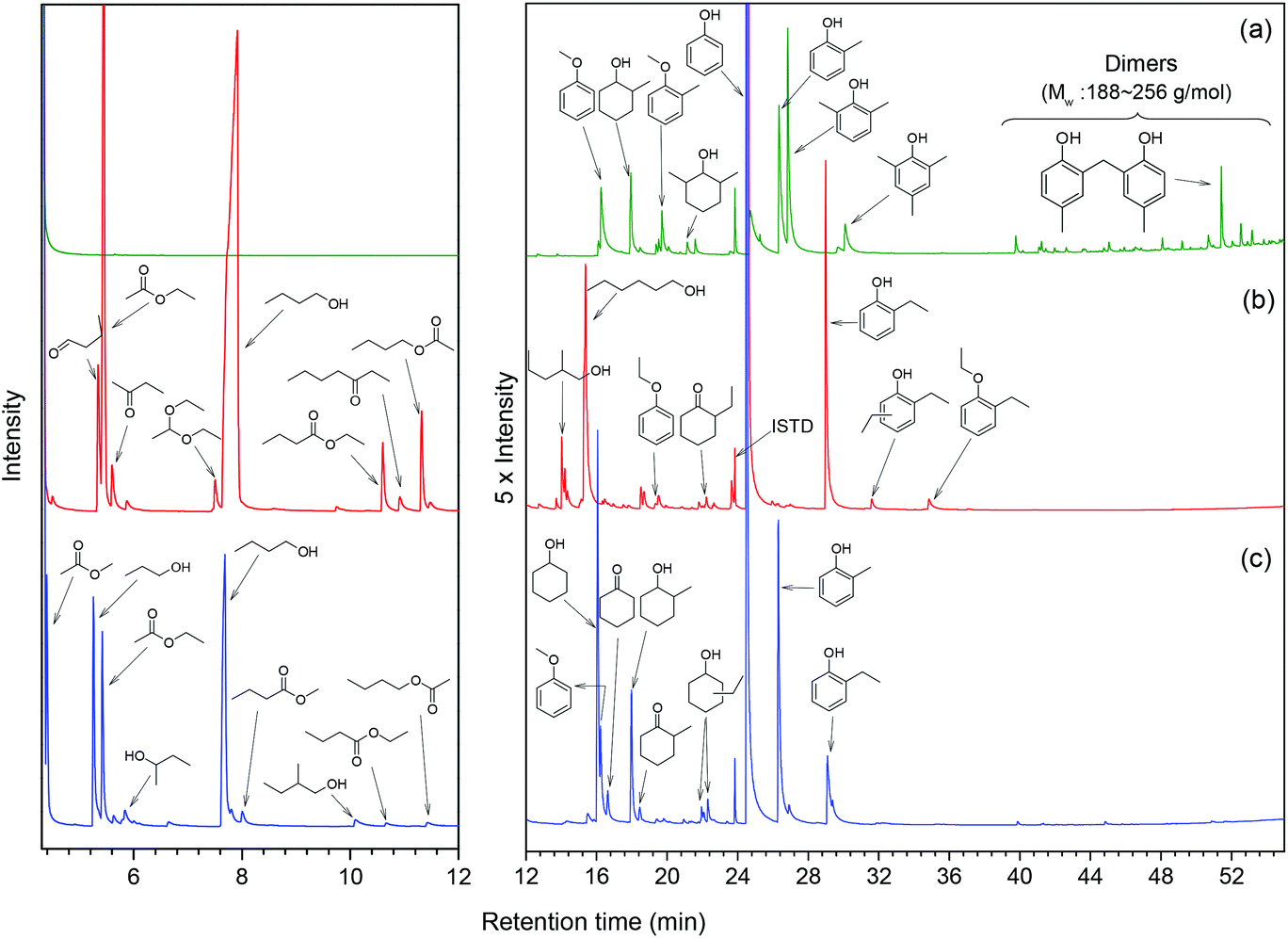
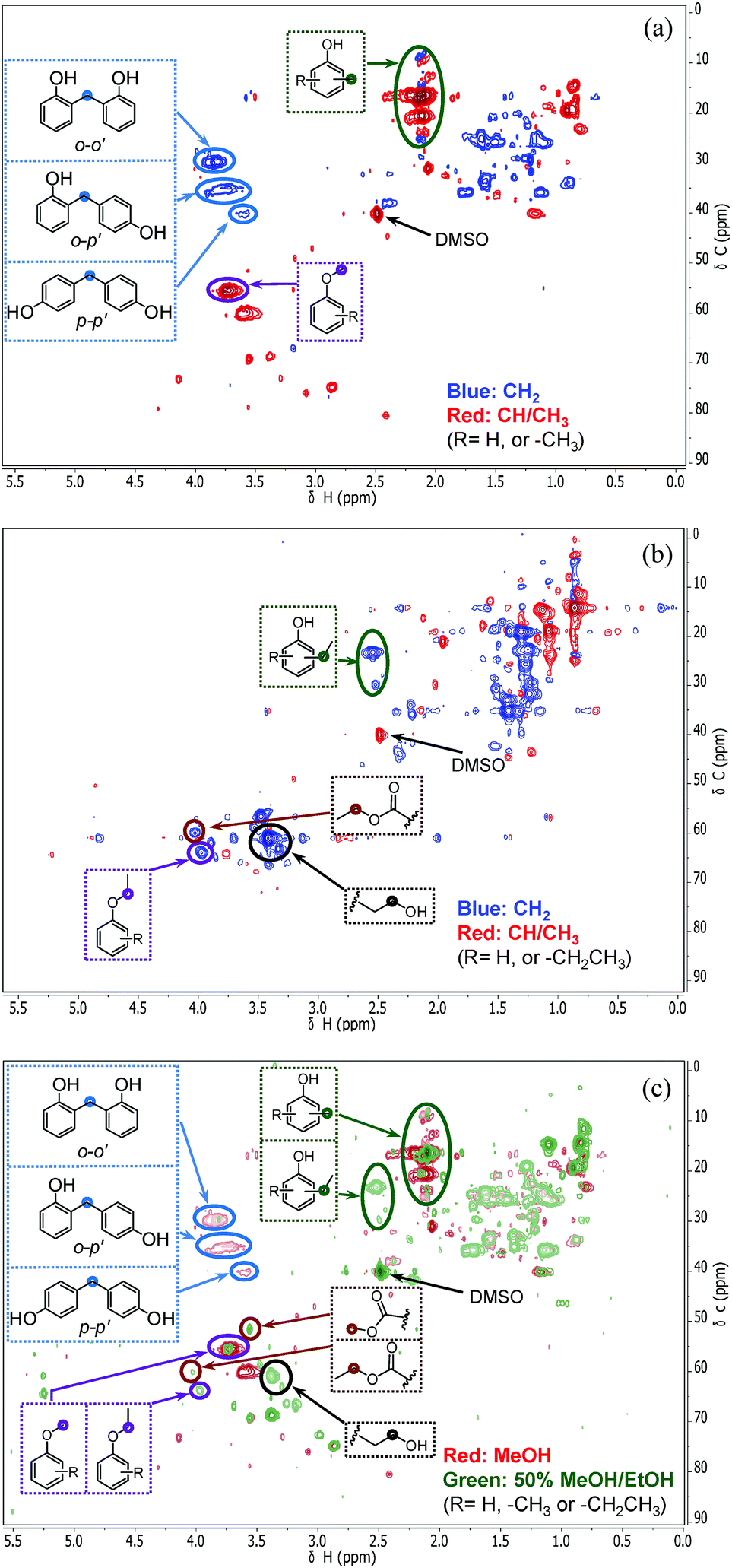
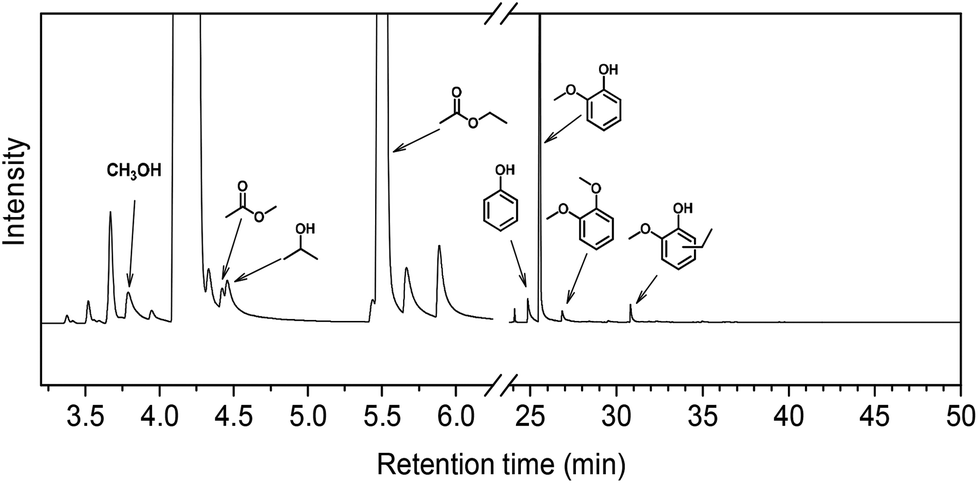
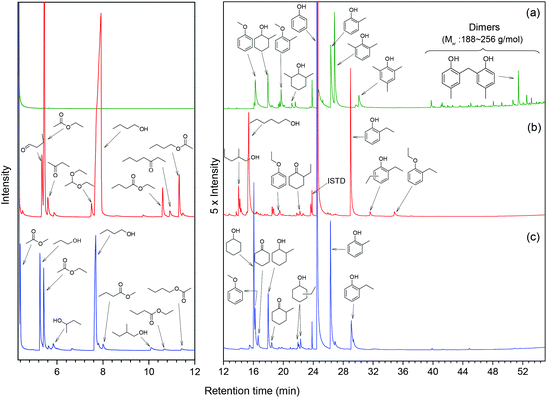
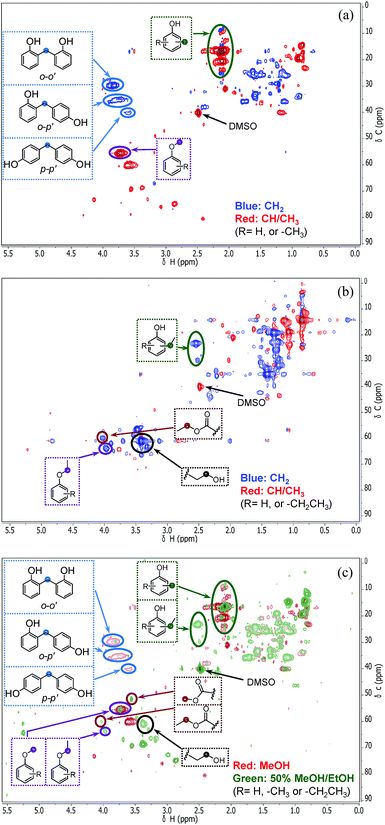
In order to understand how the solvent affects repolymerization of monomers, we compared the conversion of phenol into high-molecular-weight products in methanol and ethanol. We chose phenol because it is the basic motif in the monolignols that make up the lignin structure. When phenol was reacted in methanol at 300 °C for 1 h in the presence of the catalyst, a white resin-like residue was formed, which stuck to the reactor wall. GPC analysis revealed that the reaction mixture contained a large number of products with a broad molecular weight distribution (Fig. 2). The broad peak at early elution times is indicative of polymer formation. GC-MS analysis helps to identify the relatively light products (with molecular weights in the range from 188 to 256 g mol−1) of this polymerization reaction (Fig. 3a). Using the NIST 11 and NIST11s libraries, we identified 2,2′-methylenebis (4-methyl-phenol) among the products (Mw = 228 g mol−1). From the mass spectra, we deduce that the other high-molecular-weight products are also methylene-bridged isomers with different degrees of methylation of the aromatic ring and the phenolic hydroxyl group. Quantitative analysis based on GC-FID showed that phenol conversion was 95 wt%; the monomer product yield was only 23 wt%. The significant loss of mass balance is due to the formation of oligomers and polymers as also evidenced by GPC. We also analyzed this reaction mixture by 1H–13C heteronuclear single quantum coherence (HSQC) NMR spectrometry (Fig. 4a). The NMR spectrum contains many cross-peaks assigned to C-methylated and O-methylated products. It confirms our claim of extensive methylation. As expected, three types of methylene bridge isomers were observed: o–o′ (δC/δH at 29.3/3.86), o–p′ (δC/δH at 34.4/3.76), and p–p′ (δC/δH at 39.6/3.60).24 Among these, the o–o′ bridges were present most frequently. This cross-linking structure arises from reaction of two moles of (methylated) phenol and one mole of formaldehyde formed by dehydrogenation of methanol.30,31 The results were very different when the reaction was performed in ethanol. No resin-like polymer was observed in the reactor, and, notably, no products with molecular masses exceeding 178 g mol−1 were present as confirmed by GC-MS (Fig. 3b). GPC analysis also confirms the absence of oligomers and polymers after reaction in ethanol. The narrow GPC peak corresponds to unconverted phenol and its monomeric derivatives (Fig. 2). HSQC NMR analysis (Fig. 4b) also shows that no cross-linking reactions with formaldehyde that occurred in the reaction in methanol took place. Instead, many other cross-peaks assigned to higher alcohols, alkyl esters, as well as the C-ethylated and O-ethylated aromatic products were formed. These results point out the fact that Guerbet reactions of ethanol as well as esterification and C- and O-ethylation reactions of phenol dominated in ethanol.
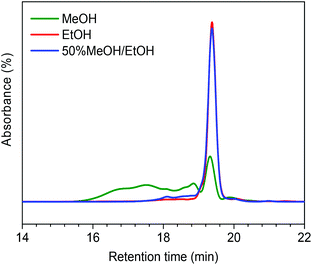 |
| | Fig. 2 GPC chromatograms of reaction mixtures obtained from the reaction of phenol at 300 °C for 1 h over the CuMgAlOx catalyst in methanol, ethanol and 50%/50% (v/v) methanol/ethanol mixture (chromatograms normalized to total peak area). | |
 |
| | Fig. 3 GC-MS chromatograms of reaction mixtures obtained from reaction of phenol at 300 °C for 1 h over the CuMgAlOx catalyst in (a) methanol, (b) ethanol, and (c) 50%/50% (v/v) methanol/ethanol solvents (GC-MS chromatograms normalized to the internal standard, ISTD). | |
 |
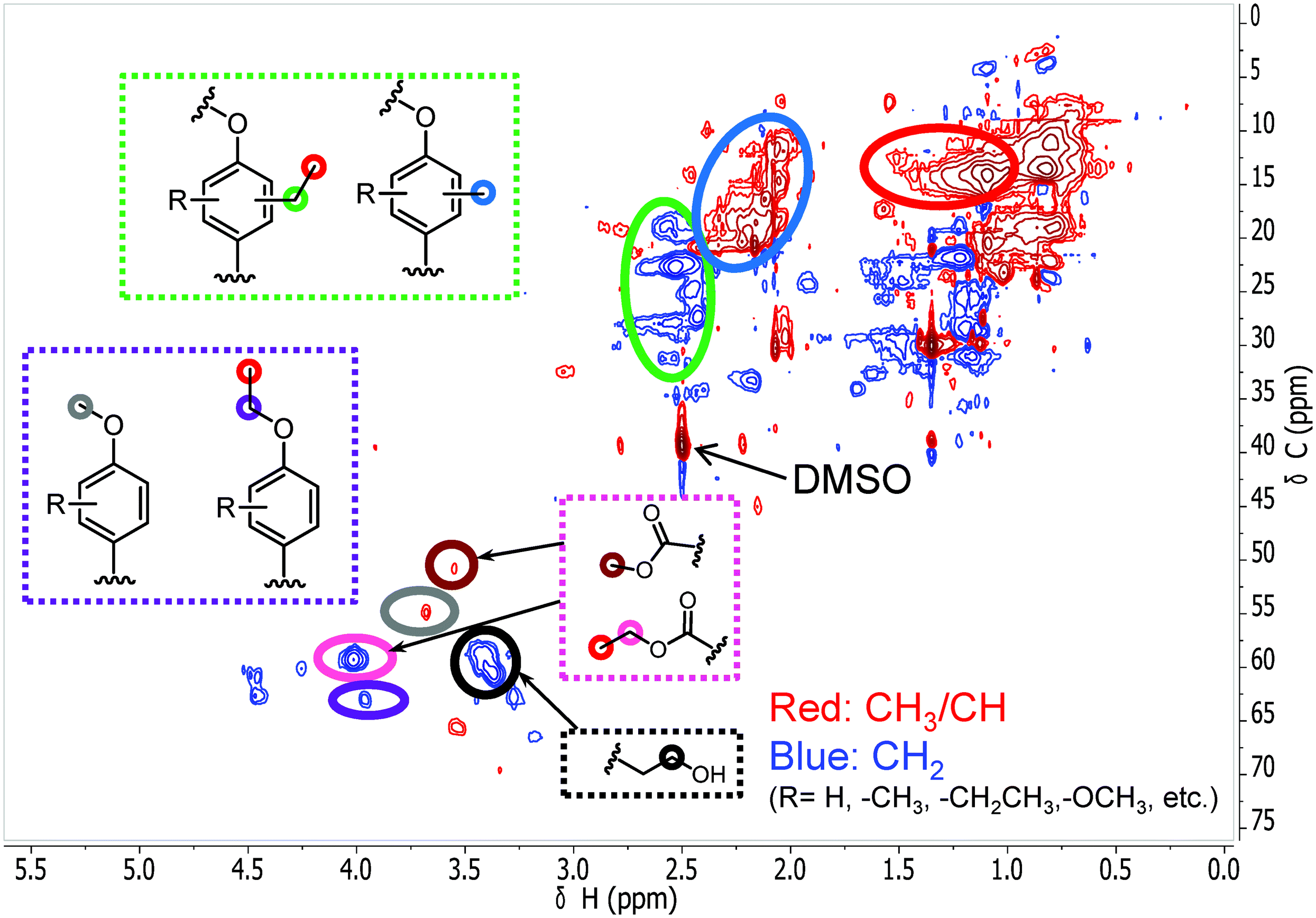
| | Fig. 4 The side-chain region of the 1H–13C HSQC NMR spectra of the reaction products of phenol conversion (300 °C; 1 h; CuMgAlOx catalyst): (a) spectrum for methanol solvent, (b) spectrum for ethanol solvent and (c) combined spectra of methanol (red) and 50%/50% (v/v) methanol/ethanol solvent (green) (The combined spectra have been normalized by the total peak volume). | |
Taken together, these results show that significant phenol oligomerization takes place in methanol, but not in ethanol. The reaction of phenol with formaldehyde for the production of phenolic resins (e.g., resoles and novolaks) is well known in the polymer industry.25,26 Methanol can be readily converted to formaldehyde in the presence of metal catalysts.27,28 Compared with the high reactivity of formaldehyde, the reaction of phenol with higher aldehydes to form resins requires strongly acidic conditions.25,26 Under base-catalyzed conditions, higher aldehydes tend to undergo aldol condensation and self-resinification reactions,30,31 as also evident from our GC-MS data that point to the formation of higher alcohols (e.g., n-butanol) and esters (e.g., ethyl acetate) (Fig. 3b). The Guerbet-type reactions between aldehydes and alcohols are known to be catalyzed by Cu-based catalysts.10 Methanol cannot self-couple through the Guerbet reaction,29 as also apparent from our GC analysis of possible methanol conversion products (Fig. 3a). We infer from these data that the monomeric products formed during lignin disassembly at elevated temperatures will react with formaldehyde formed by dehydrogenation of the methanol solvent. When the reaction is conducted in ethanol, acetaldehyde does not exhibit such behavior. During lignin depolymerization, many different phenolics will be present that will exhibit different reactivities towards formaldehyde.
On the basis of these findings, we conclude that formaldehyde plays an important role in the repolymerization of lignin decomposition products and char formation.30,31 An example of application of this property is in partially replacing phenol in phenol-formaldehyde (PF) resins production.32,33 Saisu et al. reported that the negative effect of formaldehydecan be mitigated by adding phenol as a capping agent during lignin depolymerization.30,31,34,35 They used a water–phenol mixture at 673 K to demonstrate the conversion of organosolv lignin into chemicals.31 However, the high value of phenol prohibits its use as capping agent in practice. In the present study, we observed that the Guerbet reaction and esterification of ethanol solvent occurred at very high rates. Ethanol is also known to react with methanol/formaldehyde to form higher alcohols and esters over CuMgAlOx mixed-metal oxides.27,29 Accordingly, we hypothesized that ethanol will also react with formaldehyde formed during lignin conversion. In order to confirm this supposition, we conducted another model reaction with phenol as the reactant, but this time in a 50%/50% (v/v) methanol/ethanol solvent mixture. Also in this case, no resin-like residue formed. The GPC analysis of the resulting mixture gave results very similar to those obtained in the ethanol case (Fig. 2). The GC-MS (Fig. 3c) results confirm that hardly any high-molecular-weight products formed in this alcohols mixture. HSQC NMR revealed that almost no methylene-bridged structure formed (Fig. 4c). As expected, we observed significant amounts of higher alcohols (e.g., n-propanol) and esters (e.g., methyl acetate) (Fig. 3c); this result is also supported by HSQC NMR analysis (Fig. 4c). Thus, we can firmly conclude ethanol can efficiently scavenge formaldehyde formed by methanol dehydrogenation.
The scavenging of formaldehyde by ethanol is important, because methanol and formaldehyde can be formed during lignin depolymerization process. Methoxy groups on the phenolic ring are present in the syringyl and guaiacyl monolignols.5 These methoxy groups are easily eliminated as methanol.8,15,36 Demethoxylation of lignin is confirmed by the observation that methoxy groups are removed from the parent lignin and the presence of methanol in the product mixture during the early stage of reaction (Fig. S4†). It has also been suggested that formaldehyde can be directly obtained from the γ-carbon of the alkyl side-chain in lignin during hydrolysis31,37 and pyrolysis.4,38 Given our results, we speculated that formaldehyde formation from lignin during its disassembly is a major cause of the undesired repolymerization reactions that leads to low monomer yields and char. In order to verify whether formaldehyde derived from methoxy groups in lignin can also be scavenged by ethanol, we conducted an experiment with guaiacol as the reactant. Fig. 5 shows the GC-MS result of guaiacol conversion in ethanol at 300 °C (reaction time: 2 h). Methanol and phenol are among the reaction products; the formation of methyl acetate and iso-propanol prove that ethanol reacts with formaldehyde derived from guaiacol demethoxylation. Based on these findings, we can now firmly conclude that ethanol acts as a scavenger for reactive formaldehyde intermediates. In this way, ethanol can effectively suppress repolymerization reactions involving formaldehyde derived from lignin itself during its depolymerization. During lignin depolymerization in a methanol/ethanol mixture (entry 3) we observed a significant decrease in the yield of THF-insoluble lignin residue (18 wt%) compared to the yield obtained in methanol solvent (39 wt%, entry 2). GPC analysis of this residue further evidences a lower rate of repolymerization (Fig. 1). Again, significant amounts of n-propanol and methyl acetate were obtained. These results are consistent with the phenol model reactions in this mixture. Unfortuanately, although repolymerization was substantially suppressed, the monomer yield increased only slightly from 6 wt% to 9 wt% compared with the reaction in methanol.
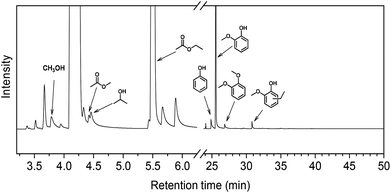 |
| | Fig. 5 GC-MS chromatograms of reaction mixtures obtained from reaction of guaiacol at 300 °C for 2 h over the CuMgAlOx catalyst in ethanol. | |
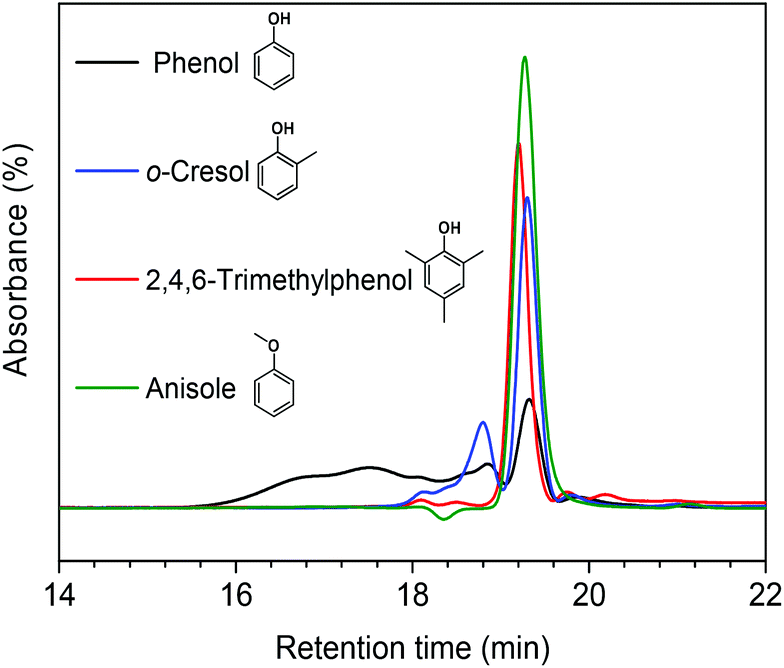
In our earlier study, we suggested that alkylation plays an important role in suppressing repolymerization. Ethanol acts as a capping agent, stabilizing the highly reactive phenolic intermediates by O-alkylating the hydroxyl groups and C-alkylating the aromatic rings.12 In order to further verify this statement, we explored in more detail how alkylation suppresses the repolymerization of model monomers. To this end, we also used o-cresol, 2,4,6-trimethylphenol, and anisole as reactants in methanol. The former two compounds are models for C-alkylated phenols, the latter one for O-alkylated phenol. Fig. 6 shows the GPC analysis results for the product mixtures. We observed a good correlation between the extent of polymerization and the degree of C-alkylation. With one methyl group present in the ortho-position (o-cresol), polymerization occurred at a much lower rate compared with phenol as the reactant. When the phenolic ring contained three methyl groups at ortho- and para-positions, almost no polymerized product was observed. Furthermore, we found that polymerization was completely suppressed when anisole was the reactant. These results point out the important role of the phenolic hydroxyl group in repolymerization processes during lignin depolymerization. Both C-alkylation and O-alkylation contribute to suppress repolymerization, which provides a solid evidence for the importance of alkylation during lignin conversion.12
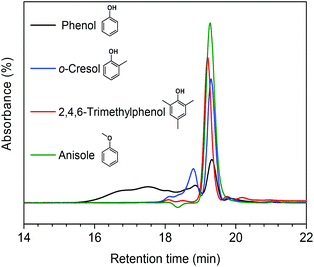 |
| | Fig. 6 GPC chromatograms of reaction mixtures obtained from the reaction at 300 °C for 1 h over the CuMgAlOx catalyst using different reactants in methanol solvent (depicted chromatograms have been normalized by the sum of the peak area). | |
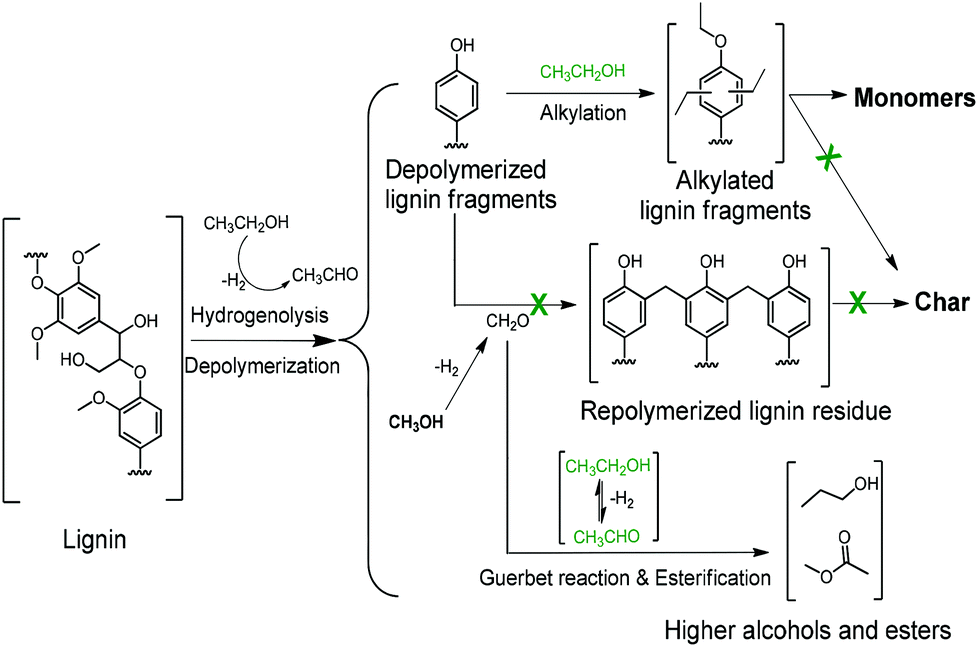
Scheme 1 summarizes the most important aspects of lignin depolymerization in supercritical ethanol. Ethanol has three important functions. First, ethanol serves as a source of hydrogen to facilitate the lignin depolymerization and deoxygenation reactions by hydrogenolysis. Hydrogen is observed among the gas-phase products of ethanol-mediated lignin depolymerization (Table S1†). Second, ethanol acts as a scavenger for formaldehyde formed by removal of methoxy groups from the lignin, thereby suppressing repolymerization reactions involving formaldehyde. Third, ethanol serves as a capping agent to stabilize the reactive phenolic intermediates by O-alkylating the phenolic hydroxyl groups and C-alkylating the aromatic rings. Given the low reactivity of BTX-like compounds, we surmise that repolymerization of oxygen-free aromatics will not occur. The latter two roles of ethanol cause the rate of repolymerization of phenolic products derived from lignin disassembly to be low, which explains the absence of char formation in ethanol solvent.
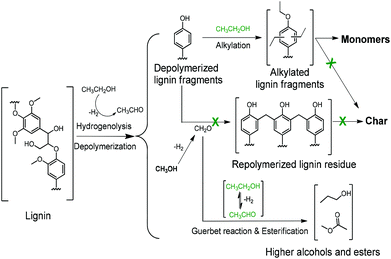 |
| | Scheme 1 The roles of alkylation, the Guerbet reaction, and esterification on suppressing char formation during lignin depolymerization over the CuMgAlOx catalyst in supercritical ethanol. | |
Lignin depolymerization at higher temperature
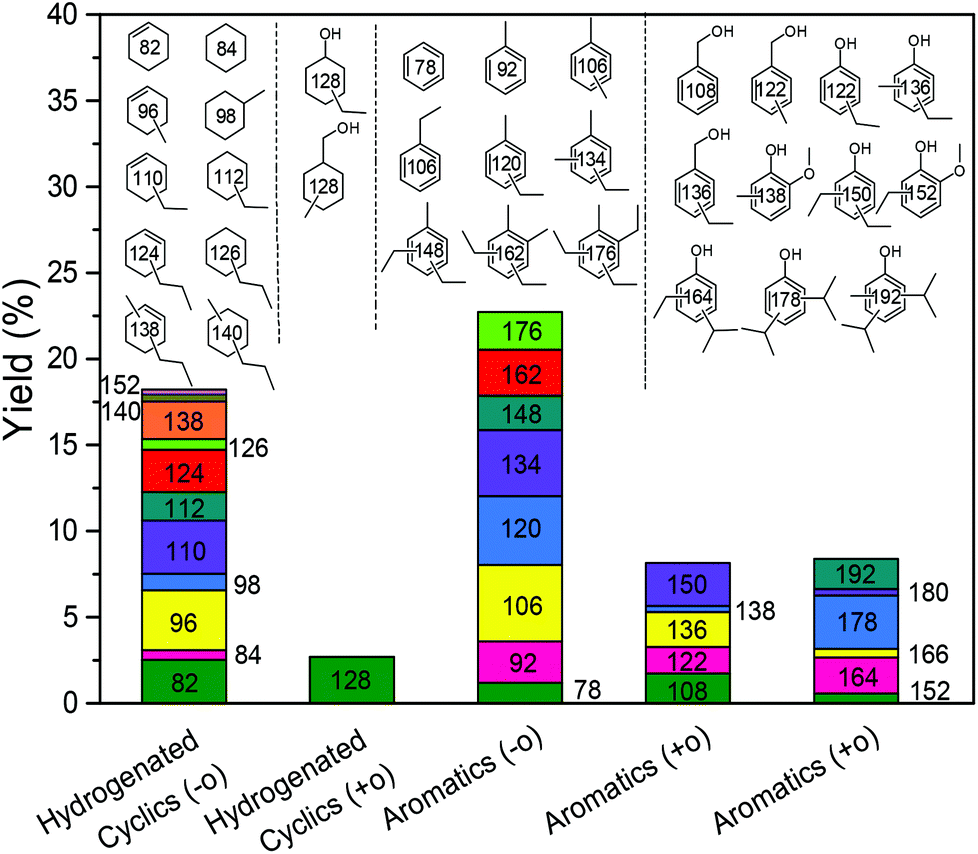
With these insights in hand, we optimized the reaction temperature for the production of monomeric aromatics. Under the optimized conditions, a monomers yield of 60 wt% was obtained after reaction for 8 h at 380 °C (entry 4). Fig. 7 shows the monomers distribution in the product mixture of this reaction. The selectivity to oxygen-free aromatics and hydrogenated cyclics (cycloalka(e)nes) is 68%, indicating that extensive deoxygenation occurred. We also found that the THF-soluble lignin residue can be further upgraded in the same way. After reaction under optimized conditions, the THF-soluble residue has been largely depolymerized (Mw = 469 g mol−1) and deoxygenated (O/C ratio = 0.09). The H/C ratio of this residue is 1.25. When this fraction is subjected to a second reaction in ethanol, more monomers are obtained (entry 5). The combined monomers yield of the two reaction experiments is 85%. Notably, much less char (3 wt%) was formed by this approach. HSQC NMR analysis of this lignin residue showed that almost no methoxy groups remained due to demethoxylation (Fig. 8). These data also show that the phenolic intermediates were stabilized by C- and O-alkylation. The formation of alkyl ester and alcohol groups reveals that the Guerbert reaction and esterification also take place between ethanol and lignin side-chains. These findings indicate that ethanol might also play role in capping the reactive side chains (e.g., aldehyde groups) of lignin, preventing condensation reactions between larger lignin fragments. These results further support the conclusion that alkylation, Guerbet reactions and esterification can suppress repolymerization reactions of larger fragments and, in this way, char formation during lignin depolymerization.
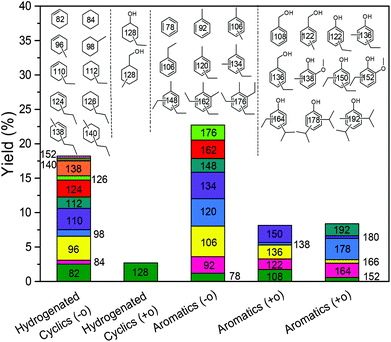 |
| | Fig. 7 Monomeric product distribution following lignin reaction at 380 °C for 8 h over the CuMgAlOx catalyst in ethanol solvent. | |
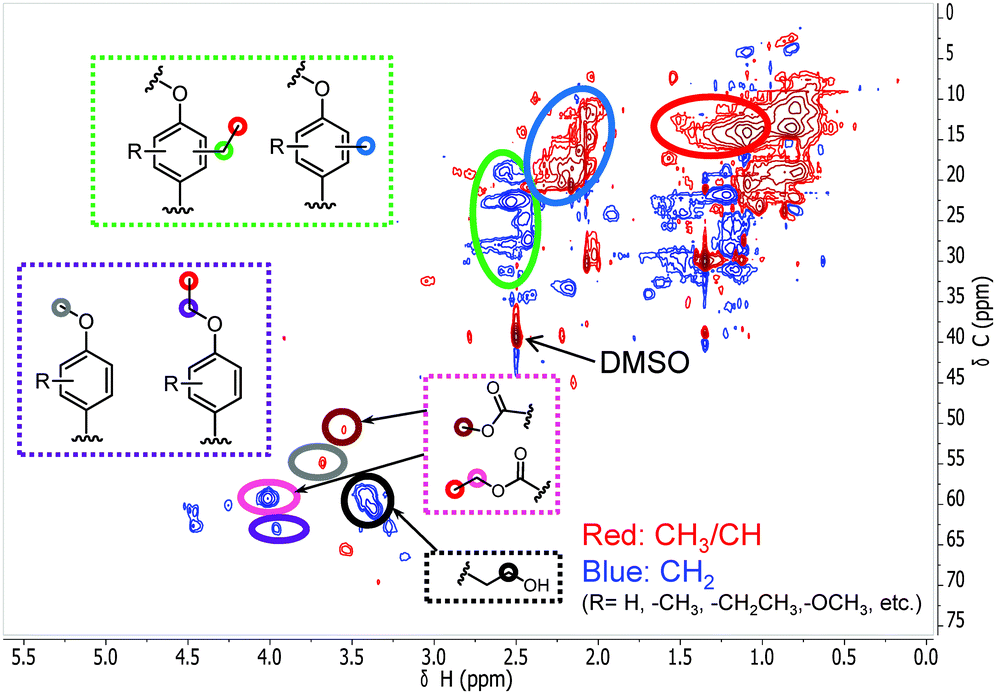 |
| | Fig. 8 The side-chain region of the 1H–13C HSQC NMR spectra of the THF-soluble lignin residue obtained from the lignin reaction at 380 °C for 8 h over the CuMgAlOx catalyst in ethanol solvent. | |
We also applied the optimized depolymerization process to other types of lignin. With organosolv lignin (Alcell, entry 6) as the feed, a similar product yield was obtained as for soda lignin. The product distribution was also quite similar (Fig. S5A†) and the amount of char was slightly lower, presumably because of the better solubility of organosolv Alcell lignin in ethanol. With Kraft lignin (entry 7), the monomers yield was 86 wt%. The yields of BTX and alkylated cycloalka(e)nes were 35 wt% and 25 wt%, respectively (Fig. S5B†). With Kraft lignin, more char was formed. We suspect that it might be due to the presence of inpurities (Na and S content: 13.1 wt% and 2.8 wt%, respectively) in Kraft lignin. Nevertheless, this result shows that the catalyst can also convert sulfur-containing lignins. The higher than 100% mass balances can be explained by the extensive alkylation of the lignin products with the solvent.
Conclusion
In summary, we have demonstrated high-yield production of monomeric aromatics from lignin using a CuMgAlOx catalyst in supercritical ethanol with little char formation. The monomeric products are mainly composed of alkylated aromatics and cycloalka(e)nes. The oxygen-free aromatics can be used as chemical building blocks and as octane boosters when blended with gasolines.39 Oxygenated aromatics may serve as valuable compounds for the chemical and polymer industry.40 They can also be used as low-sooting diesel fuel additives.41 Our approach does not require critical metals nor external molecular hydrogen, thereby greatly reducing operational costs. These aspects render the described lignin depolymerization process a viable candidate for the conversion of lignin into a range of valuable products. An additional benefit is that different types of lignin can be effectively converted. We revealed important new mechanistic insights about lignin depolymerization. Ethanol is effective as a capping agent and formaldehyde scavenger, suppressing repolymerization and char-forming reactions.
Experimental section
Chemicals and materials
Protobind 1000 alkali lignin was purchased from GreenValue. It was produced by soda pulping of wheat straw (sulfur-free lignin with less than 4 wt% carbohydrates and less than 2 wt% ash). Alcell™ Organosolv lignin was obtained from the Wageningen UR Lignin Platform. It was extracted from mixed hardwoods by an organosolv process using ethanol–water solvent. Kraft lignin was purchased from Sigma-Aldrich. All commercial chemicals were analytical reagents and were used without further purification.
Catalyst preparation
20 wt% Cu-containing MgAl mixed oxide (CuMgAlOx) catalyst was prepared by a co-precipitation method with a fixed M2+/M3+ atomic ratio of 2. For example, 6 g CuMgAlOx catalyst was prepared in the following way: 4.40 g Cu(NO3)2·2.5 H2O, 15.67 g Mg(NO3)2·6H2O, and 15.01 g Al(NO3)3·9 H2O were dissolved in 100 ml de-ionized water. This solution in parallel with 100 ml of a NaOH (9.60 g) solution were slowly added (1 drop per s) through 100 ml dropping funnels to 250 ml of Na2CO3 (5.09 g) solution in a 1000 ml necked flask at 60 °C with vigorous stirring, whilst keeping the pH of the slurry at 10. When addition was complete after ca. 45 min, the milk-like light-blue slurry was aged at 60 °C under stirring for 24 h. The precipitate was filtered and washed with distilled water until the filtrate reached a pH of 7. The solid was dried overnight at 105 °C and grinded and sieved to a particle size below 125 μm. The hydrotalcite layered structure of the obtained powder was checked and confirmed by XRD. The hydrotalcite-like precursor was calcined with a heating rate of 2 °C min−1 from 40 °C to 460 °C and kept at this temperature for 6 h in static air. The resulting catalyst was denoted by CuMgAlOx.
Catalytic reactions
50 ml AmAr stirred high-pressure autoclaves were used to study the (catalytic) conversion of lignin in (m)ethanol. Typically, the autoclave was charged with a suspension of 0.5 g catalyst and 1.0 g lignin in 20 ml solvent. An amount of 10 μl n-dodecane was added as the internal standard. The reactor was sealed and purged with nitrogen several times to remove oxygen. After leak testing, the pressure was set to 10 bar and the reaction mixture was heated to the desired reaction temperature under continuous stirring at 500 rpm within 1 h. After the reaction, the reactor was rapidly quenched to room temperature in an ice bath. For those reactions conducted at 100 ml Parr autoclaves, the same procedure was applied. The only difference is that 40 ml solvent was applied and the same amount of n-dodecane internal standard was added after reaction.
For the model compound reactions, 50 ml AmAr autoclave was used following the same procedure as the lignin reaction mentioned above. In these cases, 1.0 g feedstock (e.g., phenol, o-cresol, anisole, etc.) and 0.2 g catalyst were added in 20 ml solvent. The reactions were performed at 300 °C for 1 h. After reaction, 10 μl n-dodecane internal standard was added in the solution. The reaction mixture was collected and combined with the solution obtained from washing the autoclave and volume to 20 ml with acetone. The reaction mixture was subjected to filtration with a 0.45 μm syringe filter. The resulting solution was further subjected to GC-MS, GPC and 1H–13C HSQC NMR analysis.
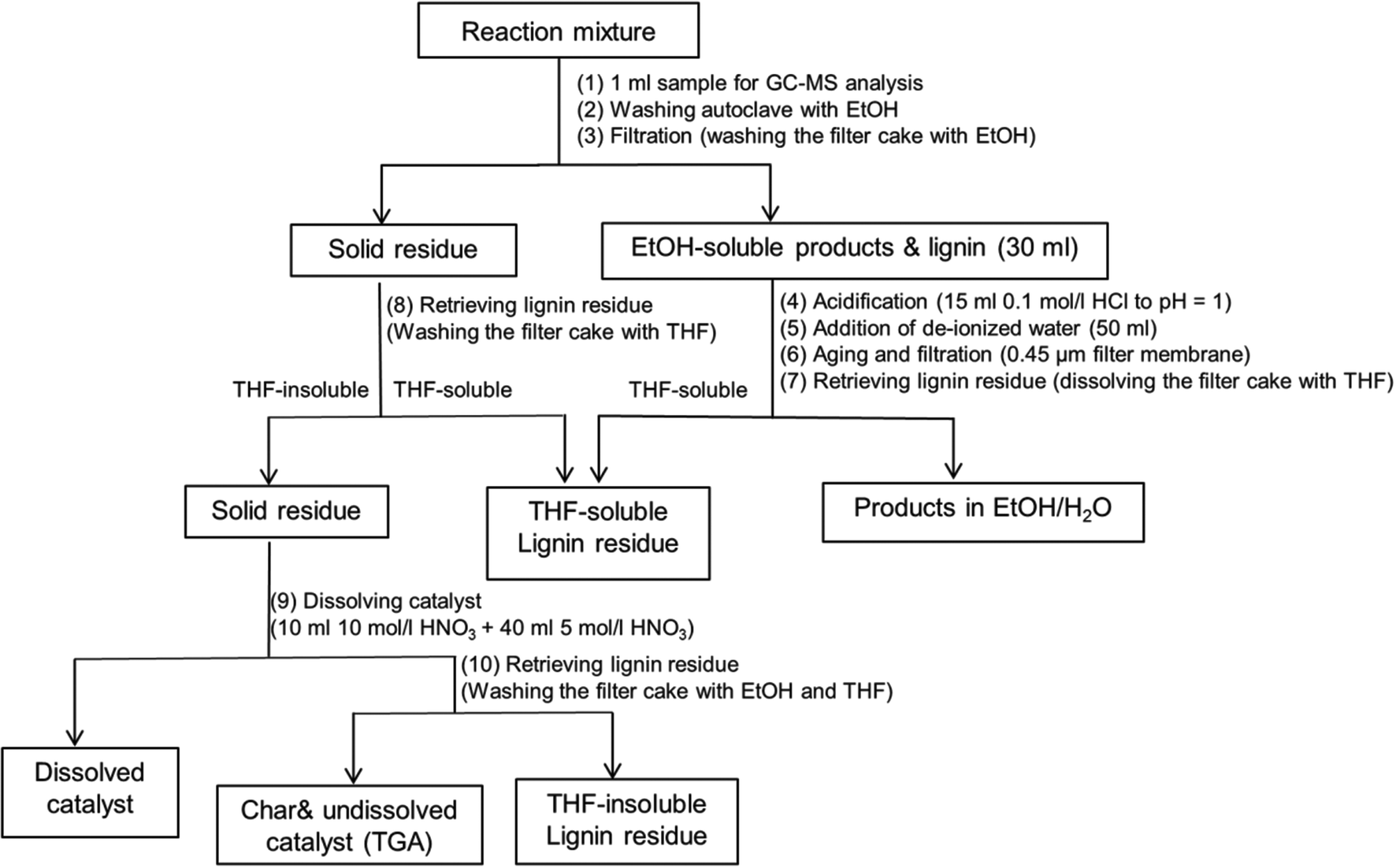
A work-up procedure as shown in Scheme 2 was developed (the numbers between brackets refer to the steps in Scheme 2). Firstly, an aliquot of 1 ml was taken from the reaction mixture and directly analyzed by GC-MS without dilution following filtration with a 0.45 μm syringe filter (1). The remaining mixture was collected and combined with the solution obtained from washing the autoclave with ethanol (2). The combined mixture was subsequently subjected to filtration and the filter cake was washed with ethanol several times (3). The filtrate volume was brought to 30 ml by blowing the reaction mixture with air at room temperature, followed by acidification by adding 15 ml of a 0.1 mol l−1 HCl solution (final pH = 1) (4), and 50 ml de-ionized water to precipitate unconverted lignin and high molecular-weight lignin fragments (5). After aging for about 30 min, the resulting mixture was filtered over a 0.45 μm filter membrane (6). The filter cake was retrieved by washing with THF (7). The solid residue from step (3) was then washed with excess THF in order to retrieve the unconverted lignin adsorbed on catalyst (8). The lignin residue was obtained by combining the two THF solutions and removing THF by rotary evaporation at 60 °C. The resulting filter cake was regarded as a mixture of catalyst and repolymerized products. In order to distinguish the yield of repolymerized product, we further dissolved the catalyst using concentrated HNO3 following the procedure reported in literature.10 0.2 g solid residue obtained from step (8) was loaded in a 50 ml flask. 10 ml 10 mol l−1 HNO3 was initially added to dissolve copper. The slurry was further treated with addition of 40 ml 5 mol l−1 HNO3 (9). The resulting mixture was filtered over a filter crucible (porosity 4). The filter cake was retrieved by washing with excess ethanol and THF (10). After removing THF solvent by rotary evaporation, another fraction of lignin residue was obtained and denoted as THF-insoluble lignin residue. The remaining filter cake was regarded as char and undissolved catalyst. Thermo gravimetric Analysis (TGA) was further applied to determine the exact amount of char.
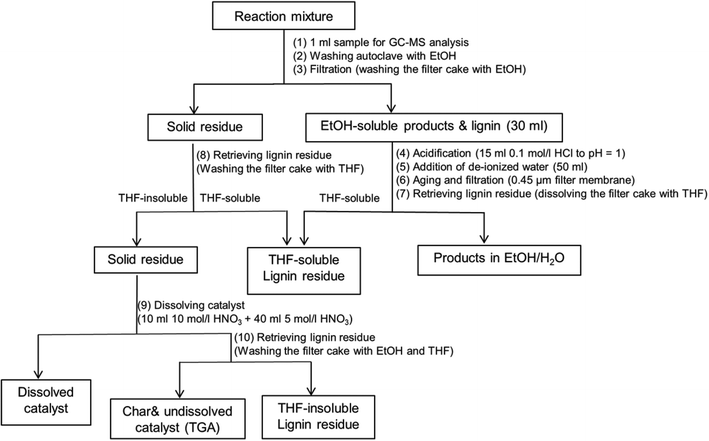 |
| | Scheme 2 Work-up procedure of reaction product mixture from lignin reaction. | |
Lignin product analysis
The liquid phase product mixture were analyzed by a Shimadzu 2010 GC-MS system equipped with a RTX – 1701 column (60 m × 0.25 mm × 0.25 μm) and a flame ionization detector (FID) together with a mass spectrometer detector. Identification of products was achieved based on a search of the MS spectra with the NIST11 and NIST11s MS libraries. The peaks with the same molecular weight (Mw) were unified and presented by the structure determined by (1D) GC-MS and/or (2D) GC × GC-MS (details can be found in our previous work (ref. 12). These products were further divided into four groups, namely hydrogenated cyclics (−o (oxygen-free)), hydrogenated cyclics (+o (oxygen-containing)), aromatics (−o) and aromatics (+o), according to the nature of the ring structure and functional groups. All the quantitative analyses of liquid phase product were based on 1D GC-FID. Experimentally determined weight response factors of cyclohexane (1.221), cyclohexanone (0.992), ethyl benzene (1.103) and ethyl guaiacol (0.803) were used for these four groups related to n-dodecane as the internal standard. The yields of lignin residue, monomers and char were calculated by using eqn (1)–(4).| |  | (1) |
| | 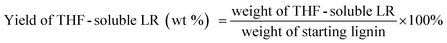 | (2) |
| | 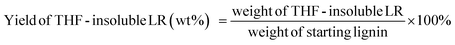 | (3) |
| |  | (4) |
1H, 13C and 1H–13C HSQC NMR analysis
All NMR spectra were recorded using a VARIAN INOVA 500 MHz spectrometer. For the model compound sample, an aliquot of 2 ml solution was taken from the reaction mixture followed by removing the solvent by an air flow at room temperature. The resulting mixture was dissolved in 0.7 ml dimethylsulfoxide-d6 (DMSO-d6). For analysis of the lignin residue, appoximately 70 mg of lignin residue was dissolved in 0.7 ml DMSO-d6. 1H–13C HSQC NMR spectra were obtained using the gHSQCAD program. Normally, 8 (model compound sample) or 16 (lignin residue sample) scans, 2 s relaxation delay, and 256 t1 increments were used. Data processing was performed using the MestReNova software.
Gel permeation chromatography
GPC analyses were performed by using a Shimadzu apparatus equipped with two columns connected in series (Mixed-C and Mixed-D, polymer Laboratories) and a UV-Vis detector at 254 nm. The column was calibrated with Polystyrene standards. Analyses were carried out at 25 °C using THF as eluent with a flow rate of 1 ml min−1. For the model compound analysis, an aliquot of 40 μl solution was taken from the reaction mixture followed by removing the solvent by blowing with air under room temperature. The sample was dissolved with 1 ml THF (the concentration is ∼2 mg ml−1). For the lignin residue analysis, the sample was prepared at a concentration of 2 mg ml−1. All the samples were filtered using 0.45 μm filter membrane prior to injection.
Elemental analysis
The carbon, hydrogen, and oxygen (CHO) content of the lignin residue were quantitatively determined by means of elemental analysis (PerkinElmer 2400 series II Elemental Analyzer, CHN mode). The lignin samples were dried overnight in a vacuum oven at 60 °C to remove residual water and solvent. Carbon and hydrogen analysis was conducted by combustion followed by thermal conductivity and infrared detection of effluent gases. The oxygen content was determined by considering that the material consisted of C, O, and H atoms.
Acknowledgements
This work was funded by the “New Energy House” project of the Eindhoven Energy Institute in collaboration with the Knowledge and Innovation Community InnoEnergy of the European Institute of Innovations and Technology.
References
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 709 CrossRef CAS PubMed.
- T. R. Brown and R. C. Brown, Biofuels, Bioprod. Biorefin., 2013, 7, 235–245 CrossRef CAS PubMed.
- P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2013, 52, 11980–11987 CrossRef CAS PubMed.
- M. P. Pandey and C. S. Kim, Chem. Eng. Technol., 2011, 34, 29–41 CrossRef CAS PubMed.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- J. E. Miller, L. Evans, A. Littlewolf and D. E. Trudell, Fuel, 1999, 78, 1363–1366 CrossRef CAS.
- V. M. Roberts, V. Stein, T. Reiner, A. Lemonidou, X. B. Li and J. A. Lercher, Chem. – Eur. J., 2011, 17, 5939–5948 CrossRef CAS PubMed.
- J. A. Onwudili and P. T. Williams, Green Chem., 2014, 16, 4740–4748 RSC.
- K. Barta, T. D. Matson, M. L. Fettig, S. L. Scott, A. V. Iretskii and P. C. Ford, Green Chem., 2010, 12, 1640–1647 RSC.
- T. D. Matson, K. Barta, A. V. Iretskii and P. C. Ford, J. Am. Chem. Soc., 2011, 133, 14090–14097 CrossRef CAS PubMed.
- Q. Song, F. Wang, J. Y. Cai, Y. H. Wang, J. J. Zhang, W. Q. Yu and J. Xu, Energy Environ. Sci., 2013, 6, 994–1007 CAS.
- X. Huang, T. I. Korányi, M. D. Boot and E. J. M. Hensen, ChemSusChem, 2014, 7, 2276–2288 CrossRef CAS PubMed.
- R. Ma, W. Y. Hao, X. L. Ma, Y. Tian and Y. D. Li, Angew. Chem., Int. Ed., 2014, 53, 7310–7315 CrossRef CAS PubMed.
- X. Y. Wang and R. Rinaldi, ChemSusChem, 2012, 5, 1455–1466 CrossRef CAS PubMed.
- J. Zakzeski and B. M. Weckhuysen, ChemSusChem, 2011, 4, 369–378 CrossRef CAS PubMed.
- A. L. Jongerius, P. C. A. Bruijnincx and B. M. Weckhuysen, Green Chem., 2013, 15, 3049–3056 RSC.
- S. N. Cheng, C. Wilks, Z. S. Yuan, M. Leitch and C. B. Xu, Polym. Degrad. Stab., 2012, 97, 839–848 CrossRef CAS PubMed.
- A. K. Deepa and P. L. Dhepe, ACS Catal., 2014, 5, 365–379 CrossRef.
- K. Barta, G. R. Warner, E. S. Beach and P. T. Anastas, Green Chem., 2014, 16, 191–196 RSC.
- W. Y. Xu, S. J. Miller, P. K. Agrawal and C. W. Jones, ChemSusChem, 2012, 5, 667–675 CrossRef CAS PubMed.
- J. Zakzeski, A. L. Jongerius, P. C. A. Bruijnincx and B. M. Weckhuysen, ChemSusChem, 2012, 5, 1602–1609 CrossRef CAS PubMed.
- R. Bal and S. Sivasanker, Appl. Catal., A, 2003, 246, 373–382 CrossRef CAS.
- S. N. Cheng, I. D'cruz, M. C. Wang, M. Leitch and C. B. Xu, Energy Fuels, 2010, 24, 4659–4667 CrossRef CAS.
- D. D. Werstler, Polymer, 1986, 27, 750–756 CrossRef CAS.
-
L. Pilato, Phenolic Resins: A Century of Progress, Springer, Berlin Heidelberg, 2010, pp. 1–151 Search PubMed.
-
N. B. o. C. Engineers, Phenolic Resins Technology Handbook, NIIR Project Consultancy Services, Delhi, 2008 Search PubMed.
- J. J. Bravo-Suarez, B. Subramaniam and R. V. Chaudhari, Appl. Catal., A, 2013, 455, 234–246 CrossRef CAS PubMed.
- C. Carlini, M. Marchionna, M. Noviello, A. M. R. Galletti, G. Sbrana, F. Basile and A. Vaccari, J. Mol. Catal. A: Chem., 2005, 232, 13–20 CrossRef CAS PubMed.
- J. T. Kozlowski and R. J. Davis, ACS Catal., 2013, 3, 1588–1600 CrossRef CAS.
- K. Okuda, M. Umetsu, S. Takami and T. Adschiri, Fuel Process. Technol., 2004, 85, 803–813 CrossRef CAS PubMed.
- M. Saisu, T. Sato, M. Watanabe, T. Adschiri and K. Arai, Energy Fuels, 2003, 17, 922–928 CrossRef CAS.
-
J. E. Holladay, J. F. White, J. J. Bozell and D. Johnson, Top Value-Added Chemicals from Biomass-Volume II—Results of Screening for Potential Candidates from Biorefinery Lignin, Pacific Northwest National Laboratory (PNNL), Richland, WA (US), 2007 Search PubMed.
- A. Tejado, G. Kortaberria, C. Peña, M. Blanco, J. Labidi, J. M. Echeverría and I. Mondragon, J. Appl. Polym. Sci., 2008, 107, 159–165 CrossRef CAS PubMed.
- Z. S. Yuan, S. N. Cheng, M. Leitch and C. B. Xu, Bioresour. Technol., 2010, 101, 9308–9313 CrossRef CAS PubMed.
- A. Toledano, L. Serrano and J. Labidi, Fuel, 2014, 116, 617–624 CrossRef CAS PubMed.
- V. N. Bui, D. Laurenti, P. Delichere and C. Geantet, Appl. Catal., B, 2011, 101, 246–255 CrossRef CAS PubMed.
- K. Lundquis and L. Ericsson, Acta Chem., Scand., 1970, 24, 3681–3686 CrossRef PubMed.
- R. J. Evans, T. A. Milne and M. N. Soltys, J. Anal. Appl. Pyrolysis, 1986, 9, 207–236 CrossRef CAS.
- Q. G. Yan, Y. W. Lu, C. X. Wan, J. Han, J. Rodriguez, J. J. Yin and F. Yu, Energy Fuels, 2014, 28, 2027–2034 CrossRef CAS.
-
i. R. Dowbenko, J. I. Kroschwitz and M. Howe-Grant, Kirk-Othmer Encyclopedia of Chemical Technology, Wiley, New York, 4th edn, 1996, vol. 2, p. 106 Search PubMed.
- L. Zhou, M. D. Boot, B. H. Johansson and J. J. E. Reijnders, Fuel, 2014, 115, 469–478 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5gc01120e |
|
| This journal is © The Royal Society of Chemistry 2015 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a