
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA monolithic device for CO2 photoreduction to generate liquid organic substances in a single-compartment reactor†
Takeo
Arai
ab,
Shunsuke
Sato
ab and
Takeshi
Morikawa
ab
aToyota Central R&D Labs., Inc., 41-1 Yokomichi, Nagakute, Aichi 480-1192, Japan. E-mail: takeo-arai@mosk.tytlabs.co.jp
bJST, ACT-C, 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan
First published on 27th May 2015
Abstract
A solar to chemical energy conversion efficiency of 4.6% was demonstrated for CO2 photoreduction to formate utilizing water as an electron donor under simulated solar light irradiation to a monolithic tablet-shaped device. The simple CO2 photoreduction system was realized by exploiting the effect of the carbon substrate on selective CO2 reduction in the presence of oxygen and selective H2O oxidation over IrOx catalysts in the presence of formate.
The development of clean energy sources and CO2 recycling are crucial approaches to the depletion of fossil resources and climate change induced by an increase in CO2 emissions; solar energy is an ideal and realistic way to achieve these goals. Direct photoconversion from CO2 to chemical fuels or organic raw materials for chemical products using water is an ultimate way to realize a sustainable carbon-neutral society. Interest in the direct solar reduction of CO2 to liquid organic substances has grown due to the higher energy density and accessible storage advantages compared with hydrogen energy. However, direct solar reduction of CO2 to liquid organics by electrons and protons extracted from water molecules, similar to photosynthesis in plants, has been considered much more difficult than the hydrogen generation reaction. The CO2 molecule is highly stable, so that challenges to the realization of photoinduced CO2 recycling using water include the higher potential required for CO2 reduction than that of hydrogen generation and the low product selectivity in the presence of water. For example, the potentials for CO2 reduction to CO and formic acid were calculated from the Gibbs free energy change to be −0.10 V (vs. SHE) and −0.17 V (vs. SHE). There have been several reports on highly efficient catalysts for selective CO2 photoreduction;1–4 however, they required a sacrificial reagent as an electron donor to facilitate CO2 reduction. More importantly, electrical coupling of reactions in a closed system, in which selective water oxidation to extract electrons and selective CO2 reduction are functionally coupled, is extremely difficult. Few studies on CO2 photoreduction utilizing water as an electron donor have been reported for semiconductor photocatalysts;5–8 however, some of them were conducted with an external electrical or chemical bias to assist the reaction, and others were active only under ultraviolet light irradiation. While in the case of complex catalysts, other molecules as sacrificial electron donors are required in place of H2O to facilitate the reaction. Recently, we reported a solar-to-chemical conversion efficiency of 0.14% for formate generation without an external electrical bias through the combination of a photocathode consisting of a p-type semiconductor, zinc-doped indium phosphide (InP), coated with a [Ru{4,4′-di(1H-pyrrolyl-3-propylcarbonate)2,2′-bipyridine}(CO)2]n polymer (RuCP)9 catalyst for CO2 reduction and a SrTiO3 photoanode for water oxidation in a two-compartment reactor separated by a proton exchange membrane, in which water is utilized as both an electron donor and a proton source.10,11 However, this system should be improved further for practical application. Solar CO2 recycling requires not only high efficiency and selectivity, but also consideration of the design of simplified low-cost systems. The reduction of CO2 to formate using a CO2 electrolyzer equipped with an indium electrode and a Si photovoltaic cell has also been reported.12 However, an applied voltage of 4–5 V is necessary to operate the CO2 electrolyzer due to the substantial negative potential for CO2 reduction over the indium electrode.12,13 The solar-to-chemical energy conversion efficiency of the system was less than 2%, while the solar power conversion efficiency was 8–9% because the combination of the CO2 electrolyzer and photovoltaic cells is accompanied by a potential drop due to various resistances (resistances in the solar cell, between the solar cell and the electrode, and permeance at the proton exchange membrane) and also requires a complicated potential transformer for impedance matching. These issues could be overcome using a monolithic tablet-shaped device, which composed of a light absorber, a cathode for CO2 reduction and an anode for H2O oxidation. The device is an independent standalone system immersed in a single-compartment reactor filled with water and CO2. Furthermore, this device is applicable on a large scale by simply setting the devices in an array configuration because the impedance loss caused by scale-up is very small and determined by the direction of tablet thickness. It is also advantageous that replacement of the catalytic components is easier and cheaper.
In the present study, we demonstrate a simple CO2 photoreduction reaction that utilizes a monolithic tablet-shaped device. The device is composed of a porous ruthenium complex polymer (p-RuCP) as a CO2 reduction catalyst, iridium oxide (IrOx) as a water oxidation catalyst, and a triple-junction of amorphous silicon-germanium (SiGe-jn, Fig. S1, ESI†) as a light absorber. p-RuCP was developed by chemical polymerization of RuCP on a porous carbon substrate (see the ESI†) and connected to the stainless steel side (narrower bandgap side of the junction) of the SiGe-jn. An IrOx nanocolloid containing no organic substances was synthesized according to a previously reported method14 and coated on the indium tin oxide (ITO) surface of the SiGe-jn (see the ESI†). These components were functionally coupled to realize CO2 photoreduction with a high solar-to-chemical conversion efficiency in a single-compartment reactor. Two essential technologies were developed to realize the monolithic tablet-shaped device for CO2 photoreduction; selective CO2 reduction, even in the presence of H2O and O2, and selective H2O oxidation, even in the presence of organic substances. Without separation functions, such as in the photosynthesis of plants, the organic substances produced from CO2 in a liquid phase can be re-oxidized and competes with the H2O oxidation to O2 reaction. This is also the case for the other side reaction, in which H+ and O2 produced from H2O can be reduced and compete with the CO2 reduction reaction in a single compartment reactor. Both these reactions cancel the products generated at both sides; therefore, selective CO2 reduction and H2O oxidation are necessary for the artificial photosynthesis system to produce liquid chemicals from CO2 and H2O in a single-compartment reactor.
The first key technology is the selective CO2 photoreduction in aqueous media, which was established using the semiconductor–metal-complex hybrid system. We have previously reported the combination of a band-controlled semiconductor for visible-light excitation with a metal-complex to catalyze selective CO2 reduction, in which photoexcited electrons are transferred from the conduction band of the semiconductor to the lowest unoccupied molecular orbital (LUMO) of the metal-complex catalyst within tens of picoseconds, which resulted in highly selective CO2 photoreduction.15,16 Because the concept of the semiconductor–metal-complex hybrid catalyst is highly versatile, we have constructed the InP/RuCP photocathode catalyst for CO2 reduction.17
The CO2 reduction potential (ECR) of p-RuCP was estimated to be −0.18 V (vs. RHE) (Fig. S2 and Table S1, ESI†), which is substantially lower than that of metal electrodes. For example, the CO2 reduction potential for formate generation over an indium electrode was reported to be −0.90 V (vs. RHE).13 This is the advantage of using a metal-complex catalyst, but there still remain issues such as the competing reduction reaction of O2 evolved simultaneously over the water-oxidation site with the CO2 reduction reaction over InP/RuCP. O2 is more easily reduced than CO2 and the elimination of O2 is thus necessary for CO2 reduction using a metal complex catalyst.
As evidence, the current efficiency ηC, for formate formation over InP/RuCP in the presence of O2 is shown in Fig. 1A (see Table S2, ESI,† for details). The InP/RuCP photocathode was used as the working electrode in a three-electrode configuration. The Pt wire and a Hg/Hg2SO4 electrode were used as counter and reference electrodes, respectively. 0.1 M phosphate buffer was used as the electrolyte. Gaseous CO2 containing various concentrations of oxygen was continuously bubbled into the reactor during the reaction, and the CO2 photoreduction reaction was conducted at +0.21 V (vs. RHE) for 1 h. ηC was significantly decreased from 93% (at 0% O2) to 6% (at 7% O2) with an increase in the oxygen concentration due to selective O2 reduction (O2 → O2−) competing with CO2 reduction. Therefore, a system was developed to enhance the CO2 reduction selectivity over RuCP, even in the presence of O2. A porous carbon cloth (CC) sheet made of carbon fiber was applied, which possesses a low activity for hydrogen generation (Fig. S3, ESI†), and a surface area that is two orders of magnitude larger than that of the flat and smooth surface of a conventional semiconductor film. ηC for formate formation over RuCP coated onto CC (CC/p-RuCP) is also shown in Fig. 1A. CO2 photoreduction reaction was conducted at +1.41 V (vs. RHE). An ηC of 76% was observed, even in the presence of 7% O2. Comparison of the current–potential characteristics of CC under Ar and CO2 atmospheres suggested preferential adsorption of CO2 on the CC (Fig. S4, ESI†). It was thus assumed that gaseous CO2 in aqueous solution was concentrated adjacent to the CC on which RuCP was polymerized. When the RuCP was applied onto the surface of the stainless-steel side (lower bandgap side) of the SiGe-jn, where H2 generation is preferential (Fig. S3, ESI†), ηC for formate generation was only 0.3%, while it significantly improved to 94 ± 5% when using CC/p-RuCP (Table S3, ESI†).
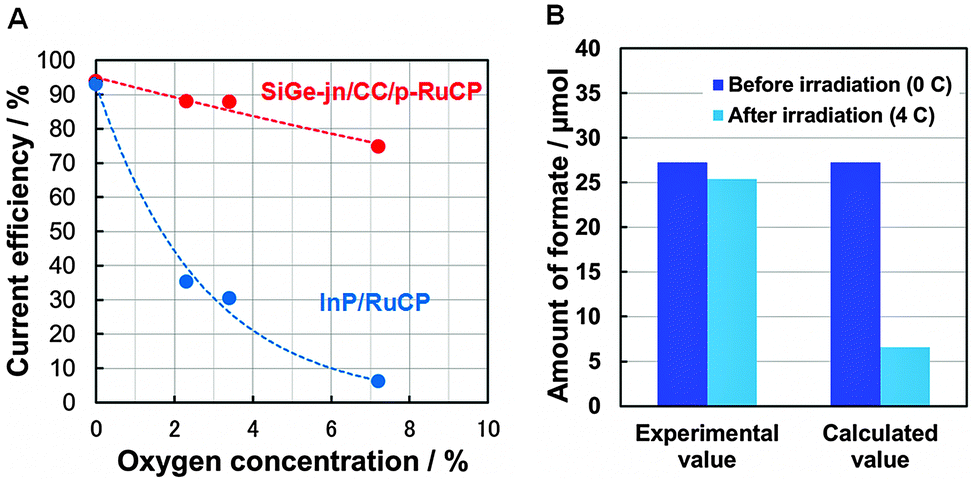 | ||
| Fig. 1 (A) Current efficiency for formate formation as a function of oxygen concentration in CO2 flow over SiGe-jn/CC/p-RuCP and InP/RuCP photocathodes under light irradiation for 1 h in 0.1 M phosphate buffer electrolyte. (B) Amount of formate observed before and after photodegradation over the IrOx/SiGe-jn photoanode. | ||
The second key technology is highly selective H2O oxidation, even in the presence of formate. Photoreduction of CO2 to liquid organic products in a single-compartment cell is difficult because the products accumulated in the liquid phase can be re-oxidized to CO2 on the surface of water oxidation catalysts such as TiO2, as previously reported.11 Iridium oxide, an excellent catalyst for H2O oxidation,18 is reported to oxidize formic acid on Ti/IrO2 electrodes in perchloric acid.19 In the present study, formate was not decomposed in the oxidation reaction over the IrOx catalyst in a phosphate buffer solution. Fig. 1B shows the photodegradation of formate over the IrOx/SiGe-jn photoanode performed in 0.1 M phosphate buffer electrolyte containing ca. 27 μmol (ca. 1.4 mM) of formate with a three-electrode configuration at −0.25 V (vs. RHE). The bias voltage was set at the operation point of the present device estimated with a two-electrode configuration (details are provided later). Even though the total charge of 4 C observed during the photoanodic reaction over the IrOx/SiGe-jn photoanode was sufficient to decompose ca. 21 μmol formate (calculated value in Fig. 1B), the amount of formate decreased was negligible, which indicates that the IrOx catalyst has very low activity for the photodegradation of formate. It was also reported that the current efficiency for formate degradation over Ti/IrO2 in a perchloric acid electrolyte decreased from over 90% to less than 10%, according to the decrease in the concentration of formate from ca. 550 mM to ca. 30 mM, which indicates that the applied current exceeds the mass transport limit of formate.17 Therefore, it is supposed that the anodic photocurrent over the IrOx/SiGe-jn photoanode also exceeded the mass transport of formate on the surface of the IrOx catalyst and generated oxygen from water. The negligible photodegradation of formate over the IrOx/SiGe-jn photoanode was also observed in sulfate, borate and carbonate solutions (Fig. S5, ESI†).
SiGe-jn was selected as the semiconductor for photoexcitation, as employed in the previous report.20 SiGe-jn has an open circuit voltage (VOC) of 2.1 V, which is thermodynamically adequate to oxidize water and extract electrons (approximately 1.4 V), and the p–i–n and tunnel junctions in SiGe-jn facilitate the charge separation and transfer of photoexcited electrons and holes.21 The conduction band minimum (ECBM) of SiGe-jn was estimated to be −0.52 V (vs. RHE) from the current–potential characteristics (Fig. S6, ESI†). The ECBM of SiGe-jn is more negative than the ECR of −0.18 V (vs. RHE, see Fig. S2, ESI†) over p-RuCP; therefore, electron transfer from SiGe-jn in a photoexcited state to p-RuCP is thermodynamically possible. Furthermore, the valence band maximum (EVBM) of SiGe-jn was estimated to be 1.58 V (vs. RHE) by subtracting VOC from ECBM. The onset potential estimated from the current–potential curve for water oxidation (EWO) over IrOx was also estimated to be 1.5 V (vs. RHE) (Fig. S7, ESI†). Thus, the EVBM of SiGe-jn is also more positive than the EWO over IrOx, so that IrOx/SiGe-jn can facilitate H2O oxidation. The semiconductor–metal-complex hybrid system employs a technical advantage of the Ru-complex catalyst, i.e., a low potential required for CO2 reduction; therefore, the system using the SiGe-jn can demonstrate CO2 reduction to formate coupled with water oxidation reaction with a very low voltage of less than 2.1 V.
Based on these two key technologies, the IrOx/SiGe-jn/CC/p-RuCP monolithic tablet-shaped device was constructed. A schematic illustration of the device is shown in Fig. 2A. The CO2 photoreduction reaction was conducted by immersing the device in an aqueous phosphate buffer solution saturated with gaseous CO2 (pH 6.4) in a single-compartment reactor under irradiation with solar simulated light (1 sun, AM1.5, Fig. S8, ESI†). Formate as a liquid organic substance was generated from only CO2 and H2O raw materials using sunlight as an energy source. The time course for the generation of formate during the CO2 photoreduction reaction using the monolithic device with the best performance under simulated solar light irradiation (through a square-shaped slit of 0.25 cm2) is shown in Fig. 2B. Formate was generated continuously during irradiation for 6 h and the solar-to-chemical conversion efficiency was calculated to be 4.6% from the rate of formic acid generation (μmol HCOOH s−1) multiplied by the change in Gibbs free energy per mole of formic acid formation from CO2 and water (at 298 K, ΔG = 270 kJ mol−1) according to a previous report.22 This result is supported by the photocurrent observed at the operation point shown in the current–potential characteristics of the IrOx/SiGe-jn photoanode and the CC/p-RuCP cathode in the three-electrode configuration (Fig. S9, ESI†). A similar photocurrent was also observed at zero bias (vs. counter electrode) with the two-electrode configuration using the IrOx/SiGe-jn and CC/p-RuCP electrodes (Fig. S10, ESI†). The solar-to-chemical conversion efficiency for the present CO2 reduction is comparable to that observed for solar hydrogen production utilizing a similar light absorber20 and also reached a level comparable to the theoretical maximal photosynthetic energy conversion efficiency for C3 crops (e.g., rice and wheat), estimated to be 4.6%.23 This reaction was also confirmed to be reproducible. The mean value of the solar-to-chemical conversion efficiency utilizing three monolithic devices was 4.3%.
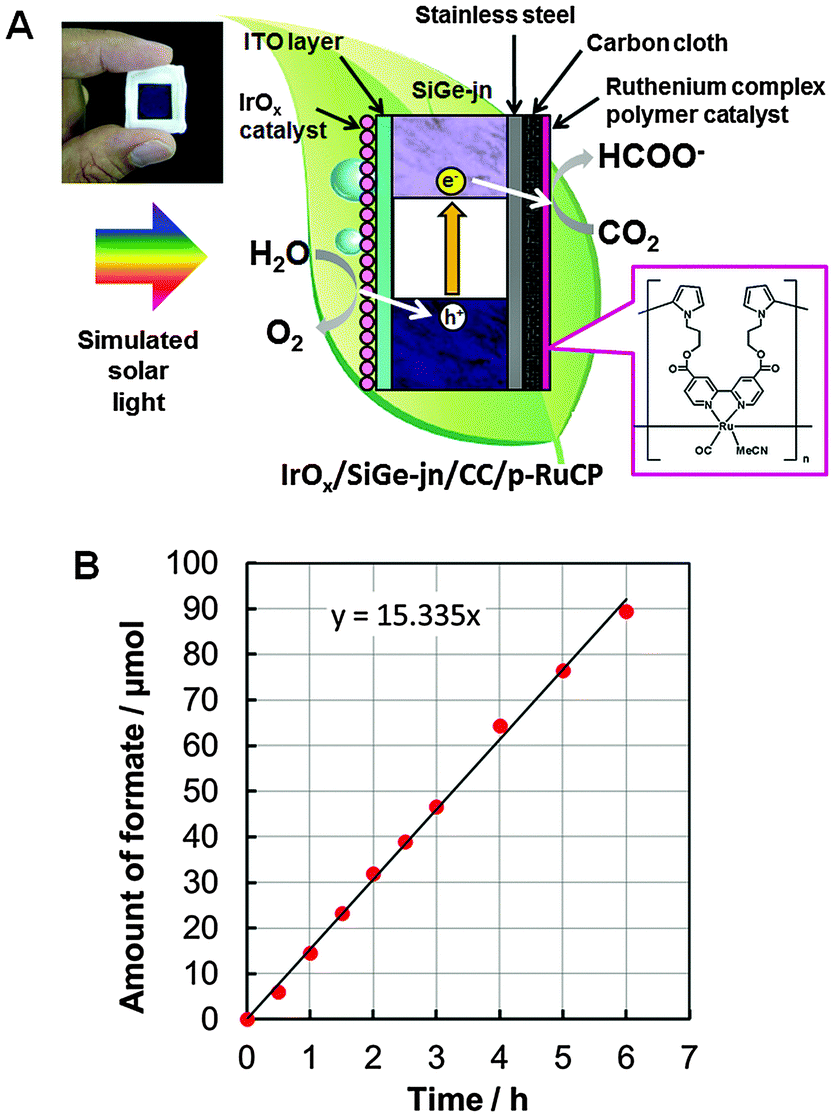 | ||
| Fig. 2 (A) Schematic illustration of the IrOx/SiGe-jn/CC/p-RuCP monolithic tablet-shaped device for CO2 photoreduction. (B) Time course for the generation of formate during the CO2 photoreduction reaction using IrOx/SiGe-jn/CC/p-RuCP under simulated solar light irradiation (1 sun, AM1.5, 0.25 cm2). The IrOx/SiGe-jn/CC/p-RuCP monolith was immersed in a single-compartment quartz reactor filled with CO2-saturated phosphate buffer solution (pH 6.4). | ||
To verify a stoichiometric reaction, the quantity of oxygen molecules generated over IrOx/SiGe-jn/CC/p-RuCP during CO2 photoreduction was determined from in situ measurements. The experiment was conducted in a flow reactor equipped with a single-compartment Pyrex glass cell and a gas chromatograph. The IrOx/SiGe-jn/CC/p-RuCP monolithic device was immersed in 0.1 M phosphate buffer saturated with gaseous CO2 (flow rate 20 mL min−1). A solar simulator equipped with an AM1.5 filter was also used as the light source. The irradiation conditions were different from that used in Fig. 2B due to the experimental setup (see the ESI† for details). During light irradiation, oxygen bubbles were clearly observed at the IrOx surface. The total amount of oxygen generated after 2 h irradiation was 26.4 μmol (Fig. 3A), which corresponds to 105.6 μmol of photoexcited holes, while 50.2 μmol of formate in the liquid phase and 3.6 μmol of hydrogen in the gas phase were generated simultaneously after 2 h irradiation, which accounts for 107.6 μmol of photoexcited electrons. The amount of electrons was approximately equal to that of photoexcited holes, which strongly suggest that stoichiometric CO2 reduction is achieved using electrons extracted from water molecules. Furthermore, the ratio of the number of electrons consumed to generate formate to that for the total reduction products was 93%, which is in good agreement with the current efficiency for formate production observed in a half reaction over the SiGe-jn/CC/p-RuCP photocathode (Table S3, ESI†).
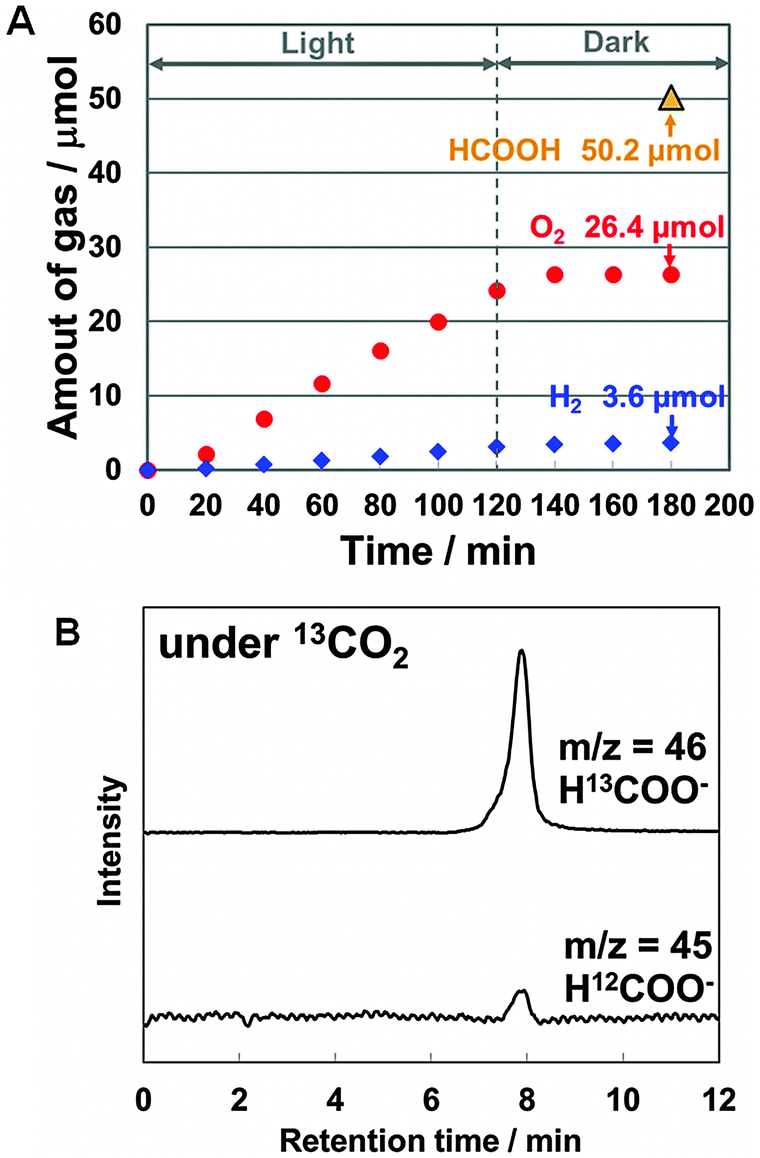 | ||
| Fig. 3 (A) Experimental verification for formate production from CO2 and water molecules; time course for oxygen and hydrogen generation during CO2 photoreduction using a tablet-shaped wireless configuration. The amount of formate was determined at the end of the photoreaction. (B) IC-TOFMS spectra from a tracer experiment utilizing 13CO2. | ||
In addition, isotope tracer analysis was conducted using the IrOx/SiGe-jn/CC/p-RuCP device in 0.1 M phosphate buffer saturated with gaseous 13CO2 (flow rate 20 mL min−1) to avoid the possibility of experimental error warned by Mul and colleagues.22 Ion chromatography interfaced with time-of-flight mass spectrometry (IC-TOFMS) was used to clarify the formation of H13COO− (m/z = 46) (Fig. 3B, mass spectra are shown in Fig. S11, ESI†), which confirmed that the carbon source for formate was the CO2 molecules. Thus, formate was generated from only CO2, H2O and solar energy over the monolithic device.
Conclusions
A monolithic tablet-shaped device was developed for CO2 photoreduction to liquid organics in a single-compartment cell. CO2 photoreduction reaction in almost neutral pH aqueous media containing gaseous CO2 under irradiation of simulated solar light (1 sun, AM1.5) was successfully demonstrated by immersing the monolithic tablet-shaped device composed of a semiconductor–metal-complex hybrid system in a single-compartment reactor. The solar-to-chemical energy conversion efficiency for formate generation reached 4.6% without external electrical and chemical bias voltages, or a membrane for the separation of products. CO2 photoreduction to formate utilizing H2O as an electron donor and a proton source was confirmed by the stoichiometry of reduction/oxidation products and isotope tracer analysis. Highly selective CO2 reduction and water oxidation in the presence of competitive substrates were identified as the primary technological keys that enable the total reaction to occur in a single-compartment reactor. The secondary key is to facilitate electron transfer from the H2O oxidation catalyst to the CO2 reduction catalyst by overcoming the slow CO2 reduction rate over the metal-complex catalyst. These technologies provide a general and paradigm-changing concept for standalone artificial photosynthesis. The efficiency of the present system can be improved by further optimizing the composition of materials and band engineering of semiconductors and metal complexes because the concept of the semiconductor–metal-complex hybrid system is highly versatile.24–28 Therefore, the development of new semiconductor materials and metal-complex catalysts is crucial for further improvement. Furthermore, since a costly iridium oxide catalyst is impractical, development of a water oxidation catalyst is still important. Formic acid, which was generated in the present system, is an industrially useful organic substance29 that has the potential to function as a liquid energy carrier with a high density for CO or H2, and as a chemical form for CO2 fixation with high density. Direct formic acid fuel cells30 and the utilization of formic acid as a hydrogen carrier for fuel cells31 have been investigated. A recent report indicated that methanol can be synthesized via disproportionation of formic acid catalyzed by a molecular iridium species.32 As an extension, appropriate replacement of the catalysts in the present system could lead to the direct generation of alcohol.33Acknowledgements
The authors thank Dr R. Asahi for useful discussion. The authors also thank M. Yamamoto, M. Asaoka, H. Uchiyama, N. Kawahara, and M. Kondo for technical assistance.Notes and references
- J. Hawecker, J.-M. Lehn and R. Ziessel, J. Chem. Soc., Chem. Commun., 1983, 536–538 RSC.
- Y. Tamaki, K. Koike, T. Morimoto and O. Ishitani, J. Catal., 2013, 304, 22–28 CrossRef CAS.
- T. Morimoto, C. Nishiura, M. Tanaka, J. Rohacova, Y. Nakagawa, Y. Funada, K. Koike, Y. Yamamoto, S. Shishido, T. Kojima, T. Saeki, T. Ozeki and O. Ishitani, J. Am. Chem. Soc., 2013, 135, 13266–13269 CrossRef CAS PubMed.
- S. Sato, T. Morikawa, T. Kajino and O. Ishitani, Angew. Chem., Int. Ed., 2013, 52, 988–992 CrossRef CAS PubMed.
- M. Halmann, Nature, 1978, 275, 115–116 CrossRef CAS.
- T. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature, 1979, 277, 637–638 CrossRef CAS.
- K. Iizuka, T. Wato, Y. Miseki, K. Saito and A. Kudo, J. Am. Chem. Soc., 2011, 133, 20863–20868 CrossRef CAS PubMed.
- S. Yotsuhashi, H. Hashiba, M. Deguchi, Y. Zenitani, R. Hinogami, Y. Yamada, M. Deura and K. Ohkawa, AIP Adv., 2012, 2, 042160 CrossRef.
- S. Chardon-Noblat, A. Deronzier, R. Ziessel and D. Zsoldos, J. Electroanal. Chem., 1998, 444, 253–260 CrossRef CAS.
- S. Sato, T. Arai, T. Morikawa, K. Uemura, T. M. Suzuki, H. Tanaka and T. Kajino, J. Am. Chem. Soc., 2011, 133, 15240–15243 CrossRef CAS PubMed.
- T. Arai, S. Sato, T. Kajino and T. Morikawa, Energy Environ. Sci., 2013, 6, 1274–1282 CAS.
- J. L. White, J. T. Herb, J. J. Kaczur, P. W. Majsztrik and A. B. Bocarsly, J. CO2 Util., 2014, 7, 1–5 CrossRef CAS.
- Z. M. Detweiler, J. L. White, S. L. Bernasek and A. B. Bocarsly, Langmuir, 2014, 30, 7593–7600 CrossRef CAS PubMed.
- Y. Zhao, E. A. Hernandez-Pagan, N. M. Vargas-Barbosa, J. L. Dysart and T. E. Mallouk, J. Phys. Chem. Lett., 2011, 2, 402–406 CrossRef CAS.
- S. Sato, T. Morikawa, S. Saeki, T. Kajino and T. Motohiro, Angew. Chem., Int. Ed., 2010, 49, 5101–5105 CrossRef CAS PubMed.
- K. Yamanaka, S. Sato, M. Iwaki, T. Kajino and T. Morikawa, J. Phys. Chem. C, 2011, 115, 18348–18353 CAS.
- T. Arai, S. Sato, K. Uemura, T. Morikawa, T. Kajino and T. Motohiro, Chem. Commun., 2010, 46, 6944–6946 RSC.
- S. Trasatti, Electrochim. Acta, 1984, 29, 1503–1512 CrossRef CAS.
- S. Fierro, L. Ouattara, E. H. Calderon, E. Passas-Lagos, H. Baltruschat and C. Comninelis, Electrochim. Acta, 2009, 54, 2053–2061 CrossRef CAS.
- S. Y. Reece, J. A. Hamel, K. Sung, T. D. Jarvi, A. J. Esswein, J. J. H. Pijpers and D. G. Nocera, Science, 2011, 334, 645–648 CrossRef CAS PubMed.
- X. Deng and E. A. Schiff, in Handbook of Photovoltaic Science and Engineering, ed. A. Luque and S. Hegedus, Wiley, Chichester, England, 2003, pp. 505–565 Search PubMed.
- Z. Chen, T. F. Jaramillo, T. G. Deutsch, A. Kleiman-Shwarsctein, A. J. Forman, N. Gaillard, R. Garland, K. Takanabe, C. Heske, M. Sunkara, E. W. McFarland, K. Domen, E. L. Miller, J. A. Turner and H. N. Dinh, J. Mater. Res., 2010, 25, 3–16 CrossRef CAS.
- X.-G. Zhu, S. P. Long and D. R. Ort, Curr. Opin. Biotechnol., 2008, 19, 153–159 CrossRef CAS PubMed.
- C.-C. Yang, Y.-H. Yu, B. van der Linden, J. C. S. Wu and G. Mul, J. Am. Chem. Soc., 2010, 132, 8398–8406 CrossRef CAS PubMed.
- K. Sekizawa, K. Maeda, K. Koike, K. Domen and O. Ishitani, J. Am. Chem. Soc., 2013, 135, 4596–4599 CrossRef CAS PubMed.
- K. Maeda, K. Sekizawa and O. Ishitani, Chem. Commun., 2013, 49, 10127–10129 RSC.
- M. Schreier, P. Gao, M. T. Mayer, J. Luo, T. Moehl, M. K. Nazeeruddin, S. D. Tilley and M. Grätzel, Energy Environ. Sci., 2015, 8, 855–861 CAS.
- B. Kumar, J. M. Smieja, A. F. Sasayama and C. P. Kubiak, Chem. Commun., 2012, 48, 272–274 RSC.
- W. Leitner, Angew. Chem., Int. Ed. Engl., 1995, 34, 2207–2221 CrossRef CAS.
- J. Jiang and A. Wieckowski, Electrochem. Commun., 2012, 18, 41–43 CrossRef CAS.
- J. F. Hull, Y. Himeda, W.-H. Wang, B. Hashiguchi, R. Periana, D. J. Szalda, J. T. Muckerman and E. Fujita, Nat. Chem., 2012, 4, 383–388 CrossRef CAS PubMed.
- A. J. M. Miller, D. M. Heinekey, J. M. Mayer and K. I. Goldberg, Angew. Chem., Int. Ed., 2013, 52, 3981–3984 CrossRef CAS PubMed.
- D. J. Boston, C. Xu, D. W. Armstrong and F. M. MacDonnell, J. Am. Chem. Soc., 2013, 135, 16252–16255 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure, current–potential characteristics for various measurements, current efficiency for formate production and a simulated solar light spectrum. See DOI: 10.1039/c5ee01314c |
| This journal is © The Royal Society of Chemistry 2015 |