
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA modular DNA origami-based enzyme cascade nanoreactor†
Veikko
Linko‡
ab,
Marika
Eerikäinen‡
ab and
Mauri A.
Kostiainen
*a
aBiohybrid Materials, Department of Biotechnology and Chemical Technology, Aalto University, FI-00076 Aalto, Finland. E-mail: mauri.kostiainen@aalto.fi
bMolecular Materials, Department of Applied Physics, Aalto University, FI-00076 Aalto, Finland
First published on 12th January 2015
Abstract
In this communication, we present a nanoscale reactor assembled from tuneable and spatially addressable tubular DNA origami units. We can controllably combine separate origami units equipped with glucose oxidase (GOx) and horseradish peroxidase (HRP), and demonstrate efficient GOx/HRP enzyme cascade reaction inside the tube. The reactor could be utilized as a nanoscale diagnostic tool, and modularity of the proposed system would further enable more complex reactions.
Nanoscale engineering has shown substantial potential to revolutionize a wide range of scientific fields making e.g. novel medical treatments1 and miniaturizing electronics possible.2 For biochemical applications, precise organization of materials on nanoscale could enable customized machinery that are able to mimic complex natural systems found in living cells.3 Numerous parallel multistep reactions can go on in the cells with exceptional efficiency and specificity including catalytic cycles. This is achieved via compartmentalization:3 enzymes are appropriately arranged in the micro-/nanoreactors, which control the flow of molecules through these domains and also separate different reaction compounds from each other.
Various materials and approaches can be used for encapsulating catalysts, such as sol–gel materials,4 and efficient catalytic reactions have been realized by utilizing for example porous polymersomes,5,6 carbon nanotubes,7,8 viruses,9,10 inorganic nanocrystal–protein complexes11 and nanosized ferrous matrices12 as scaffolds for the catalysts. However, during recent years, the possibility to create exact and complex biocompatible nanoarchitectures by using DNA as a building material has markedly emerged.13–15 Especially the ‘DNA origami’ technique has become a widely used method to fabricate arbitrary spatially well-controlled two- (2D)16 and three-dimensional (3D) nanostructures.17 The customized shapes and the nanoscale addressability of materials on DNA structures through rational design have yielded various interesting bionanotechnological applications including sophisticated drug delivery vehicles,18 artificial ion channels,19 gatekeepers for solid-state nanopores20–22 molecular scale electronic circuit boards,23–25 and plasmonic devices.26,27
A DNA origami technique could be equally utilized in assembling enzyme systems for designed cascade reactions and studying the enzyme functions28 and reaction pathways.29 There exist a variety of examples of DNA-based enzyme systems30–32 but only the very recent approaches have taken advantage of the superior addressability of the DNA origami technique. These origami-based enzyme cascade arrangements include a distance-adjustable glucose oxidase (GOx)–horseradish peroxidase (HRP) pair assembled on a rectangular origami33 and a similar system, where the 2D flat DNA sheet equipped with the enzyme pair was rolled into a confined tube, thus resulting in the encapsulation of the catalysts.34 Lately, a multi-enzyme reaction with a swinging arm geometry was built and demonstrated on a DNA-tile substrate.35
In this communication, we propose a modular enzyme cascade nanoreactor that is comprised of robust 3D DNA origami building blocks (see Fig. 1A). Each DNA origami unit can act as a building block hosting a chosen catalyst (or any other desired function). These blocks can be further controllably assembled together in any desired order thus forming a defined-size tubular nanofactory with a tailored assembly line. Here, we demonstrated the feasibility of the method by using two distinct units with either GOx- or HRP-enzymes anchored inside the origami compartment (Fig. 1B). The units were fabricated and purified separately, and efficiently glued together via programmable DNA base pairing. Finally, the catalytic activity of a two-unit nanoreactor was monitored in the environment containing D-glucose as a reactant and 3,3′,5,5′-tetramethylbenzidine (TMB) as a reporter (Fig. 1B).
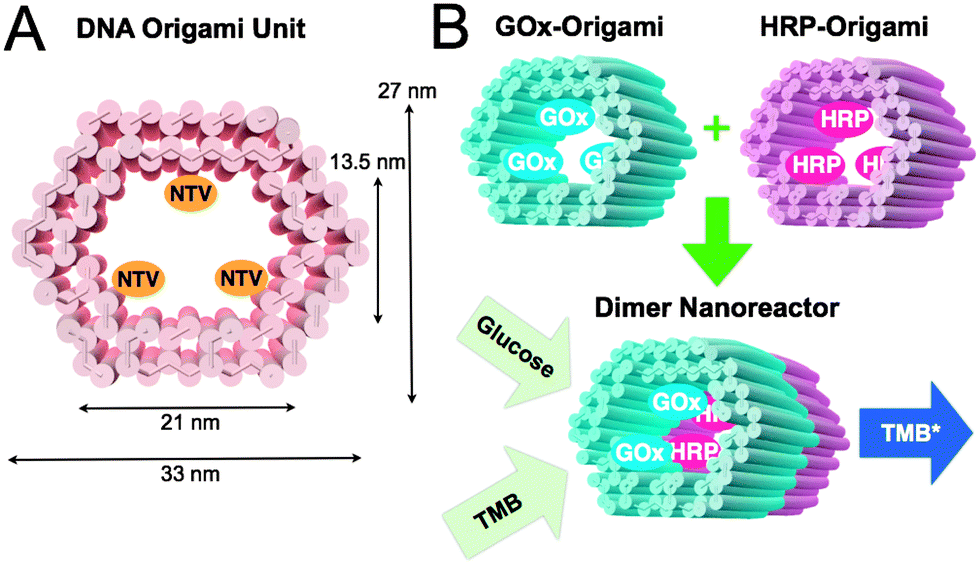 | ||
| Fig. 1 (A) CanDo-simulated36 shape and dimensions of a DNA origami unit used as a building block for the nanoreactor. The length of an origami is approximately 30 nm. NTVs indicate neutravidins, which are anchored to the inner surface of a tubular origami via biotinylated strands protruding from the origami. NTV acts as a binding site for biotinylated enzymes. (B) A schematic working principle of the nanoreactor. Two separately fabricated origami units are equipped with biotinylated glucose oxidase (GOx) or horseradish peroxidase (HRP) through biotin–avidin interaction. The units are linked together via base-pairing resulting in a nanoreactor that is able to perform an enzyme cascade reaction: (1) D-glucose enters the nanoreactor. In the presence of oxygen a hydrogen peroxide (H2O2) is formed and released at the GOx enzyme site. (2) 3,3′,5,5′-tetramethylbenzidine (TMB) is oxidized at the HRP enzyme as the diffused H2O2 is reduced to water. The formation of TMB diimine (TMB*) is detected using a spectrophotometer (absorbance at 650 nm). | ||
The modular nanoreactor was assembled in a stepwise process, starting with the preparation of DNA origami building blocks. Two structurally different units were fabricated by annealing an M13mp18 scaffold strand with the set of either 187 (GOx-origami) or 183 staple strands (HRP-origami). Each of the units contained 3 strands having biotin protruding from the inner surface of the tubular structure. NeutrAvidins (NTVs) were added to the units via biotin–avidin interaction in order to facilitate further binding of enzymes. A unit loaded with NTVs is presented in Fig. 1A. After NTVs were incorporated into the origamis, biotinylated enzymes (B-GOx or B-HRP) were attached to these units through the NTV binding sites (Fig. 1B). Between each step excess amounts of staple strands and unbound NTVs were removed by spin-filtering. In addition, an excess amount of HRP was removed using the same technique. See ESI† for the details.
The formed GOx- and HRP-origamis were connected together by hybridizing 32 short (3–6 bases) sequences. The short sequences sticking out at the end of one unit were paired with free scaffold sites located at the edge of another unit.37 In order to prevent the formation of multimers, the other end of the origami unit was passivated by overhanging single-stranded poly-T sequences (TTTTTTTT). Transmission electron microscopy (TEM) images of monomer units and the dimers assembled from the equal amounts of monomers within 1 day incubation at room temperature are shown in Fig. 2A and B. Fig. 2C displays agarose gel electrophoresed monomers and dimers revealing a high yield of the dimer formation (after the incubation of monomers, nearly 90% of all objects observed under TEM were correctly formed dimers, see ESI†). By choosing the strands that connect the units uniquely, this programmable method could be generalized to well-defined modular multimers, thus enabling customized and more complex assembly lines.
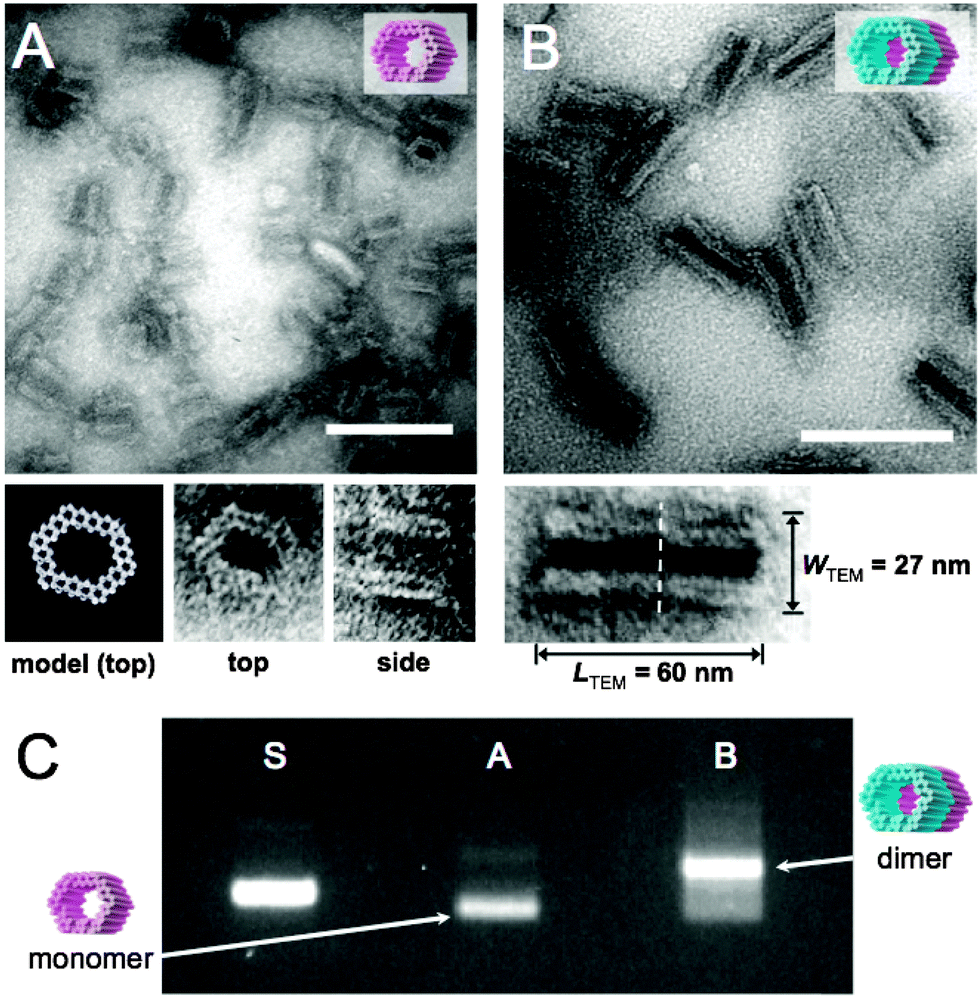 | ||
| Fig. 2 (A) TEM micrograph of single DNA origami units. Close-up images of two orthogonal orientations (top and side) of the DNA origami unit correspond well with the CanDo36-predicted model of the structure. (B) TEM micrograph of dimer nanoreactors. Close-up image of a dimer shows the interface of two units (dashed line) and the dimensions of the dimer. After 1 day incubation of monomer units, 86% of all the observed objects were correctly formed dimers (calculated from TEM images, see ESI†). Scale bars in A and B are 100 nm. (C) Agarose gel electrophoresis: ‘S’ indicates M13mp18 scaffold reference, lane ‘A’ contains single origami units (monomers) and ‘B’ is two units attached to each other (dimers), similarly as in subfigures (A) and (B). Monomers are decently folded (lane A) and the intense additional band in lane B indicates a high yield of dimer formation. | ||
Before studying the catalytic activity of a dimer nanoreactor, the units with enzymes were tested separately. In the experiments, the concentration increase of the final product TMB* (see Fig. 1B) produced by a purified (spin-filtered) DNA origami unit equipped with either HRP (substrate containing TMB and H2O2) or GOx (substrate with sodium acetate, TMB, D-glucose and B-HRP) was characterized. The activity of these units was compared to samples that were fabricated and treated similarly but did not contain NTV binding sites. The results indicate that both units are indeed able to catalyze reactions, and furthermore, that the units show significantly higher maximum reaction rates than the controls (see ESI† for initial rates and Fig. 4 for details).
After the performance of single units was verified, the activity of the dimer nanoreactor equipped with the GOx–HRP cascade was explored by mixing the dimer solution (initial concentration ∼1 nM, final concentration in the measurement ∼100 pM) with sodium acetate-based (pH 5, 2.5 mM) substrate containing TMB (250 μM) and D-glucose (20 mM). The reactant glucose and the reporter TMB were added in excess amounts in order to achieve a reaction that is restricted by the diffusion rate of the intermediate product H2O2. The activity of the purified dimer nanoreactor (excess amount of HRP removed by spin-filtering) was compared to the reference dimer, which did not contain NTV binding sites (the samples were treated identically with enzymes and equally spin-filtered) (see Fig. 3). In addition, just the substrate without any enzymes or origamis (blank sample) was used as a control. The results show that the assembled nanoreactor has significantly higher activity than the blank control sample, similar to that reported in previous studies.33,34
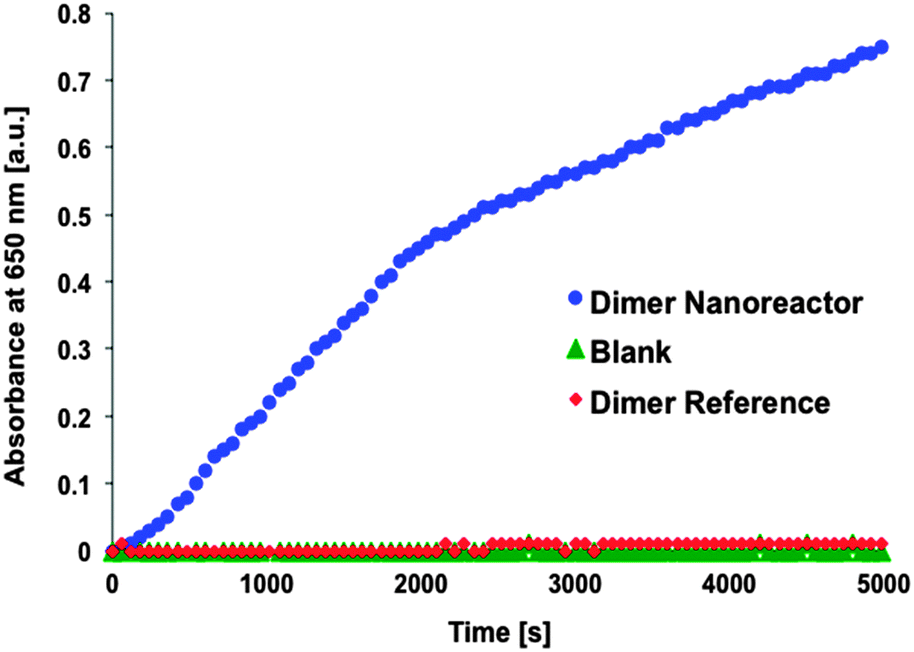 | ||
| Fig. 3 Typical progress curves. Product concentration (absorbance of TMB* in arbitrary units) as a function of time for enzymatic dimer nanoreactor (blue dots), dimer reference reactor (dimer fabricated similarly but without NTVs, red diamonds) and an enzyme substrate only (blank sample, green triangles). The data shows that the activity of the assembled nanoreactor can clearly outdo the activity of the reference dimer. Reference does not indicate any significant activity but rather follows the progress curve of a blank sample. In this measurement 40 μl of dimer samples (approx. 1 nM) were mixed with 260 μl of the substrate (3.5 mM sodium acetate (pH 5), 150 μM TMB and 10 mM D-glucose) resulting in the final concentration of ∼100 pM of origamis. | ||
Our experiments additionally prove that unspecific binding between enzymes and origami structures is insignificant, since the dimer fabricated without NTV binding sites (dimer reference sample) shows negligible catalytic activity. The same trend can be clearly seen when the maximum rate of reactions for the single origami units is compared to the reference samples (see Fig. 4). Both origami units equipped with enzymes can outdo the activity of the reference samples, and the effect is even more pronounced in the case of a dimer nanoreactor. The purity of the nanoreactor is undoubtedly an important feature for the attainable diagnostic uses, and here we demonstrate that it can be achieved via a straightforward purification step of the origami units; unbound HRPs can be efficiently removed from the solution by spin-filtering (see also ESI†). Therefore, by taking into account the high yield of dimer formation and the absence of free HRPs in the solution, the observed catalytic activity of the nanoreactor sample is predominantly resulting from the enzyme cascades located inside the properly assembled dimers. In addition, compared to previous studies,33,34 we have managed to significantly reduce the background activity of the free enzymes by purifying the units.
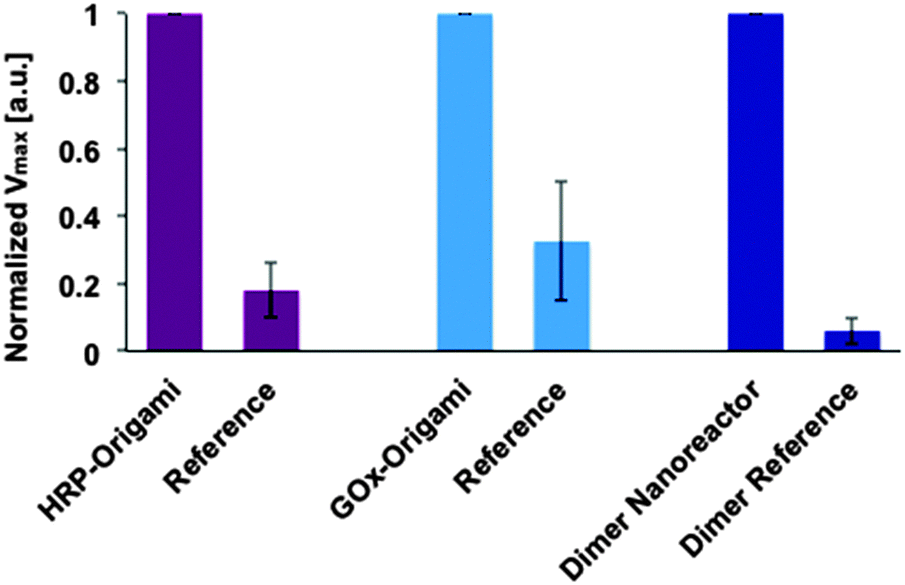 | ||
Fig. 4 Maximum rate of reactions (Vmax) at the studied substrate concentration for enzymes attached to DNA origami units and for the assembled dimer nanoreactor. The maximal rate of reaction (formation of TMB*) for the sample is normalized to 1 in each case (for independent samples), and the performance of the sample is compared to a reference, which is fabricated and treated similarly but does not contain NTV binding sites. The DNA origamis were mixed in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100–1:10 ratio with the substrate (final DNA origami concentration was typically ∼100 pM in each measurement, see ESI† for the details). :100–1:10 ratio with the substrate (final DNA origami concentration was typically ∼100 pM in each measurement, see ESI† for the details). | ||
In summary, we have reported the successful formation of a modular DNA-origami-based nanoreactor that can efficiently perform a designed enzyme cascade reaction. We believe that the presented method could be equally used for more complex reactions since the number of the units in the reactor is not limited. Moreover, the compartmentalization of the enzymes inside the robust tubular origami could presumably enhance the molecular reaction rates similar to that previously observed for more flexible origami tubes.34 That would be the case especially for larger molecules unable to diffuse through the barriers of the origami unit. The nanoreactor could be considered analogous to porous zeolites (molecular sieves) and on the other hand, the nanoreactor could efficiently process materials similar to holoenzymes. In addition, the tubular DNA vessels could be used for transporting cargo or an incorporated functional device into cells. This could be realized e.g. via virus-38 or lipid bilayer39 encapsulation of DNA units. Thus, the proposed system could open up a cornucopia of opportunities for intriguing applications in synthetic biology and bionanotechnology.
We thank Christian Wachauf and Hendrik Dietz for discussions. Financial support from the Academy of Finland (grants 263504, 267497, 273645), Biocentrum Helsinki and Emil Aaltonen Foundation is gratefully acknowledged. This work was carried out under the Academy of Finland's Centres of Excellence Programme (2014–2019) and made use of the Aalto University Nanomicroscopy Centre (Aalto NMC).
Notes and references
- M. M. Stevens and J. H. George, Science, 2005, 310, 1135–1138 CrossRef CAS PubMed.
- W. Lu and C. M. Lieber, Nat. Mater., 2007, 6, 841–850 CrossRef CAS PubMed.
- D. M. Vriezema, M. Comellas Aragonès, J. A. A. W. Elemans, J. J. L. M. Cornelissen, A. E. Rowan and R. J. M. Nolte, Chem. Rev., 2005, 105, 1445–1489 CrossRef CAS PubMed.
- D. Avnir, T. Coradin, O. Lev and J. Livage, J. Mater. Chem., 2006, 16, 1013–1030 RSC.
- D. M. Vriezema, P. M. L. Garcia, N. S. Oltra, N. S. Hatzakis, S. M. Kuiper, R. J. M. Nolte, A. E. Rowan and J. C. M. van Hest, Angew. Chem., Int. Ed., 2007, 46, 7378–7382 CrossRef CAS PubMed.
- S. F. M. van Dongen, M. Nallani, J. J. L. M. Corneliessen, R. J. M. Nolte and J. C. M. van Hest, Chem. – Eur. J., 2009, 15, 1107–1114 CrossRef CAS PubMed.
- X. Pan, Z. Fan, W. Chen, Y. Ding, H. Luo and X. Bao, Nat. Mater., 2007, 6, 507–511 CrossRef CAS PubMed.
- T. W. Chamberlain, J. H. Earley, D. P. Anderson, A. N. Khlobystov and R. A. Bourne, Chem. Commun., 2014, 50, 5200–5202 RSC.
- N. Carette, H. Engelkamp, E. Akpa, S. J. Pierre, N. R. Cameron, P. C. M. Christianen, J. C. Maan, J. C. Thies, W. Weberskirch, A. E. Rowan, R. J. M. Nolte, T. Michon and J. C. M. van Hest, Nat. Nanotechnol., 2007, 2, 226–229 CrossRef CAS PubMed.
- V. Liljeström, J. Mikkilä and M. A. Kostiainen, Nat. Commun., 2014, 5, 4445 Search PubMed.
- Z. Li, Y. Zhang, Y. Su, P. Ouyang, J. Ge and Z. Liu, Chem. Commun., 2014, 50, 12465–12468 RSC.
- Q. Fu, W.-X. Li, Y. Yao, H. Liu, H.-Y. Su, D. Ma, X.-K. Gu, L. Chen, Z. Wang, H. Zhang, B. Wang and X. Bao, Science, 2010, 328, 1141–1144 CrossRef CAS PubMed.
- N. C. Seeman, Annu. Rev. Biochem., 2010, 79, 65–87 CrossRef CAS PubMed.
- V. Linko and H. Dietz, Curr. Opin. Biotechnol., 2013, 24, 555–561 CrossRef CAS PubMed.
- F. Zhang, J. Nangreave and H. Yan, J. Am. Chem. Soc., 2014, 136, 11198–11211 CrossRef CAS PubMed.
- P. W. K. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS PubMed.
- S. M. Douglas, H. Dietz, T. Liedl, B. Högberg, F. Graf and W. M. Shih, Nature, 2009, 459, 414–418 CrossRef CAS PubMed.
- S. M. Douglas, I. Bachelet and G. M. Church, Science, 2012, 335, 831–834 CrossRef CAS PubMed.
- M. Langecker, V. Arnaut, T. G. Martin, J. List, S. Renner, M. Mayer, H. Dietz and F. C. Simmel, Science, 2012, 338, 932–936 CrossRef CAS PubMed.
- N. A. W. Bell, C. R. Engst, M. Ablay, G. Divitini, C. Ducati, T. Liedl and U. F. Keyser, Nano Lett., 2012, 12, 512–517 CrossRef CAS PubMed.
- R. Wei, T. G. Martin, U. Rant and H. Dietz, Angew. Chem., Int. Ed., 2012, 124, 4948–4951 CrossRef.
- C. Plesa, A. N. Ananth, V. Linko, C. Gülcher, A. J. Katan, H. Dietz and C. Dekker, ACS Nano, 2014, 8, 35–43 CrossRef CAS PubMed.
- V. Linko, S.-T. Paasonen, A. Kuzyk, P. Törmä and J. J. Toppari, Small, 2009, 5, 2382–2386 CrossRef CAS PubMed.
- H. T. Maune, S.-P. Han, R. D. Barish, M. Bockrath, W. A. Goddard III, P. W. K. Rothemund and E. Winfree, Nat. Nanotechnol., 2010, 5, 61–66 CrossRef CAS PubMed.
- V. Linko, J. Leppiniemi, S.-T. Paasonen, V. P. Hytönen and J. J. Toppari, Nanotechnology, 2011, 22, 275610 CrossRef PubMed.
- A. Kuzyk, R. Schreiber, Z. Fan, G. Pardatscher, E.-M. Roller, A. Högele, F. C. Simmel, A. O. Govorov and T. Liedl, Nature, 2012, 483, 311–314 CrossRef CAS PubMed.
- A.-P. Eskelinen, R. J. Moerland, M. A. Kostiainen and P. Törmä, Small, 2014, 10, 1057–1062 CrossRef CAS.
- R. Subramani, S. Juul, A. Rotaru, F. F. Andersen, K. V. Gothelf, W. Mamdouh, F. Besenbacher, M. Dong and B. R. Knudsen, ACS Nano, 2010, 4, 5969–5977 CrossRef CAS PubMed.
- F. C. Simmel, Curr. Opin. Biotechnol., 2012, 23, 516–521 CrossRef CAS PubMed.
- O. I. Wilner, Y. Weizmann, R. Gill, O. Lioubashevski, R. Freeman and I. Willner, Nat. Nanotechnol., 2009, 4, 249–254 CrossRef CAS PubMed.
- M. Erkelenz, C.-H. Kuo and C. M. Niemeyer, J. Am. Chem. Soc., 2011, 133, 16111–16118 CrossRef CAS PubMed.
- S. Juul, F. Iacovelli, M. Falconi, S. L. Kragh, B. Christensen, R. Frøhlich, O. Franch, E. L. Kristoffersen, M. Stougaard, K. W. Leong, Y.-P. Ho, E. S. Sørensen, V. Birkedal, A. Desideri and B. R. Knudsen, ACS Nano, 2013, 7, 9724–9734 CrossRef CAS PubMed.
- J. Fu, M. Liu, Y. Liu, N. W. Woodbury and H. Yan, J. Am. Chem. Soc., 2012, 134, 5516–5519 CrossRef CAS PubMed.
- Y. Fu, D. Zeng, J. Chao, Y. Jin, Z. Zhang, H. Liu, D. Li, H. Ma, Q. Huang, K. V. Gothelf and C. Fan, J. Am. Chem. Soc., 2013, 135, 696–702 CrossRef CAS PubMed.
- J. Fu, Y. R. Yang, A. Johnson-Buck, M. Liu, Y. Liu, N. G. Walter, N. W. Woodbury and H. Yan, Nat. Nanotechnol., 2014, 9, 531–536 CrossRef CAS PubMed.
- C. E. Castro, F. Kilchherr, D.-N. Kim, E. L. Shiao, T. Wauer, P. Wortmann, M. Bathe and H. Dietz, Nat. Methods, 2011, 8, 221–229 CrossRef CAS PubMed.
- R. Jungmann, M. Scheible, A. Kuzyk, G. Pardatscher, C. E. Castro and F. C. Simmel, Nanotechnology, 2011, 22, 275301 CrossRef PubMed.
- J. Mikkilä, A.-P. Eskelinen, E. H. Niemelä, V. Linko, M. J. Frilander, P. Törmä and M. A. Kostiainen, Nano Lett., 2014, 14, 2196–2200 CrossRef PubMed.
- S. D. Perrault and W. M. Shih, ACS Nano, 2014, 8, 5132–5140 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, additional TEM image of dimer formation, progress curves for DNA origami units, DNA origami designs and DNA strand sequences. See DOI: 10.1039/c4cc08472a |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2015 |