
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring glutathione lyases as biocatalysts: paving the way for enzymatic lignin depolymerization and future stereoselective applications†
Pere
Picart
a,
Marc
Sevenich
a,
Pablo
Domínguez de María
bc and
Anett
Schallmey‡
*a
aJunior Professorship for Biocatalysis, Institute of Biotechnology, RWTH Aachen University, Worringerweg 3, 52074 Aachen, Germany. E-mail: a.schallmey@tu-braunschweig.de; Tel: +49 531 391 5803
bInstitut für Technische und Makromolekulare Chemie (ITMC), RWTH Aachen University, Worringerweg 1, 52074 Aachen, Germany
cSustainable Momentum, SL. Ap. Correos 3517. 35004, Las Palmas de Gran Canaria, Canary Islands, Spain Web: http://www.sustainable-momentum.net
First published on 20th July 2015
Abstract
Glutathione-dependent β-etherases and glutathione lyases are key-enzymes for the biocatalytic depolymerization of lignin. In the first step, the nucleophilic attack of glutathione to the common β-O-4-aryl-ether motif in lignin is catalyzed by β-etherases and afterwards the glutathione is removed again by the action of glutathione lyases. Given their potential impact for lignin valorization, in this paper novel glutathione lyases are reported and biocatalytically characterized based on lignin model compounds. As a result, an enzyme exhibiting increased thermostability and lowered enantioselectivity – key features for implementation of glutathione lyases in enzymatic lignin depolymerization processes – was identified. Furthermore, first mutational studies of these enzymes revealed the possibility to further alter the activity as well as enantioselectivity of glutathione lyases by means of protein engineering. From a practical perspective, one-pot multi-step processes combining β-etherases and glutathione lyases are successfully set-up, giving hints on the potential that the implementation of these biocatalysts may bring for biorefinery purposes.
Introduction
Glutathione-S-transferases (GSTs; EC 2.5.1.18) constitute a protein superfamily involved in a broad range of detoxifying processes, ranging from protection against chemical and oxidative stress to the biodegradation of xenobiotics and lignin.1 GSTs catalyze the conjugation of the tripeptide glutathione (GSH) to compounds bearing an electrophilic center, generally increasing the solubility and diminishing the toxicity of the resulting compounds.2 The highly reactive GSH thiolate anion is stabilized by a serinyl or tyrosinyl residue conserved in the thioredoxin-like N-terminal domain, involved in GSH binding (G-site) in GSTs. Likewise, a more variable helix-rich C-terminal domain is responsible for the interaction with the hydrophobic substrates (H-site).3 Besides that, one type of GSTs, namely the omega-class GSTs, though exhibiting the canonical GST fold, carry a cysteine residue in their active site catalyzing a range of thiol transfer and reduction reactions that are not catalyzed by other members.4With regard to lignin catabolism – a fundamental step for the earth's carbon cycle, several involved enzymes have been identified.5–9 Among them, GSTs (namely β-etherases and glutathione lyases) play a central role.10 For instance, Sphingobium sp. SYK-6 degrades a wide variety of lignin model compounds including β-O-4 aryl ethers.8,9 The catabolic pathway starts with at least four NADH-dependent stereospecific alcohol dehydrogenases (LigD, LigL, LigN and LigO), which catalyze the initial oxidation of the α-hydroxyl group in guaiacyl-α-veratrylglycerol (GVL) to the corresponding ketone (Fig. 1). Subsequently, a reductive ether bond cleavage via β-etherase-catalyzed reaction takes place. LigE, LigF and LigP (β-etherases) are stereoselective GSTs that catalyze the nucleophilic attack of the tripeptide glutathione (GSH) to the β-position of substrates containing β-aryl ether linkages, such as β-guaiacyl-α-veratrylglycerone (βGVG), to produce guaiacol and a GSH-conjugated aromatic compound (Fig. 1). β-Etherase catalysis triggers the stereochemical inversion, converting the β(R)-GVG substrate to GS-β(S)-VG (LigE and LigP), and the β(S)-GVG to GS-β(R)-VG (LigF).9 Subsequently, the last step of the catalytic pathway is conducted by LigG, a glutathione lyase catalyzing the elimination of GSH from the conjugate generated by the action of the β-etherases. LigG catalyzes β-S-thioether cleavage with high stereospecificity for the (R)-enantiomer of GS-βVG (produced by the β-etherase LigF) in the presence of GSH, to render glutathione disulfide (GSSG) and β-deoxy-α-veratrylglycerone (VG), which can be subsequently utilized by the bacterium as C-source.8,9 Conversely, LigG possesses only little activity towards the (S)-enantiomer of GS-βVG (produced by the action of β-etherases LigE and LigP). It has been suggested that both GS-βVG enantiomers could be interconverted by a racemase,11 and thus only LigG would be required to cleave the thioether in GS-βVG (to by-pass the need of another (S)-selective glutathione lyase that could not be identified in the genome of Sphingobium sp. SYK-6).
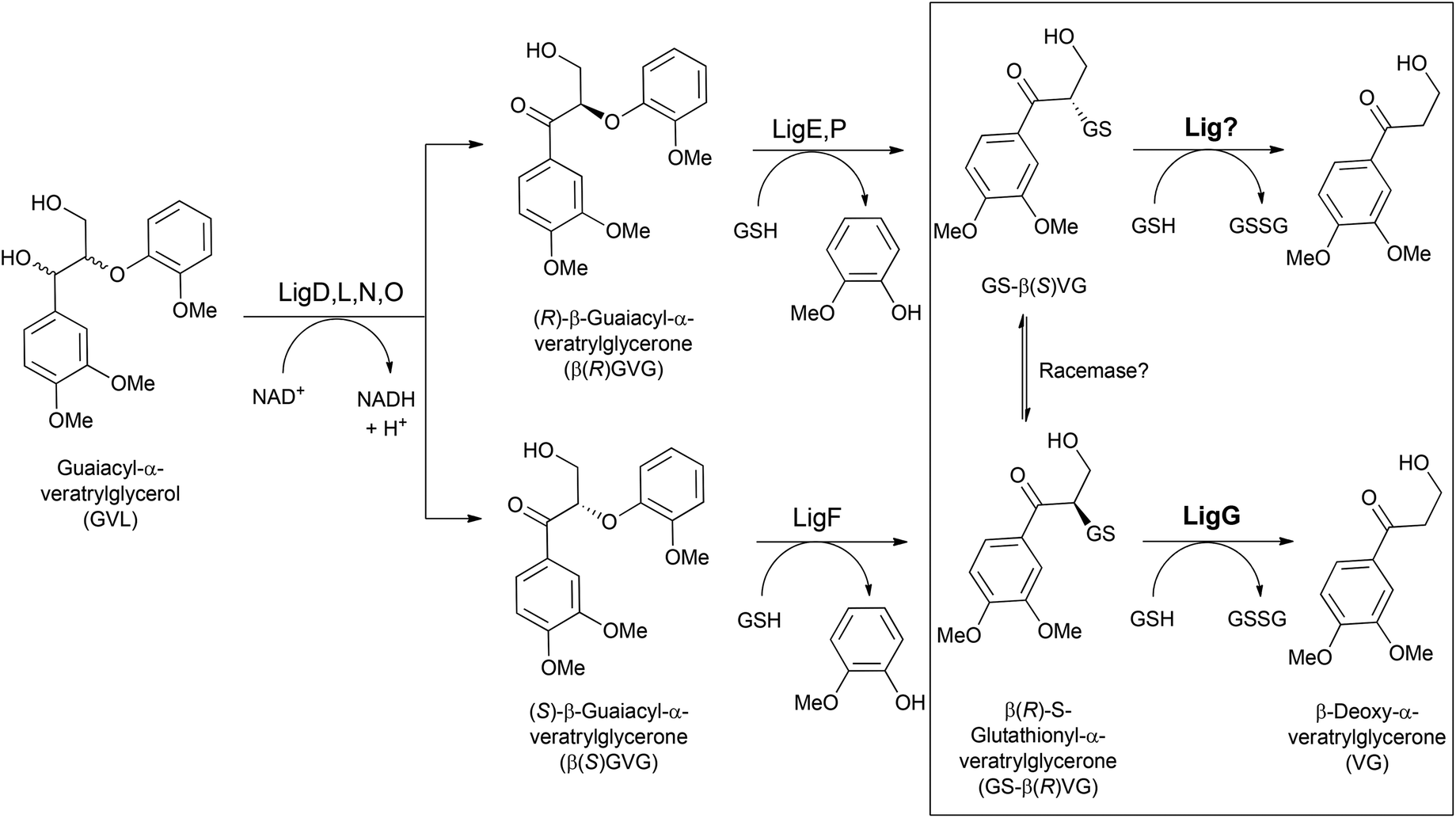 | ||
| Fig. 1 Postulated catalytic pathway for GVL in Sphingobium sp. SYK-6. NADH-dependent LigD, LigL, LigN and LigO oxidize the Cα hydroxyl group of GVL. LigE, LigF and LigP are β-etherases cleaving the two GVG enantiomers produced, yielding two enantiomeric GSH-conjugates (GS-βVG). In the last reaction step – the object of study of this paper (highlighted with a square), LigG is the glutathione lyase that uses GSH to liberate veratrylglycerone from GS-β(S)VG by formation of GSSG. In contrast, the further conversion of the GSH conjugate produced by β-etherases LigE and LigP (GS-β(S)VG) is still unknown. | ||
From a biocatalytic viewpoint, the reaction catalyzed by LigG has been scarcely studied and would represent a promising novel approach for enzymatic reactions, both covering aspects related to lignin valorization – presently an increasingly important trend,12–14 as well as incorporating potentially new approaches in biocatalytic asymmetric synthesis (e.g. performing non-natural enantioselective C–S cleavage within a multichiral active site). Therefore, given that high potential and limited development reported so far, in this work the quest for novel glutathione lyases together with their further preliminary biocatalytic characterization are reported. Moreover, structural models for the newly identified LigGs have been created and thoroughly studied, thus showing a high conservation of the active sites of these biocatalysts for the first time. Based on that structural knowledge, first glutathione lyase variants with improved activity and altered stereoselectivity have been created as well. Overall, the present work broadens the potential use of glutathione lyases in biocatalysis, paving the way for their practical use in different (fine) synthetic procedures.
Results and discussion
Identification of novel glutathione lyases (LigG): database mining, expression and purification of potential candidates
Recently, a sequence analysis and protein structure comparison of cysteine-containing glutathione transferases was performed,15 revealing that LigG [GenBank: BAK65542] from Sphingobium sp. SYK-6 possesses five homologues which form an independent cluster among cysteine-containing GSTs (omega class GSTs). Based on these findings, and to identify further potential LigG homologues exhibiting deglutathionylation activity on lignin-derived compounds, the publicly available GenBank nr database of NCBI (release 200) was screened by BLAST with the amino acid sequence of the known glutathione lyase LigG (GenBank: BAK65542.1) of Sphingobium sp. SYK6 as query. Several hits sharing up to 77% similarity to LigG were identified. Highest homology to LigG was observed for LigG-NS from Novosphingobium sp. PP1Y (GenBank: YP_004533907.1) with 66% sequence identity on protein level. In contrast, sequence identities of all other putative LigG homologues were below 53% (Fig. S1 in the ESI†). Accordingly, phylogenetic analysis of the sequences revealed that enzyme LigG clustered together with LigG-NS, whereas the other putative glutathione lyases were located on different clades in the phylogenetic tree (Fig. S2 in the ESI†). Hence, putative glutathione lyases LigG-NS (the closest LigG homologue) and LigG-TD from Thiobacillus denitrificans ATCC 25259 (GenBank: AAZ97003.1) (a more distantly related LigG homologue) were chosen in this study together with already known LigG for a detailed biochemical characterization to determine substrate specificities and stereoselectivities towards several lignin model compounds. To this end, the glutathione lyase genes ligG, ligG-TD and ligG-NS were obtained as synthetic genes with codon optimization for E. coli. Each gene was cloned into vector pET28a(+), resulting in N-terminal His-tag fusion under control of a T7 promoter, and transferred into E. coli BL21(DE3) cells. All three genes were overexpressed in E. coli BL21 (DE3) and the resulting enzymes purified via their N-terminal His-tag by affinity chromatography. All proteins were obtained in high purity (as confirmed by SDS-PAGE analysis) with yields of 30 to 200 mg L−1 cell culture (Fig. S3 in the ESI†).Biocatalytic assessment of different glutathione lyases for lignin degradative multi-step pathways
For the biocatalytic characterization of the novel glutathione lyases, the achiral model substrate GS-βVE (3) was synthetized using β-etherase LigF from Sphingobium sp. SYK-6 as biocatalyst and βGVE (1) as substrate (Fig. 2). After subsequent extractive purification with ethyl acetate to remove guaiacol, glutathione lyase substrate 3 was obtained for further biocatalytic assessments (Fig. S4 in the ESI†). Similarly, racemic 2,6-MP-VG (2) was incubated separately with β-etherases LigF or LigE in the presence of GSH to afford the two enantiomers of GSH adduct 4 (GS-β(R)-VG and GS-β(S)-VG) and 2,6-dimethoxyphenol as by-product (Fig. 2 and S4 in the ESI†).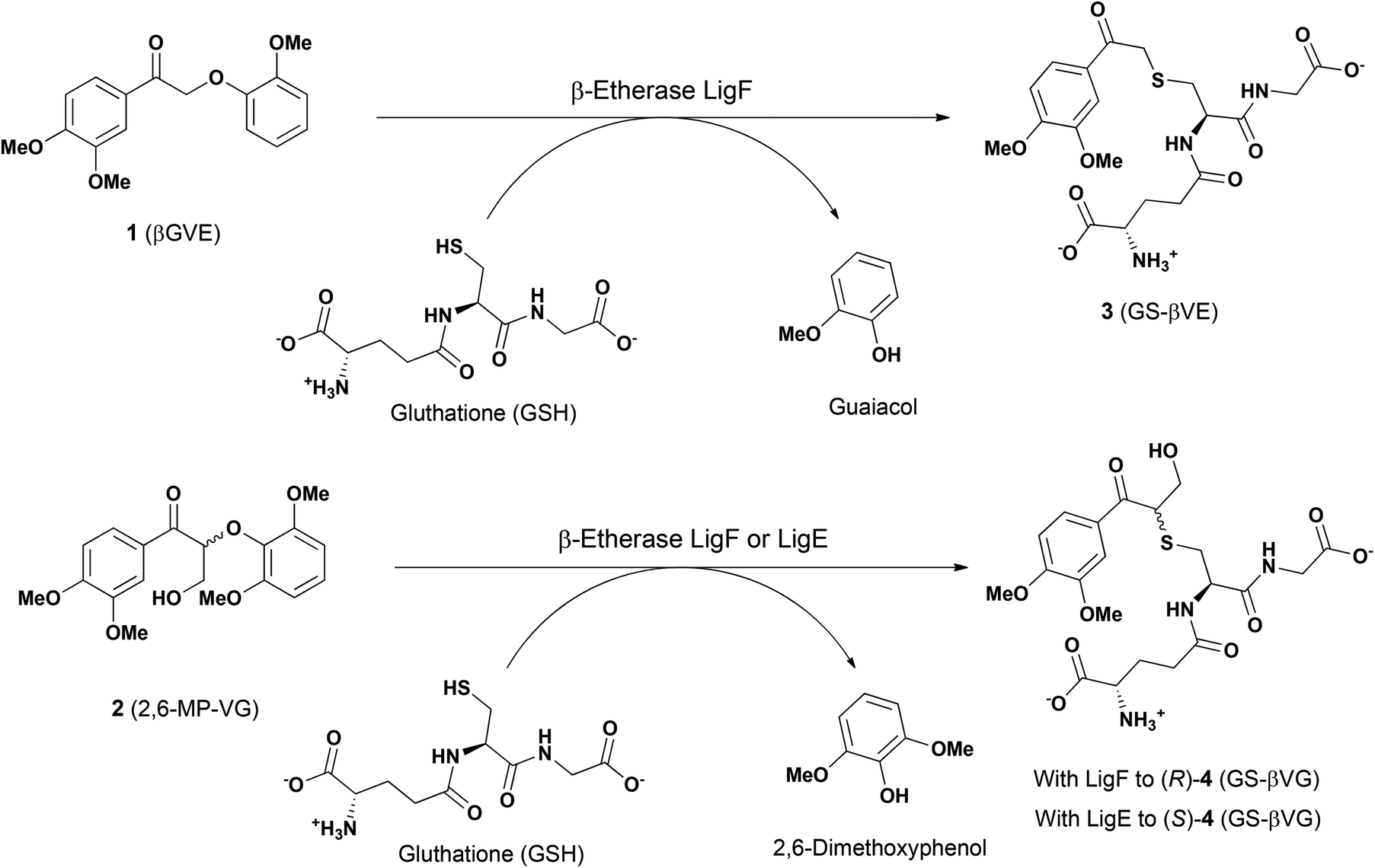 | ||
| Fig. 2 Enzymatic synthesis of the model compounds GS-βVE (3), GS-β(R)-VG ((R)-4) and GS-β(S)-VG ((S)-4), employed in this work. | ||
In a first set of experiments, achiral GS-βVE (3) was used to determine the temperature and pH profiles for each purified glutathione lyase by quantifying the amount of released product VE. As depicted in Fig. 3, the three glutathione lyases exhibited the highest activity towards GS-βVE (3) at room temperature (20 °C) (Fig. 3C) and at slightly alkaline pH 8.5–9.5 (Fig. 3B).16 Remarkably, whereas LigG and LigG-NS rapidly lost their activity when incubated at temperatures higher than 30 °C, LigG-TD still displayed about 80% of its maximum activity when incubated at 45 °C (Fig. 3C). This is remarkable as such thermostability usually goes in hand with increased enzyme stability also under the actually applied process conditions, which is a key prerequisite for the biotechnological application of enzymes.17,18 Hence, also the thermal resistance of all three glutathione lyases was studied in-depth by incubating the biocatalysts for 30 minutes at different temperatures. Afterwards, residual enzyme activity was determined in reactions with 3 under optimum reaction conditions for the glutathione lyases (Fig. 4). As a result, LigG-TD was able to retain almost full activity after 30 min incubation at even 55 °C, whereas LigG-NS and LigG rapidly lost activity upon incubation at temperatures above 40 and 45 °C, respectively. This aspect, combined with a broad pH range of activity for LigG-TD (Fig. 3B), provides promising features of this biocatalyst for practical applications.
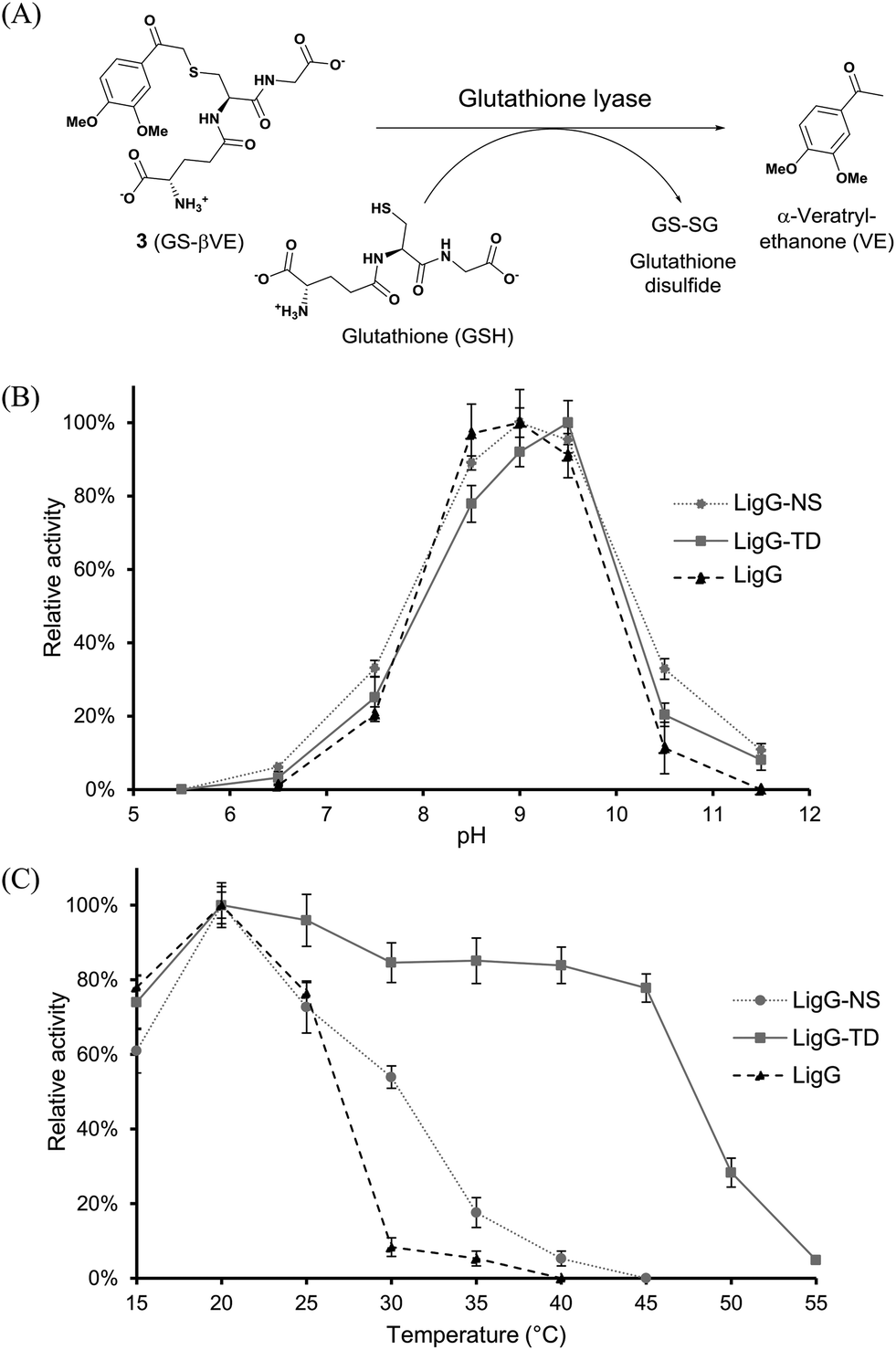 | ||
| Fig. 3 pH (B) and temperature profiles (C) of LigG homologues in the conversion of GS-βVE to afford α-veratrylethanone (A). Reactions were performed in triplicate. Values are given as % relative activity with the highest measured activity set to 100%, as determined by HPLC. Reaction conditions: 50 mM buffer, 0.4 mM GS-βVE (3), 1 mM reduced glutathione and different amounts of purified glutathione lyase (LigG 0.1 μg, LigG-NS 4 μg and LigG-TD 8 μg). | ||
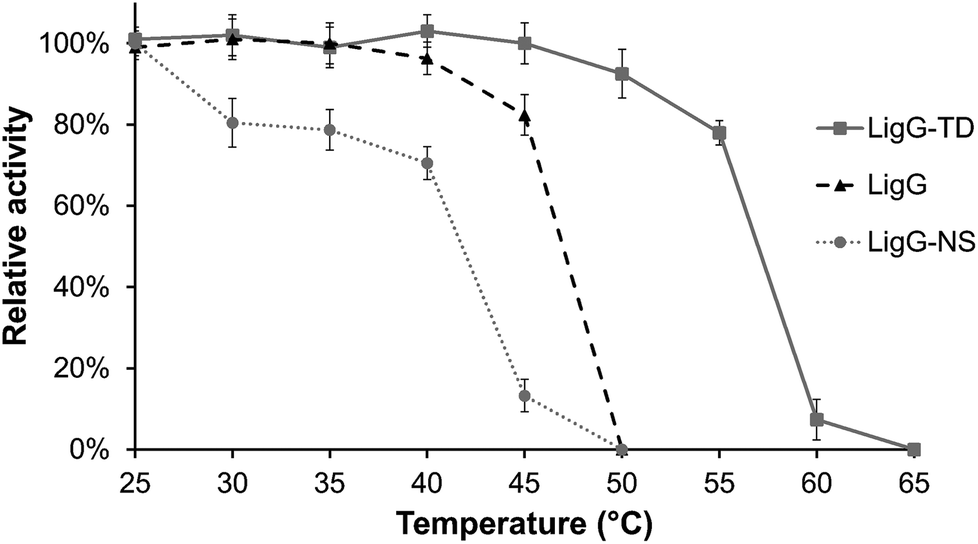 | ||
| Fig. 4 Thermal resistance of LigG, LigG-NS and LigG-TD. Glutathione lyase preparations in 50 mM glycine/NaOH buffer, pH 9.5, were incubated at different temperatures between 25 and 65 °C for 30 minutes. The samples were afterwards assayed for residual glutathione lyase activity. Reaction conditions: 50 mM glycine/NaOH buffer, pH 9.5, 0.4 mM GS-βVE (3), 1 mM reduced glutathione and different amounts of purified glutathione lyases (LigG 0.1 μg, LigG-NS 4 μg and LigG-TD 8 μg) incubated at 20 °C for 30 minutes. Reactions were performed in triplicate. | ||
To further assess the biocatalytic performance of the novel glutathione lyases, kinetic studies using 3 as substrate were conducted, affording VE as product (Fig. 5). Gratifyingly, full conversion was reached with all three enzymes, albeit at very different catalytic rates, based on their specific activities. Thus, when using glutathione in excess, LigG displayed the highest specific activity towards 3 (710 U mg−1), ca. 120-fold higher than the specific activity observed with LigG-NS (6.02 U mg−1) and 3800-fold higher than that of LigG-TD (0.185 U mg−1) (Table 2, below). Intrigued by these data, also kinetic constants KM and kcat of the three glutathione lyases for the conversion of 3 were determined (Table 1). Hence, similar to the specific activity data, the highest kcat was observed for LigG while respective values for LigG-NS and LigG-TD were three to four orders of magnitude lower. In contrast, KM values for all three glutathione lyases towards 3 were rather similar. To study also the influence of GSH concentration on enzyme activity, reactions with 0.4 mM GS-βVE (3) were repeated and specific activities of the three glutathione lyases were determined, but this time applying only equal amounts of GSH (0.4 mM). As a result, full conversion of 3 was still reached with all three enzymes, however, at lower rates. Whereas specific activity of LigG was reduced by half (352 U mg−1) under these conditions, specific activities of LigG-NS (4.21 U mg−1) and LigG-TD (0.153 U mg−1) were less affected, indicating that the latter enzymes might exhibit a lower KM for GSH as compared to LigG. Overall, LigG is a highly active catalyst in the conversion of 3 though exhibiting reduced stability, whereas LigG-TD is less reactive but offers much higher stability. In any case, being the glutathione lyases active biocatalysts for the intended processes, the set-up of in vitro evolutionary approaches might surely deliver improved variants with desired characteristics for biocatalysis (see also below).
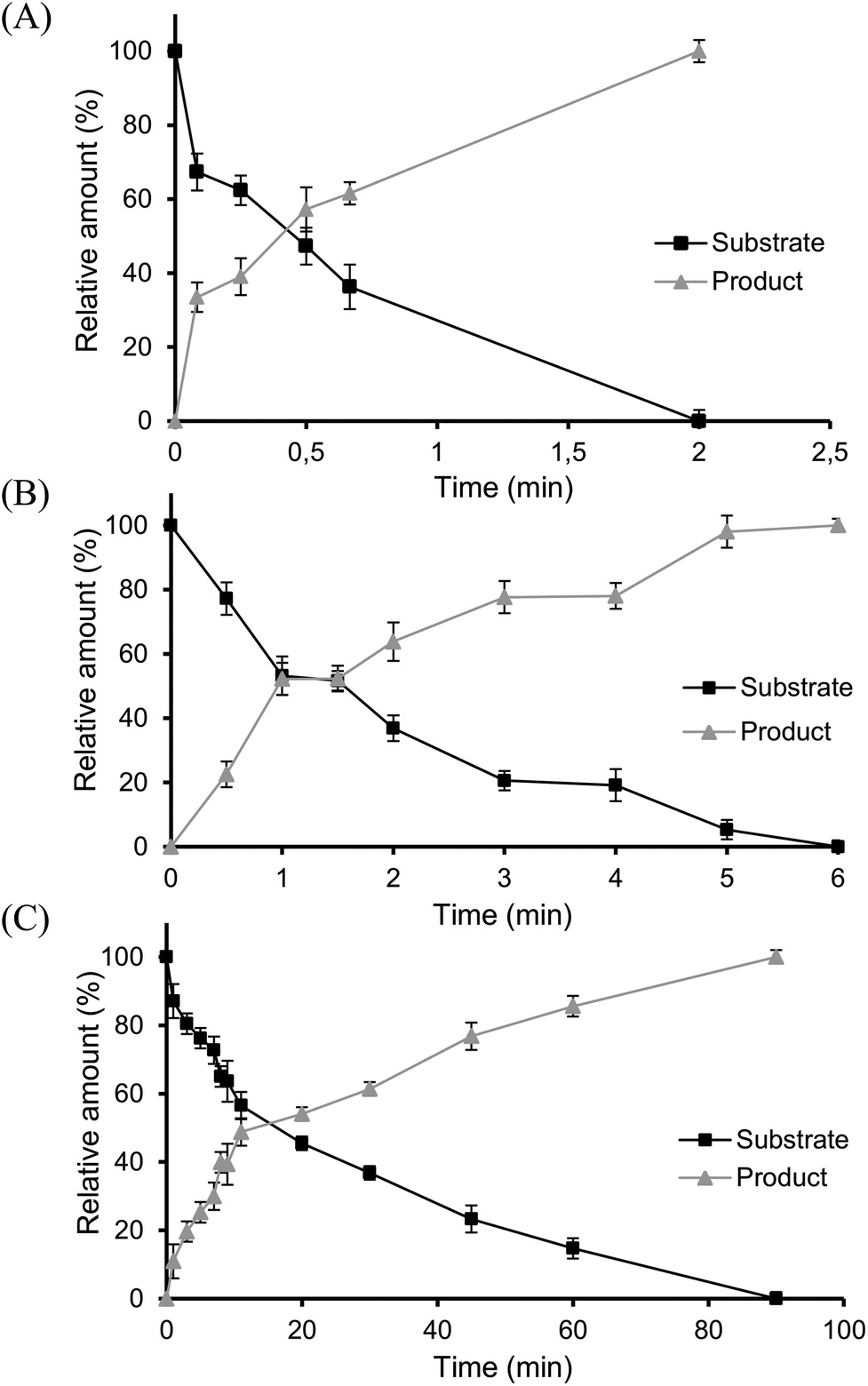 | ||
| Fig. 5 Kinetic studies of the glutathione lyases LigG (A), LigG-NS (B) and LigG-TD (C) using 3 as substrate. Reaction conditions: 50 mM buffer, 0.2 mM GS-βVE (3), 1 mM reduced glutathione and different amounts of purified LigG enzymes (LigG 0.1 μg, LigG-NS 4 μg and LigG-TD 8 μg) at 20 °C. Reactions were performed in triplicate. | ||
| Glutathione lyase | K M (mM) | k cat (s−1) |
|---|---|---|
| LigG | 0.17 | 2042 |
| LigG-NS | 0.25 | 9.5 |
| LigG-TD | 0.36 | 0.3 |
| Glutathione lyase | GS-β(R)-VG | GS-β(S)-VG | Rate R/Rate S | GS-βVE |
|---|---|---|---|---|
| LigG | 1.738 | 0.094 | 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 489 489 |
710 |
| LigG_M167A | 1.732 (100%) | 0.071 (75%) | 24394 |
5.84 (1%) |
| LigG_S109A | 1.844 (106%) | 0.107 (117%) | 17233 |
780 (110%) |
| LigG_L117A | 1.850 (106%) | 0.124 (132%) | 14919 |
291 (41%) |
| LigG-NS | 71.4 | 0.022 | 3227 | 6.02 |
| LigG-NS_M173A | 70.5 (99%) | 0.018 (82%) | 3916 | 0.091 (2%) |
| LigG-NS_T116A | 430 (603%) | 0.029 (132%) | 14827 |
2.87 (48%) |
| LigG-TD | 36.2 | 0.117 | 309 | 0.185 |
| LigG-TD_F165A | 16.0 (44%) | 0.093 (78%) | 172 | 0.090 (47%) |
| LigG-TD_F165M | 18.3 (51%) | 0.100 (83%) | 183 | 0.102 (53%) |
| LigG-TD_V108A | 34.2 (94%) | 0.127 (108%) | 269 | 0.169 (90%) |
| LigG-TD_V11G | 21.1 (58%) | 0.115 (100%) | 183 | 0.143 (74%) |
| LigG-TD_M116A | 44.9 (124%) | 0.099 (83%) | 462 | 0.346 (187%) |
Exploring the stereoselectivity of the novel glutathione lyases via multi-step one-pot enzymatic processes
The above-described temperature and pH profiles of the glutathione lyases are analogous to those reported previously for the respective β-etherases.19 In this sense, envisaging further practical options for biocatalysis, the set-up of combined one-pot processes might become of certain relevance for environmentally-friendly processes, leading to less waste formation upon reduction of downstream units. Herein, one-pot, multi-step processes by combination of β-etherases and glutathione lyases were studied. Moreover, to get insights on the enantioselectivity that the novel glutathione lyases may display, chiral substrates were introduced in the evaluation as well. In this area, recently it was reported that LigG stereospecifically cleaves GS-β(R)VG in the presence of GSH producing glutathione disulfide (GSSG) and VG.11Therefore, to investigate and compare the stereoselectivities of the novel glutathione lyases, the deglutathionylation activity on the β(S)- and β(R)-enantiomers of GS-βVG ((S)-4 and (R)-4) were assessed along a two-step process (without intermediate work-up procedures). To that end, substrate 2 was first subjected to β-etherase catalysis for 60 min, using either LigF or LigE, resulting in the formation of GS-β(R)-VG and GS-β(S)-VG, respectively.11 Afterwards, glutathione lyase as well as fresh GSH were added to the reaction mixtures containing either (R)-4 or (S)-4 and reactions were incubated at optimal pH and temperature (pH 9 or 9.5 and 20 °C). As a result, selective conversion of the GS-βVG enantiomers by all applied glutathione lyases was observed (Fig. 6).
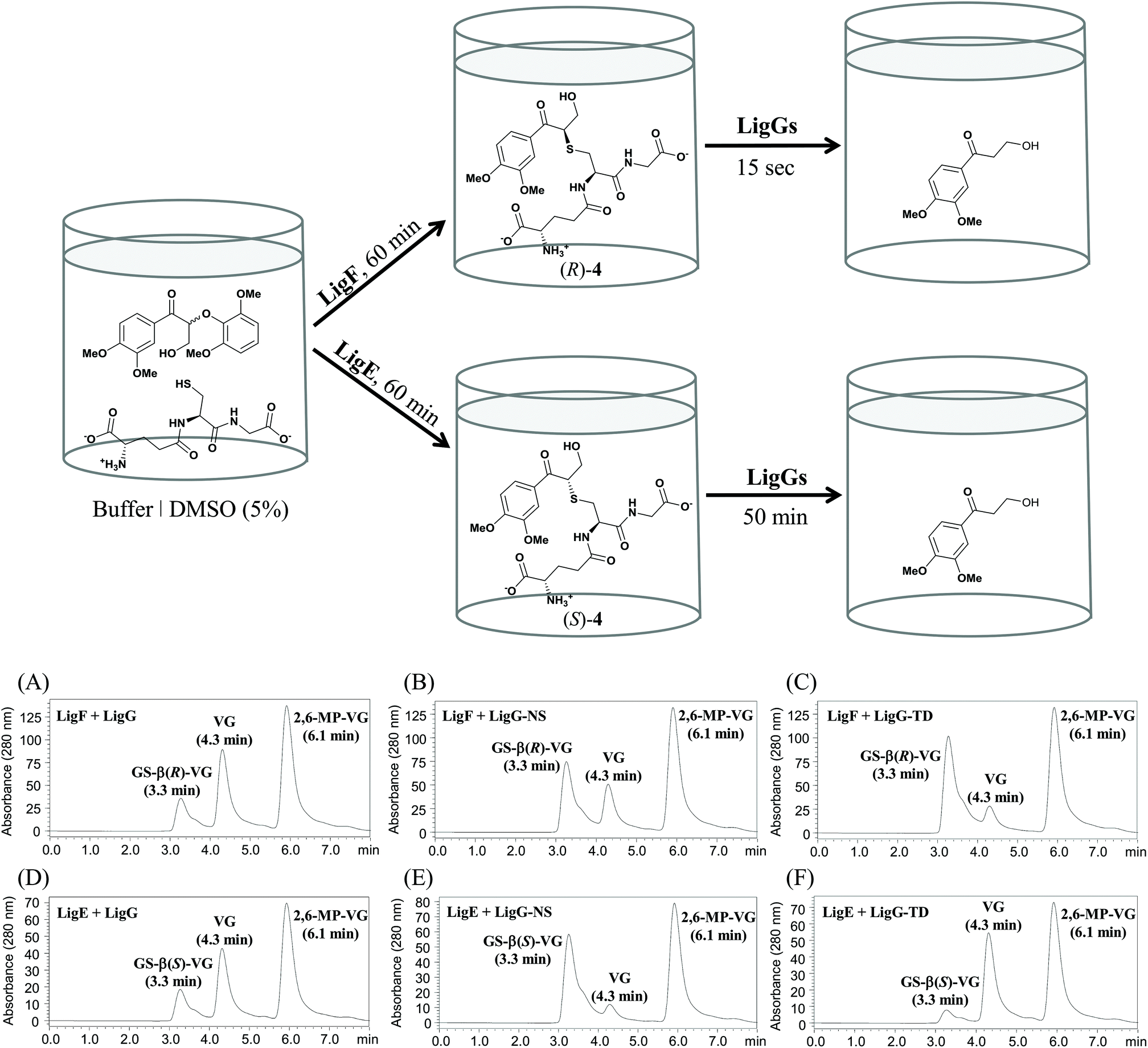 | ||
| Fig. 6 Above: enantioselective one-pot multi-step process combining β-etherases and glutathione lyases. Conditions: 20 mM glycine/NaOH buffer, pH 9 or 9.5, 1 mM reduced glutathione, 0.8 or 0.4 mM substrate 2 and 20 μg purified β-etherase LigF or LigE, respectively. Reactions were incubated at 25 °C for 60 min before adding different amounts of purified glutathione lyases as well as fresh GSH (1 mM) and further incubation at 20 °C for different incubation times. Below: HPLC chromatograms of conversions of substrate 4 catalyzed by different glutathione lyases. Peaks are labeled with the respective retention time and compound name. (A–C) Reactions of 0.4 mM (R)-4, obtained with β-etherase LigF, and different amounts of glutathione lyase enzymes (LigG (A): 0.1 μg; LigG-NS (B) and LigG-TD (C): each 1 μg) for 15 s. (D–F) Reactions of 0.2 mM (S)-4, obtained with β-etherase LigE, and 30 μg of different glutathione lyase enzymes (D: LigG, E: LigG-NS, F: LigG-TD) for 50 min. | ||
Analogous to LigG, LigG-NS and LigG-TD preferentially catalyzed the thioether cleavage of the GS-β(R)-VG enantiomer, produced by the action of the β-etherase LigF, resulting in full conversion to the product VG after rather short incubation times of 1 minute. In this multi-step process, LigG displayed the highest specific activity towards the (R)-enantiomer (1738 U mg−1), ca. 24-fold higher than the specific activity observed with LigG-NS (71.4 U mg−1) and 48-fold higher than that of LigG-TD (36.2 U mg−1) (Table 2, below). Conversely, the reactions conducted with the (S)-enantiomer of GS-βVG – formed by β-etherase LigE – and the different glutathione lyases showed rather low conversions (Fig. 6D–F). In those processes, LigG-TD displayed the highest specific activity on GS-β(S)-VG (0.117 U mg−1) followed by LigG (0.094 U mg−1). Overall, LigG exhibited the highest stereoselectivity of the three glutathione lyases converting GS-β(R)-VG 18500 times faster than GS-β(S)-VG. Remarkably, LigG-TD turned out to be the least selective, though still converting GS-β(R)-VG 300 times faster than GS-β(S)-VG. Nevertheless, full conversion of the (S)-enantiomer of GS-βVG could be achieved with LigG-TD after longer incubation times (3 h). In this context, the efficient purification of low-molecular mass lignins produced by the action of β-etherases requires an efficient deglutathionylation reaction of the lignin hydrolysis products. Given the fact that lignin contains racemic centers – due to its random radical biosynthesis20,21 – LigG-TD seems an attractive candidate to be explored in further lignin-based valorization processes. Interestingly, LigG, LigG-NS and LigG-TD displayed significantly higher specific activities on the chiral substrate 4 (specifically the (R)-enantiomer) than the achiral substrate 3, indicating that the side chain indeed seems to have a significant influence on the deglutathionylation activity. An analogous pattern has been observed earlier for β-etherases,19 where most of the enzymes are preferentially active on substrate 2 as compared to substrate 1.
Homology models and mutational studies of the glutathione lyases to reveal determinants of stereoselectivity
As mentioned earlier, efficient application of glutathione lyases together with β-etherases in lignin depolymerization ideally requires enzymes exhibiting no stereoselectivity towards their substrates. On the other hand, the existence of enantioselectivity in glutathione lyases may also open novel biocatalytic research avenues for asymmetric synthesis apart from lignin biocracking. Hence, to find possible explanations for the differences in stereoselectivity of the three glutathione lyases, structural insights into the enzymes’ active sites are required. Recently, the crystal structure of LigG with GSH bound in the active site was reported (PDB: 4G10) giving also first insights on how the substrate molecule could be bound.15 Based on this crystal structure, homology models for LigG-NS and LigG-TD were generated using the automated structure-modelling program I-tasser, an advanced computer algorithm for protein structure and function predictions.22 Interestingly, alignment of the resulting homology models with the structure of LigG and careful inspection of the enzymes’ active sites revealed a high conservation of most active site residues. Only four positions on the H-site, the region of the active site of GSTs where the non-GSH moiety of a glutathionylated substrate would bind, where found to differ among the three enzymes (or at least between LigG and LigG-TD) (Fig. 7). These residues are Gly14, Ser109, Leu117 and Met167 in the amino acid sequence of LigG. While LigG-TD differs in all four positions as compared to LigG, LigG-NS carries identical amino acids to LigG in three of the mentioned positions (Fig. S1 in the ESI†).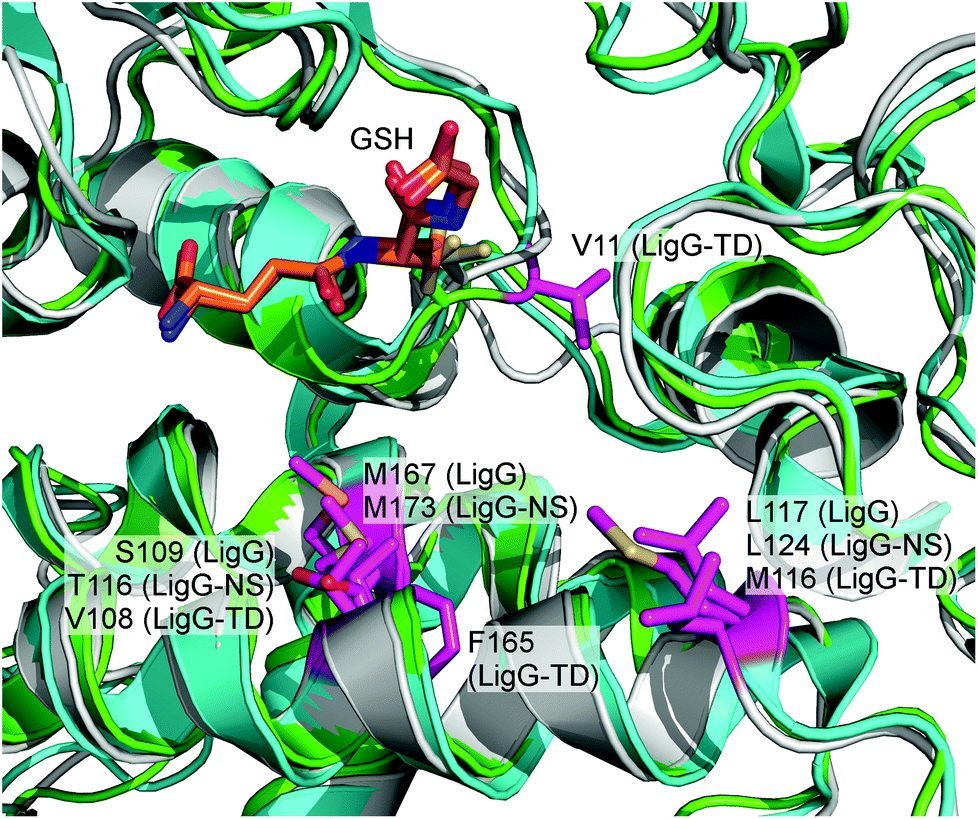 | ||
| Fig. 7 Overlay of LigG crystal structure (PDB: 4G10) and homology models of LigG-NS and LigG-TD. The GSH molecule is shown as orange sticks. The active-site residues in which the three enzymes differ are labeled and colored in pink. The LigG structure is shown in cyan, the LigG-NS model in grey and the LigG-TD model in green. | ||
To investigate a possible influence of these amino acid residues on the stereoselectivity of the glutathione lyases in the deglutathionylation of GS-βVG, specific mutations were introduced in the respective gene sequences by site-directed mutagenesis. The resulting mutants, LigG_S109A, LigG_L117A, LigG_M167A, LigG-NS_T116A, LigG-NS_M173A, LigG-TD_V11G, LigG-TD_V108A, LigG-TD_M116A, LigG-TD_F165A and LigG-TD_F165M, were expressed and purified to homogeneity as previously described for the wild-type enzymes. Afterwards, the mutants were characterized regarding their specific activities in the conversion of enantiomers GS-β(R)-VG ((R)-4) and GS-β(S)-VG ((S)-4) as well as the achiral substrate GS-βVE (3) and compared to the results using wild-type glutathione lyases (Table 2).
As it can be observed, mutagenesis of amino acid positions 167 and 173 in LigG and LigG-NS, respectively, as well as positions 109 and 116 in LigG and LigG-NS, respectively, had a similar effect. Thus, while mutation Met → Ala decreased just slightly the enzymes’ activity towards GS-β(S)VG – making them even more selective for the (R)-enantiomer, the activity for the achiral substrate 3 was virtually negligible in the variants, whereas in case of mutants LigG_S109A and LigG-NS_T116A, enzyme activity in the conversion of 4 was generally increased. For LigG-NS_T116A, the improvement towards (R)-4 was much higher (6-fold) as compared to (S)-4 yielding an even more (R)-selective glutathione lyase. Contrarily, mutagenesis of LigG-TD led to completely different outcomes. Thus, while mutation V108A (equivalent to position S109 in LigG) hardly changed activity as compared to the wild-type enzyme, mutation F165A (as well as F165M) led to a significantly decreased activity, especially towards (R)-4 as well as 3. Additionally, position V11 in LigG-TD was changed to glycine as LigG as well as LigG-NS carry a glycine at the equivalent amino acid position. Interestingly, mutant LigG-TD_V11G still exhibited the same specific activity towards (S)-4 while the enzyme's activity towards the (R)-enantiomer was decreased by half, yielding an even less (R)-selective glutathione lyase. Furthermore, when mutating the equivalent amino acid positions 116 and 117 in LigG-TD and LigG, respectively, again opposite results were obtained. While mutant LigG-TD_M116A was suddenly more (R)-selective as compared to wild-type, mutation L117A in LigG resulted in increased activity towards the (S)-enantiomer and hence reduced (R)-selectivity. Additionally, activity of LigG-TD_M116A towards 3 was significantly increased while LigG_L117A exhibited reduced activity with this achiral substrate. Overall, these results point towards a different binding mode for substrate 4 (as well as 3) within the active sites of LigG and LigG-TD whereas binding of these substrates by LigG and LigG-NS can be assumed to be very similar. Furthermore, our results confirm that the four active-site residues, which were found to differ among glutathione lyases LigG, LigG-NS and LigG-TD, indeed have a significant impact on the enzymes’ activity and selectivity allowing to further engineer these enzyme characteristics in the future. To gain more detailed insights on the structural determinants for differences in stereoselectivity in LigG and LigG-TD, however, determination of LigG-TD's crystal structure will be essential.
Experimental
Identification of novel glutathione lyases
Glutathione lyase-encoding sequences were identified by screening the publicly available GenBank nr database (release 200) of NCBI by using the basic local alignment search tool (BLAST) algorithm. The database was screened by using the amino acid sequence of LigG (GenBank: BAK65542.1) from Sphingobium sp. SYK-6. Protein sequences obtained were analyzed and aligned by using ClustalX,23 and a phylogenetic tree was constructed by using the neighbor-joining method.24 The closest homologue to LigG (LigG-NS from Novosphingobium sp. PP1Y, GenBank: YP_004533907.1) as well as a more distantly related sequence (LigG-TD from Thiobacillus denitrificans ATCC 25259, GenBank: AAZ97003.1) were selected for further studies.Cloning and site-directed mutagenesis of glutathione lyases
The genes coding for LigG, LigG-NS and LigG-TD were synthesized by Geneart GmbH (Regensburg/Germany) with codon optimization for E. coli, and cloned into a pET28a(+) vector using restriction sites NdeI and HindIII resulting in the addition of a N-terminal His-tag. The primers for PCR-based site-directed mutagenesis of the glutathione lyase gene sequences were designed according to the QuikChange™ Mutagenesis manual (Agilent Technologies, CA) and are listed in Table S1 in the ESI.† QuikChange™ site-directed mutagenesis was performed according to the manufacturer's instructions to generate the following mutants: LigG_M167A, LigG L117A and LigG_S109A; LigG-NS_M173A and LigG-NS_T116A; LigG-TD_F165A, LigG-TD_F165M, LigG-TD_V108A, LigG-TD_M116A and LigG-TD_V11G.Enzyme expression and purification
Expression of all enzymes was performed in E. coli BL21 (DE3). Strains for all experiments were grown in terrific broth medium (TB) with kanamycin (50 mg L−1) to an OD of about 0.6 at 37 °C. After induction with 0.1 mM isopropyl-β-D-thiogalactopyranoside (IPTG), expression was performed overnight at 25 °C. For the purification of enzymes, glutathione lyase-harbouring cells were re-suspended in binding buffer (20 mM sodium phosphate buffer, pH 7.4, 500 mM NaCl, 20 mM imidazole) and disrupted by sonication. After removal of cell debris by centrifugation, the resulting cell lysate was applied on a Ni2+-NTA column for purification of the His-tagged enzymes with an ÄKTA purification system (GE Healthcare, U.S.) at a flow rate of 1 mL min−1. His-tagged enzymes were eluted from the column using elution buffer (20 mM sodium phosphate buffer, pH 7.4, 500 mM NaCl, 500 mM imidazole). Protein fractions were analyzed by SDS-PAGE and Coomassie staining to check the presence and purity of recombinant enzymes. Fractions containing a purified glutathione lyase were combined, desalted using PD-10 columns (GE Healthcare) and quantified by the Bradford assay.25β-Etherases LigE and LigF from Sphingobium sp. SYK-6 were also recombinantly expressed in E. coli BL21 (DE3) and purified as described earlier.19
Synthesis of model substrates
Different lignin-type model substrates were used in this study (Fig. 2). Substrates 1, β-guaiacyl-α-veratrylethanone (βGVE), and 2, β-(2,6-methoxyphenoxy)-α-veratrylglycerone (2,6-MP-VG), were synthesized as described previously.19 The achiral β-etherase substrate 1 (βGVE) was used to produce the glutathione lyase substrate GS-βVE (3) enzymatically. The 10 mL assay mixture contained 0.8 mM substrate 1, 1 mM glutathione (reduced) and 20 μg of purified β-etherase LigF in 20 mM glycine/NaOH buffer, pH 9. Reactions were carried out at 25 °C for 60 min. After incubation, a portion of the reaction mixture (1 mL) was taken for HPLC analysis to confirm that 1 was completely converted to GS-βVE as previously reported.11,19 The remaining reaction mixture (total volume: 9 mL) was then extracted three times with 4.5 mL ethyl acetate to remove guaiacol from the mixture. The resulting aqueous phase containing 0.8 mM GS-βVE was collected and analyzed on a Prominence modular HPLC system (Shimadzu) equipped with a Nucleosil 100-5 C18 column (4.6 by 150 mm; Macherey-Nagel, Germany) to check the purity of GS-βVE. For HPLC analysis, a mixture of water (49%), acetonitrile (50%), and phosphoric acid (1.0%) was used as mobile phase with a flow rate of 1.0 mL min−1. The aqueous stock solution of GS-βVE (3) served as substrate to study the pH and temperature profiles as well as kinetics of the glutathione lyases analyzed in this work. GS-βVE, VE and substrate 1 eluted from the HPLC column with retention times of 3.1, 5.4 and 9.2 min, respectively. All compounds were detected at 280 nm. Substrates 1 and 2 were dissolved in pure dimethylsulfoxide (DMSO) before their use in the enzyme assays.Activity assays
Activities of the three glutathione lyases towards substrate 3 were determined using a 1 mL assay mixture containing 50 mM buffer, 0.4 mM GS-βVE (3) as substrate, 1 mM reduced glutathione and different amounts of purified glutathione lyase (LigG 0.1 μg, LigG-NS 4 μg and LigG-TD 8 μg). Temperature and pH profiles were studied in a temperature range of 15 °C to 55 °C and a pH range of 5.5 to 11.5, respectively. The thermal resistance of glutathione lyases was studied by incubating the purified enzyme preparations in 50 mM glycine/NaOH buffer, pH 9.0 (LigG and LigG-NS) or 9.5 (LigG-TD), at 25, 30, 35, 40, 45, 50, 55, 60 and 65 °C for 30 min. Withdrawn samples were assayed for residual glutathione lyase activity at optimal conditions (pH 9.0 or 9.5 and 20 °C) and compared to the controls (glutathione lyases kept on ice), which were set to 100%. The effect of GSH concentration on glutathione lyase activity was analyzed by performing reactions with 0.4 mM GS-βVE (3) and only 0.4 mM GSH in 50 mM glycine/NaOH buffer at optimal pH and temperature of the glutathione lyases. Reactions were stopped by the addition of methanol (final concentration, 25% v/v) after different incubation times and precipitated protein was removed by centrifugation. The deglutathionylation activity was determined by quantifying the amount of released α-veratrylethanone (VE) by HPLC using a calibration curve of commercial VE (Sigma-Aldrich). One unit is defined as the amount of enzyme that converts 1 μmol of substrate min−1. Specific activity is expressed as units per milligram of protein.For the determination of kinetic parameters KM and kcat of the three glutathione lyases for the conversion of GS-βVE (3), 0.1 mL reactions in 50 mM glycine/NaOH buffer were carried out at optimal temperature and pH conditions for each enzyme using different amounts of 3 (between 0.05 and 1.6 mM), 1 mM GSH and purified glutathione lyase (LigG: 0.025 μg, LigG-NS: 0.5 μg, LigG-TD: 50 μg). Reactions were stopped after 15 s (LigG), 2 min (LigG-NS) or 5 min (LigG-TD) by the addition of methanol. Specific deglutathionylation activity was determined for each reaction as described above. Resulting activities were plotted against the applied substrate concentration and data fitted to the Michaelis-Menten equation using Origin.
To analyze the activity and stereoselectivity of the glutathione lyases on the chiral substrate GS-βVG (4), either β(R)-S-glutathionyl-α-veratrylglycerone (GS-β(R)-VG) or β(S)-S-glutathionyl-α-veratrylglycerone (GS-β(S)-VG) were generated by β-etherase treatment of substrate 2 using β-etherases LigF and LigE, respectively.19 First, calibration curves for quantification of GS-βVG (4) by HPLC were generated using 1 mL reactions containing 20 mM glycine/NaOH buffer, pH 9, between 0.05 mM to 0.8 mM substrate 2, 1 mM reduced glutathione and 20 μg purified β-etherase LigF or LigE. Reactions were incubated at 25 °C for 60 min to allow for 50% conversion of substrate 2 (full conversion of one enantiomer). Reactions were stopped by the addition of methanol (final concentration, 25% v/v), precipitated protein was removed by centrifugation (15000g for 5 min) and the supernatants were analyzed by HPLC as described above. In order to determine LigG, LigG-NS and LigG-TD activity on (R)- and (S)-4, reaction mixtures containing 0.8 mM of 2 in 20 mM glycine/NaOH buffer, pH 9, and 1 mM reduced glutathione were incubated with 20 μg of either LigF or LigE as described above, followed by addition of fresh GSH (1 mM) and incubation with one of the three glutathione lyases studied in this work. Reactions were carried out at optimal temperature conditions for each enzyme and incubated for different times. After incubation with glutathione lyase, reactions were stopped by the addition of methanol, precipitated protein was removed by centrifugation and supernatants were analyzed by HPLC using the conditions mentioned above. Thus, specific deglutathionylation activities towards both enantiomers GS-β(R)-VG and GS-β(S)-VG were determined by measuring substrate GS-βVG consumption after incubation with each glutathione lyase. GS-βVG (4), VG and substrate 2 eluted from the HPLC column with retention times of 3.3, 4.3 and 6.0 min, respectively.
Homology models
3-D models of LigG-NS and LigG-TD protein sequences were generated by I-TASSER, a web based server,21,26 based on the crystal structure of LigG (PDB: 4G10).15 Visualization of protein structures was carried out using the PyMOL software (http://www.pymol.org/).Conclusions
Enzymatic cascade reactions for lignin depolymerization could pave the way for an efficient and selective lignin valorization. In that respect, β-etherases hold great potential for the selective ether bond cleavage in lignin-like polymers. However, the GSH-adduct formed by the action of β-etherases must be further cleaved by the action of glutathione lyases to render the aromatic compounds by releasing the glutathione group attached to the GSH-conjugate. The existence of β(R)- and β(S)-configurations in native plant lignins would need both β(R)- and β(S)-stereospecific β-etherases and glutathione lyases – or unselective enzymes – to maximize the efficiency of the cascade reaction. In this paper, we have described that novel glutathione lyases LigG-NS and LigG-TD as well as known LigG exhibit a remarkable stereospecificity to structurally related lignin model compounds with enantiopreference towards the β(R)-configuration, although they still kept low activity on the β(S)-configuration. In addition to that, we provided first evidence that stereospecificity of these enzymes can be modulated by active-site mutations. Thus, mutant LigG-NS_T116A showed a remarkable increase of its activity and stereospecificity towards the (R)-enantiomer whereas the equivalent mutation in LigG-TD_V108A increased the activity for the (S)-enantiomer resulting in a less (R)-selective enzyme. This highlights the importance of specific amino acid residues in the stereoselectivity of the glutathione lyases and demonstrates the possibility to alter the enzymes’ selectivity (as well as activity) by protein engineering. Last but not least, the combination of β-etherases and glutathione lyases in one-pot processes provides promising prognoses for the future application of these biocatalysts in biorefineries.Acknowledgements
We thank Jakob Mottweiler and Prof. Carsten Bolm (Institute of Organic Chemistry, RWTH Aachen University) for the generous provision of lignin model substrates 1 and 2. This work was performed as part of the Cluster of Excellence “Tailor-Made Fuels from Biomass”, which is funded by the Excellence Initiative of the German Research Foundation to promote science and research at German universities.References
- J. D. Hayes, J. U. Flanagan and I. R. Jowsey, Annu. Rev. Pharmacol. Toxicol., 2005, 45, 51–88 CrossRef CAS PubMed.
- N. Allocati, L. Federici, M. Masulli and C. Di Ilio, FEBS J., 2009, 276, 58–75 CrossRef CAS PubMed.
- A. Oakley, Drug Metab. Rev., 2011, 43, 138–151 CrossRef CAS PubMed.
- P. G. Board, Drug Metab. Rev., 2011, 43, 226–235 CrossRef CAS PubMed.
- E. Masai, Y. Katayama, S. Nishikawa, M. Yamasaki, N. Morohoshi and T. Haraguchi, FEBS Lett., 1989, 249, 348–352 CrossRef CAS PubMed.
- E. Masai, Y. Katayama, S. Kubota, S. Kawai, M. Yamasaki and N. Morohoshi, FEBS Lett., 1993, 24, 135–140 CrossRef.
- E. Masai, Y. Katayama, S. Nishikawa and M. Fukuda, J. Ind. Microbiol. Biotechnol., 1999, 23, 364–373 CrossRef CAS PubMed.
- E. Masai, Y. Katayama and M. Fukuda, Biosci., Biotechnol., Biochem., 2007, 71, 1–15 CrossRef CAS PubMed.
- E. Masai, A. Ichimura, Y. Sato, K. Miyauchi, Y. Katayama and M. Fukuda, J. Bacteriol., 2003, 185, 1768–1775 CrossRef CAS PubMed.
- The studied β-etherases and glutathione lyases were not shown to be extracellular enzymes and no secretion signal sequence can be found at the N-terminus of the respective enzymes encoded by Sphingobium sp. SYK-6 and the other Sphingomonads. Masai et al.5 initially found that the β-etherase enzyme from Sphingobium sp. SYK-6 was tightly associated with the cellular membrane. Moreover, the genome sequence of that strain carries a large number of genes encoding major facilitator superfamily transporters. Hence, it was suggested that these transporters are required for the uptake of a large variety of lignin-derived compounds. See also: E. Masai, N. Kamimura, D. Kasai, A. Oguchi, A. Ankai, S. Fukui, M. Takahashib, I. Yashirob, H. Sasakib, T. Haradab, S. Nakamurab, Y. Katanob, S. Narita-Yamadab, H. Nakazawab, H. Harac, Y. Katayamad, M. Fukudaa, S. Yamazakib and N. Fujita, J. Bacteriol., 2012, 194, 534–535 CrossRef CAS PubMed.
- D. L. Gall, H. Kim, F. Lu, T. J. Donohue, D. R. Noguera and J. Ralph, J. Biol. Chem., 2014, 289, 8656–8667 CrossRef CAS PubMed.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 1246843 CrossRef PubMed.
- J. G. Linger, D. R. Vardon, M. T. Guarnieri, E. M. Karp, G. B. Hunsinger, M. A. Franden, C. W. Johnson, G. Chupka, T. J. Strathmann, P. T. Pienkos and G. T. Beckham, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 12013–12018 CrossRef CAS PubMed.
- Z. Strassberger, S. Tanase and G. Rothenberg, RSC Adv., 2014, 4, 25310–25318 RSC.
- E. Meux, P. Prosper, E. Masai, G. Mulliert, S. Dumarçay, M. Morel, C. Didierjean, E. Gelhaye and F. Favier, FEBS Lett., 2012, 586, 3944–3950 CrossRef CAS PubMed.
- The alkaline pH optimum of glutathione lyases is analogous to the one reported for β-etherases and is possibly influenced by the pKa of the thiol side chain of GSH, which might be required to be deprotonated for catalysis. According to literature, this pKa should be around 9.1 to 9.6. See, for instance, (a) The Merck Index, 11th ed., 1989, entry# 4369; (b) R. M. C. Dawson, D. C. Elliot, W. H. Elliot and K. M. Jones, Data for Biochemical Research, Oxford University Press, New York, 3rd edn, 1987, pp. 16–17 Search PubMed.
- L. D. Unsworth, J. van der Oost and S. Koutsopoulos, FEBS J., 2007, 274, 4044–4056 CrossRef CAS PubMed.
- M. de Champdoré, M. Staiano, M. Rossi and S. D'Auria, J. R. Soc. Interface, 2007, 22, 183–191 CrossRef PubMed.
- P. Picart, C. Müller, J. Mottweiler, L. Wiermans, C. Bolm, P. Domínguez de María and A. Schallmey, ChemSusChem, 2014, 7, 3164–3171 CrossRef CAS PubMed.
- T. Bugg, M. Ahmad, E. Hardiman and R. Rahmanpour, Nat. Prod. Rep., 2011, 28, 1883–1896 RSC.
- M. E. Brown and M. C. Chang, Curr. Opin. Chem. Biol., 2014, 19, 1–7 CrossRef CAS PubMed.
- Y. Zhang, BMC Bioinformatics, 2008, 9, 40 CrossRef PubMed.
- J. D. Thompson, T. J. Gibson, F. Plewniak, F. Jeanmougin and D. G. Higgins, Nucleic Acids Res., 1997, 25, 4876–4882 CrossRef CAS PubMed.
- N. Saitou and M. Nei, Mol. Biol. Evol., 1987, 4, 406–425 CAS.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS PubMed.
- R. Ambrish, A. Kucukural and Y. Zhang, Nat. Protocols, 2010, 5, 725–738 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5gc01078k |
| ‡ Present address: Institute for Biochemistry, Biotechnology and Bioinformatics, Technische Universität Braunschweig, Spielmannstr. 7, 38106 Braunschweig, Germany. |
| This journal is © The Royal Society of Chemistry 2015 |