
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Functional native disulfide bridging enables delivery of a potent, stable and targeted antibody–drug conjugate (ADC)†
João P. M.
Nunes
a,
Maurício
Morais
a,
Vessela
Vassileva
b,
Eifion
Robinson
a,
Vineeth S.
Rajkumar
b,
Mark E. B.
Smith
a,
R. Barbara
Pedley
b,
Stephen
Caddick
a,
James R.
Baker
*a and
Vijay
Chudasama
*a
aDepartment of Chemistry, University College London, London, WC1H 0AJ, UK. E-mail: j.r.baker@ucl.ac.uk; v.chudasama@ucl.ac.uk; Tel: +44 (0)20 7679 2653 Tel: +44 (0)20 7679 2077
bUCL Cancer Institute, 72 Huntley Street, London, UK
First published on 1st June 2015
Abstract
Herein we report the use of next generation maleimides (NGMs) for the construction of a potent antibody–drug conjugate (ADC) via functional disulfide bridging. The linker has excellent stability in blood serum and the ADC, armed with monomethyl auristatin E (MMAE), shows excellent potency and cancer cell selectivity in vitro.
Antibody–drug conjugates (ADCs) are comprised of antibodies that are armed with warheads using appropriate linker technologies.1,2 They combine the exquisite targeting ability of antibodies (i.e. allowing for discrimination between healthy and diseased tissue) with the cell-killing ability of cytotoxic drugs. This class of targeted therapy has shown considerable promise in the treatment of various cancers in recent years with two FDA-approved ADCs coming onto the market recently (i.e. Adcetris™ and Kadcyla™) and with over 30 ADCs in the clinic.3 However, in order for ADCs to deliver their full potential, sophisticated conjugation technologies to connect the warhead to the antibody are vital.4 Conjugation to antibodies is typically achieved through either multiple lysine modification or by functionalisation of thiols generated by reduction of interchain disulfide bonds; neither of these is ideal (Fig. 1).4 Lysine modification is sub-optimal as it generates heterogeneous ADCs, which have been shown to have a narrow therapeutic window relative to homogeneous ADCs.5 Cysteine modification, following interchain disulfide reduction, results in the permanent loss of structural disulfide bonds, which may reduce the stability of the ADC in vivo.4 It also generates heterogeneous mixtures when targeting typical drug-to-antibody ratios (DARs) of 2–4. Other approaches using cysteine-based site-directed mutagenesis and unnatural amino acids should also be highlighted.5,6 However they also have limitations, e.g. disulfide scrambling post-reduction and high cost combined with relatively low expression yields, respectively.
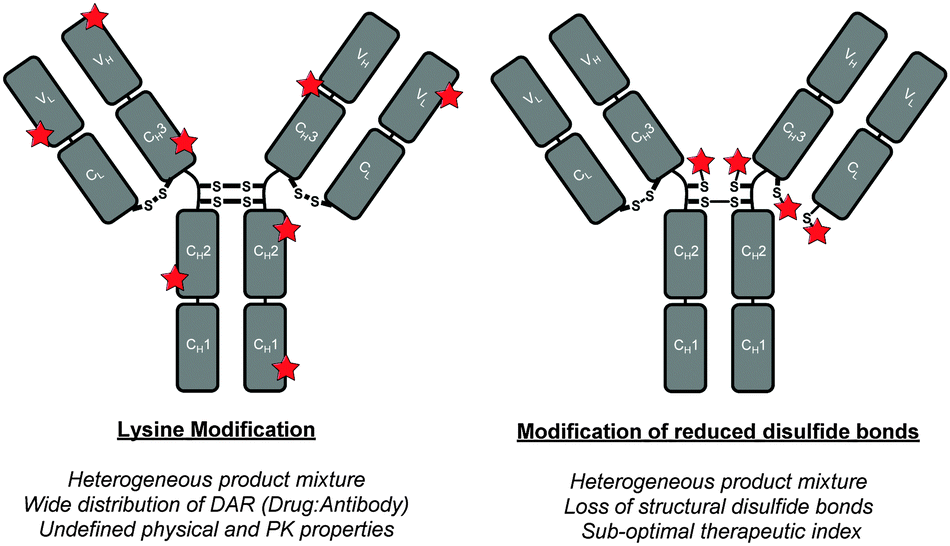 | ||
| Fig. 1 Classical approaches to ADC construction. | ||
Recently, we have described methods for the insertion of next generation maleimides (NGMs) into disulfide bonds in various proteins, engineered antibody single-chain variable fragments (ScFv), a fragment antigen-binding (Fab) construct and a full antibody to yield site-selectively modified NGM conjugates (Scheme 1).7 The NGM conjugate may be hydrolysed to a maleamic acid.7b Having optimised the chemistry to modify proteins with NGMs, herein we describe the construction of an industry relevant ADC (i.e. an antibody functionalised with a highly potent drug) and evaluate its biological relevance in terms of stability in blood serum, and efficacy plus cancer cell selectivity in vitro.
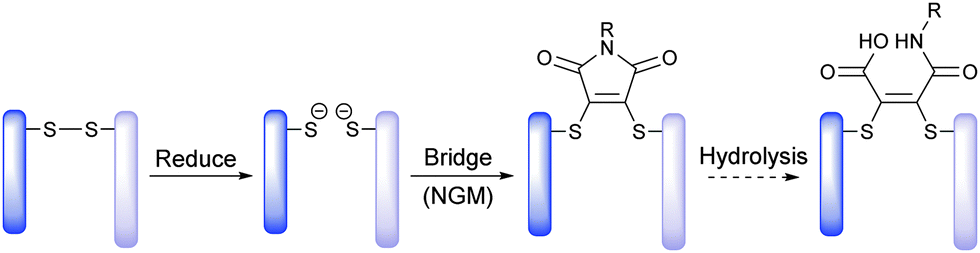 | ||
| Scheme 1 Exemplification of NGM technology for insertion into a disulfide bond and optional hydrolysis. | ||
Initially, we had to select a suitable full antibody platform to appraise the application of our technology. Trastuzumab (Herceptin™) is a monoclonal IgG1 antibody that targets the internalising HER2 receptor and has been used successfully in the treatment of HER2+ breast cancer.8 Furthermore, it is the antibody component of trastuzumab emtansine (Kadcyla™), a recently FDA-approved ADC therapy for the same indication.9 We therefore chose trastuzumab as the full antibody targeting system on which to evaluate this research. We also needed to select a suitable warhead for this study and decided to use anti-cancer drug monomethyl auristatin E (MMAE) as its suitability as the drug component of ADCs is well-established.3,10 Finally, we needed to select a suitable fluorophore to appraise the blood serum stability of the NGM linkage. To this end, we selected Alexa Fluor® 488 owing to its established use in this context.10b
Our study began by testing the blood serum stability of NGM–trastuzumab antibody conjugates against a classical maleimide analogue. It has been reported that classical maleimide conjugates are unstable in serum, due to their propensity to undergo retro-conjugate additions, leading to transfer of the attached cargo onto blood thiols; particularly albumin.11 It is vital in the next generation of ADCs that this vulnerability is avoided, i.e. to limit off-target toxicity associated with premature drug release or drug transfer. Whilst spontaneous retro-conjugate additions are not mechanistically possible in NGM conjugates, cleavage by a thiol addition–elimination mechanism is.7f However, this mode of reactivity can be precluded by hydrolysis to maleamic acids, which are unreactive to thiols.7b To test whether NGM conjugates (non-hydrolysed and hydrolysed) offer more suitable platforms for serum stability than classical maleimides we constructed fluorescent trastuzumab antibody conjugates 1–3. This was achieved by conjugation of reagents 4 and 5 to reduced trastuzumab and carrying out CuAAC click attachments of Alexa Fluor 488® azide. This afforded conjugates 1 and 2. It was notable that conjugate 2 was obtained with a fluorophore-to-antibody ratio of 3.91, consistent with conjugation of an NGM reagent to each of the four accessible interchain disulfide bonds. Conjugate 3 was obtained by hydrolysis of conjugate 2, and retained a fluorophore loading of ca. 4 (see ESI† for further details).
We appraised the blood serum stability of conjugates 1–3 by analysing fluorescence (post-HPLC separation) of antibody, albumin and “free” fluorophore as a function of days of incubation (Fig. 2, see ESI† for further details). As expected, the study with classical maleimide conjugate 1 revealed substantial transfer of the fluorophore to albumin after 4 days. Interestingly, dithiomaleimide-derived conjugate 2 gave a similar profile, suggesting that, albeit via a different mechanism, thiol exchange was occurring with albumin's cysteine 34 at a similar rate. In sharp contrast, maleamic acid conjugate 3 was completely stable, even over a prolonged period of 7 days. From this data it can be concluded that the optimum NGM–ADC platform would incorporate a maleamic acid linker to ensure robust serum stability.
 | ||
| Fig. 2 Blood serum stability of conjugates 1–3 and structure of thiol reactive reagents 4 and 5. | ||
Following on from the successful blood serum stability experiments with a maleamic acid linker, we synthesised NGM–MMAE construct 6 for direct conjugation onto trastuzumab (Fig. 3). To do this, dithiomaleimide acid 7 was coupled to amine-PEG12-tert-butylester, followed by TFA deprotection to afford dithiomaleimide-PEG12-acid 8. This was then coupled to MMAE to form NGM–MMAE 6.
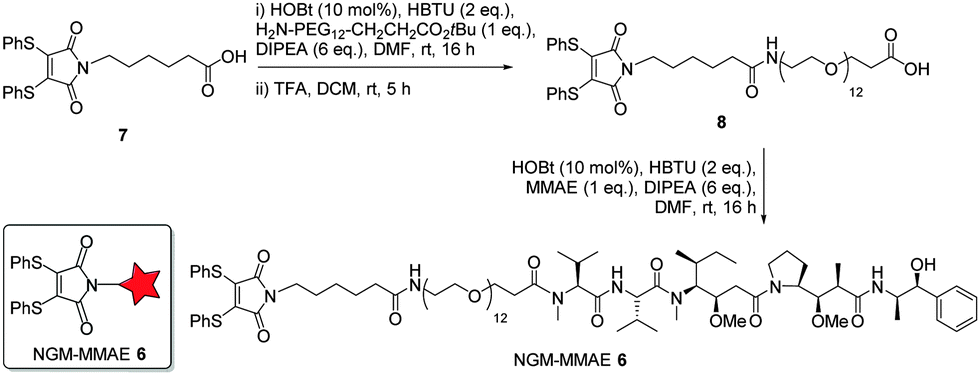 | ||
| Fig. 3 Synthesis of MMAE–NGM construct 6. | ||
With NGM–MMAE 6 in-hand, we applied our optimised conditions for the insertion of NGMs into the full antibody system of trastuzumab with the aim of functionally re-bridging its native interchain disulfide bonds. To do this, we only used a small excess of reducing agent (i.e. TCEP, 6 eq.) and NGM–MMAE (5 eq.) to ensure complete reaction and to target a drug-to-antibody ratio (DAR) of ca. 4. This was followed by hydrolysis at pH 8.4 at 20 °C for 72 h to form thiol-stable NGM–MMAE–ADC 9. This gave an excellent average DAR of 3.89 by HIC analysis (Fig. 4, see ESI† for details on analysis), which is consistent with our Alexa Fluor® 488 loading. The ADC therapeutic was formed in good yield and in an efficient manner with retention of binding confirmed by ELISA analysis (see ESI† for details).
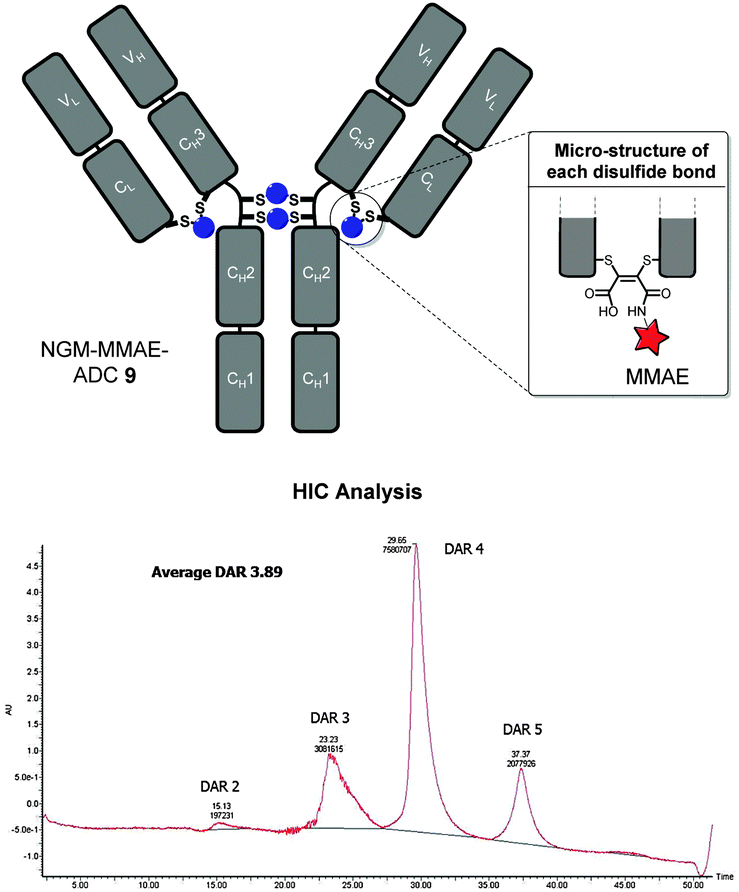 | ||
| Fig. 4 NGM–MMAE–ADC 9 with HIC analysis. | ||
With an industry relevant ADC in-hand, we appraised its effectiveness in vitro. To appraise the selectivity and potency of the constructs we used breast cancer cell lines SKBR-3 (HER2-positive) and MCF-7 (HER2-negative). As a comparable reduction in cell viability is observed for both cell lines at the same concentration of MMAE (IC50 = 0.23 nM and 0.50 nM for SKBR-3 and MCF-7 respectively),10b the cell lines are appropriate for the study, thus paving the way for cytotoxicity studies with NGM–MMAE–ADC 9 (Fig. 5). Gratifyingly, SKBR-3 cell viability was reduced significantly when incubated with the conjugate (IC50 = 0.33 nM), especially when compared to the control of native Herceptin where the reduction in cell survival was minimal at relatively high concentrations. Additionally, highlighting the targeted delivery aspect of the work, no toxicity was observed in analogous studies with MCF-7 cells incubated with NGM–MMAE–ADC 9. These results clearly highlight the selectivity of our conjugate over MMAE alone, and indicate that the toxic drug in our conjugates is delivered by an HER2-dependent internalisation mechanism.
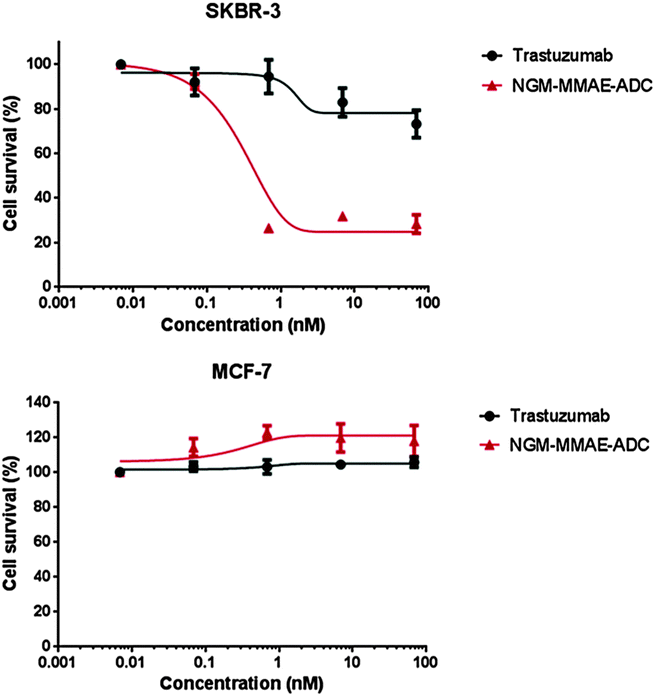 | ||
| Fig. 5 Inhibition of cell proliferation in cancer cell lines SKBR-3 (HER2-positive) and MCF-7 (HER2-negative) for NGM–MMAE–ADC 9 with trastuzumab control. The error bars are the standard error of the mean. | ||
In conclusion, NGM linkages have been shown to functionally re-bridge the native disulfide bonds of a full antibody, to be stable in blood serum post-hydrolysis and successfully used for the preparation of an industry relevant NGM–MMAE–ADC. The NGM–MMAE–ADC was shown to selectively target and kill HER2+ cells, thus highlighting the potential of our chemistry for the production of efficacious ADCs. We will next seek to evaluate NGM–ADCs in an in vivo setting, alongside our antibody fragment and antibody bispecific platforms.
The authors gratefully acknowledge the EPSRC, BBSRC, BRC, Wellcome Trust, HEFCE, SBC, MRC, UCL and UCLB for support of our programme. Dr Rajkumar is funded by the CCIC collaboration -CRUK & EPSRC Comprehensive Cancer Imaging Centre at KCL & UCL, which is jointly funded by Cancer Research UK and the Engineering and Physical Sciences Research Council (EPSRC).
Notes and references
- (a) P. Sapra, A. T. Hooper, C. J. O'Donnell and H.-P. Gerber, Expert Opin. Invest. Drugs, 2011, 20, 1131 CrossRef CAS PubMed; (b) S. C. Alley, N. M. Okeley and P. D. Senter, Curr. Opin. Chem. Biol., 2010, 14, 529 CrossRef CAS PubMed.
- J. R. Adair, P. W. Howard, J. A. Hartley, D. G. Williams and K. A. Chester, Expert Opin. Biol. Ther., 2012, 12, 1191 CrossRef CAS PubMed.
- A. Mullard, Nat. Rev. Drug Discovery, 2013, 12, 329 CrossRef CAS PubMed.
- (a) J. A. Flygare, T. H. Pillow and P. Aristoff, Chem. Biol. Drug Des., 2013, 81, 113 CrossRef CAS PubMed; (b) L. Ducry and B. Stump, Bioconjugate Chem., 2010, 21, 5 CrossRef CAS PubMed.
- (a) J. R. Junutula, K. M. Flagella, R. A. Graham, K. L. Parsons, E. Ha, H. Raab, S. Bhakta, T. Nguyen, D. L. Dugger, G. Li, E. Mai, G. D. Lewis Phillips, H. Hiraragi, R. N. Fuji, J. Tibbitts, R. Vandlen, S. D. Spencer, R. H. Scheller, P. Polakis and M. X. Sliwkowski, Clin. Cancer Res., 2010, 16, 4769 CrossRef CAS PubMed; (b) J. R. Junutula, H. Raab, S. Clark, S. Bhakta, D. D. Leipold, S. Weir, Y. Chen, M. Simpson, S. Ping Tsai, M. S. Dennis, Y. Lu, Y. G. Meng, C. Ng, J. Yang, C. C. Lee, E. Duenas, J. Gorrell, V. Katta, A. Kim, K. McDorman, K. Flagella, R. Venook, S. Ross, S. D. Spencer, W. Lee Wong, H. B. Lowman, R. Vandlen, M. X. Sliwkowski, R. H. Scheller, P. Polakis and W. Mallet, Nat. Biotechnol., 2008, 26, 925 CrossRef CAS PubMed.
- E. S. Zimmerman, T. H. Heibeck, A. Gill, X. Li, C. J. Murray, M. R. Madlansacay, C. Tran, N. T. Uter, G. Yin, P. J. Rivers, A. Y. Yam, W. D. Wang, A. R. Steiner, S. U. Bajad, K. Penta, W. Yang, T. J. Hallam, C. D. Thanos and A. K. Sato, Bioconjugate Chem., 2014, 25, 351 CrossRef CAS PubMed.
- (a) F. F. Schumacher, V. A. Sanchania, B. Tolner, Z. V. F. Wright, C. P. Ryan, M. E. B. Smith, J. M. Ward, S. Caddick, C. W. M. Kay, G. Aeppli, K. A. Chester and J. R. Baker, Sci. Rep., 2013, 3, 1525 Search PubMed; (b) C. P. Ryan, M. E. B. Smith, F. F. Schumacher, D. Grohmann, D. Papaioannou, G. Waksman, F. Werner, J. R. Baker and S. Caddick, Chem. Commun., 2011, 47, 5452 RSC; (c) F. F. Schumacher, M. Nobles, C. P. Ryan, M. E. B. Smith, A. Tinker, S. Caddick and J. R. Baker, Bioconjugate Chem., 2011, 22, 132 CrossRef CAS PubMed; (d) M. E. B. Smith, F. F. Schumacher, C. P. Ryan, L. M. Tedaldi, D. Papaioannou, G. Waksman, S. Caddick and J. R. Baker, J. Am. Chem. Soc., 2010, 132, 1960 CrossRef CAS PubMed; (e) L. Castañeda, A. Maruani, F. F. Schumacher, E. Miranda, V. Chudasama, K. A. Chester, J. R. Baker, M. E. B. Smith and S. Caddick, Chem. Commun., 2013, 49, 8187 RSC; (f) V. Chudasama, M. E. B. Smith, F. F. Schumacher, D. Papaioannou, G. Waksman, J. R. Baker and S. Caddick, Chem. Commun., 2011, 47, 8781 RSC; (g) F. Bryden, A. Maruani, H. Savoie, V. Chudasama, M. E. B. Smith, S. Caddick and R. W. Boyle, Bioconjugate Chem., 2014, 25, 611 CrossRef CAS PubMed; (h) F. F. Schumacher, J. P. M. Nunes, A. Maruani, V. Chudasama, M. E. B. Smith, K. A. Chester, J. R. Baker and S. Caddick, Org. Biomol. Chem., 2014, 12, 7261 RSC; (i) A. Maruani, S. Alom, P. Canavelli, M. T. W. Lee, R. E. Morgan, V. Chudasama and S. Caddick, Chem. Commun., 2015, 51, 5279 RSC; (j) A. Maruani, M. E. B. Smith, E. Miranda, K. A. Chester, V. Chudasama and S. Caddick, Nat. Commun., 2015, 6, 6645 CrossRef CAS PubMed.
- C. A. Hudis, N. Engl. J. Med., 2007, 357, 39 CrossRef CAS PubMed.
- S. Verma, D. Miles, L. Gianni, I. E. Krop, M. Welslau, J. Baselga, M. Pegram, D.-Y. Oh, V. Diéras, E. Guardino, L. Fang, M. W. Lu, S. Olsen and K. Blackwell, N. Engl. J. Med., 2012, 367, 1783 CrossRef CAS PubMed.
- (a) S. O. Doronina, B. E. Toki, M. Y. Torgov, B. A. Mendelsohn, C. G. Cerveny, D. F. Chace, R. L. DeBlanc, R. P. Gearing, T. D. Bovee, C. B. Siegall, J. A. Francisco, A. F. Wahl, D. L. Meyer and P. D. Senter, Nat. Biotechnol., 2003, 21, 778 CrossRef CAS PubMed; (b) G. Badescu, P. Bryant, M. Bird, K. Henseleit, J. Swierkosz, V. Parekh, R. Tommasi, E. Pawlisz, K. Jurlewicz, M. Farys, N. Camper, X. Sheng, M. Fisher, R. Grygorash, A. Kyle, A. Abhilash, M. Frigerio, J. Edwards and A. Godwin, Bioconjugate Chem., 2014, 25, 1124 CrossRef CAS PubMed.
- B.-Q. Shen, K. Xu, L. Liu and H. Raab, et al. , Nat. Biotechnol., 2012, 30, 184 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra for all small molecules, SDS-PAGE gels for bioconjugates. See DOI: 10.1039/c5cc03557k |
| This journal is © The Royal Society of Chemistry 2015 |