
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthetic enzyme supercomplexes: co-immobilization of enzyme cascades
F.
Kazenwadel
*,
M.
Franzreb
and
B. E.
Rapp
Karlsruhe Institute of Technology, Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany. E-mail: franziska.kazenwadel@kit.edu
First published on 22nd April 2015
Abstract
A sustainable alternative to traditional chemical synthesis is the use of enzymes as biocatalysts. Using enzymes, different advantages such as mild reaction conditions and high turnover rates are combined. However, the approach of using soluble enzymes suffers from the fact that enzymes have to be separated from the product post-synthesis and can be inactivated by this process. Therefore, enzymes are often immobilized to solid carriers to enable easy separation from the product as well as stabilization of the enzyme structure. In order to mimic the metabolic pathways of living cells and thus to create more complex bioproducts in a cell-free manner, a series of consecutive reactions can be realized by applying whole enzyme cascades. As enzymes from different host organisms can be combined, this offers enormous opportunities for creating advanced metabolic pathways that do not occur in nature. When immobilizing this enzyme cascades in a co-localized pattern a further advantage emerges: as the product of the previous enzyme is directly transferred to its co-immobilized subsequent catalyst, the overall performance of the cascade can be enhanced. Furthermore when enzymes are in close proximity to each other, the generation of by-products is reduced and obstructive effects like product inhibition and unfavorable kinetics can be disabled. This review gives an overview of the current state of the art in the application of enzyme cascades in immobilized forms. Furthermore it focuses on different immobilization techniques for structured immobilizates and the use of enzyme cascade in specially designed (microfluidic) reactor devices.
Introduction
The use of enzymes for the fermentation of food and beverages as well as in medicinal applications is almost as old as mankind. Even in the bible (Isaiah, 2nd Book of Kings) it is reported, that a wound was healed by applying a patch of fig. Nowadays we know that considerable amounts of the enzyme ficain were responsible for the healing effect.1 Since the beginning of the 20th century, single enzymes are specifically isolated from crude materials and used in industrial fields such as food, pharmacology and textile industry, for production of fine chemicals, for electricity generation in biofuel cells and in diagnosis. Compared to classical chemical synthesis, the use of enzymes offers crucial advantages:2 while organic synthesis is often conducted in pollutive organic solvents, most enzyme reactions take place under mild pH and temperature conditions in aqueous milieu as given in the natural environment – inside a cell. Most enzymes show high specificity both to their substrates and products, which reduces the formation of unwanted byproducts that subsequently have to be separated from the product. Furthermore by protein engineering, specificity, stability and enzyme characteristics can be modified according to specific industrial applications.3,4 However, if enzymes are applied in soluble form, they have to be separated from the product post-synthesis. This process is often expensive, time-consuming and the catalysts are mostly inactivated. The immobilization of enzymes can solve this problem: by converting the enzymes to an insoluble form, they can be easily separated from the reaction solution.5 Enzymes can be cross-linked to each other,6 entrapped into matrices7,8 or attached to solid supports such as microparticles, fibers or other functionalized surfaces. Thus, the activity and stability of the enzymes can be enhanced and their selectivity can even be tuned, depending on the immobilization strategy.9 However, in some cases the enzyme activity can also be reduced by immobilization, for example if the flexibility of the enzyme is disturbed or the binding occurs in or near the active center of the enzyme required for substrate conversion. Therefore an optimal immobilization protocol has to be developed for each enzyme. For the production of more complex products, one reaction step may not be enough. Thus, a series of consecutive enzyme catalyzed reactions may be required. In this case whole enzyme cascades are implemented. In 2011 Ricca et al. excellently reviewed the advantages of using enzyme cascades for the one-pot production of chiral chemicals, such as alcohols, amines and amino acids.10 The co-localized immobilization of enzyme cascades consisting of two or more types of biocatalysts offers further advantages: a specific arrangement of the enzymes that enables close proximity to each other leads to an effect called substrate channeling: the product of an enzyme is directly transferred to the co-localized following enzyme, where it can act in turn as substrate.11,12 Those short diffusional distances accelerate the speed of the reaction and lead to an enhanced performance of the cascade compared to their soluble form.13 Another advantage compared to whole-cell-catalysis is the opportunity of building up artificial enzyme complexes whose components can be derived from an immense variety of different host organisms with distinct characteristics and advantages. Thus, artificial metabolic pathways can be engineered that do not occur in nature.14There are different approaches for the realization of enzyme cascades. Enzymes can be co-immobilized by different techniques in a more or less specific pattern, which leads to close proximity to each other and facilitates substrate channeling. Biocatalysts can also be separated into different reaction compartments that the product stream passes subsequently. As in separated reaction compartments the process parameters can be adapted, this approach is favored if the selected enzymes differ in their requirements concerning optimal process conditions.
Examples of an important enzymatic systems
The most prominent example of an enzyme cascade used technically is the bi-enzymatic system for sugar detection consisting of Glucose Oxidase (GOx) and a peroxidase, mostly derived from horseradish (HRP, Armoracia rusticana). Glucose is oxidized by the GOx using ambient oxygen and gluconolactone while hydrogen peroxide (H2O2) is generated. The hydrogen peroxide is used by the peroxidase to oxidize a dye that is added to the substrate solution from its colourless to its coloured form. This leads to a quantifiable absorbance signal which is in case of constant reaction term proportional to the glucose concentration in the medium. Carr et al. published already in 1946 the use of this enzyme system for blood sugar detection in the form of a rapid bedside test for diabetic patients.15 The whole history of monitoring blood glucose using enzyme based biosensors, as well as the main aspects concerning technical improvements, standardized analytics and performance levels were reviewed by Yoo and Lee in 2010.16 If further enzymes are added to the cascade, the detection spectrum can be extended to other types of sugar molecules. For instance, van Dongen et al. published in 2000 the extension of the system by the Lipase B from Candida antarctica (CalB), which enables the system to convert an acetate-protected glucose to glucose and its subsequent detection.17 Fornera et al. and Böhm et al. extended the system by β-galactosidase which enables the detection of lactose.18,19A second important field, in which enzyme cascades are used, is the production of electricity using enzyme based biofuel cells. Enzymes used for this type of application normally belong to the family of oxidoreductased. The topic was very well reviewed in 2007 by Minteer et al. highlighting the advantages and disadvantages of enzyme fuel cells compared to microbial fuel cells. Advantages are the higher power densities that can be achieved by the immobilization of the biocatalysts and their higher specificity. However, they suffer from short life times (7–10 days) and only partly oxidized fuel substrates.20 The first enzyme cascade for electricity generation was already applied in 1998 by Palmore et al. The authors used alcohol dehydrogenase, aldehyde dehydrogenase and formate dehydrogenase for the oxidation of methanol to carbon dioxide.21 Another work was published by Akers et al. in 2004, where ethanol was oxidized in a two-step reaction to acetate by combining an alcohol dehydrogenase and an aldehyde dehydrogenase.
Related work
There have been many important contributions reviewing the generation and application of enzyme cascades whereby the most important ones will be briefly outlined in this section. In 2010, Betancor and Luckarift reviewed the application of enzyme cascades in biosensing and production.22 The excellent paper by Schoffelen and van Hest published in 2013 summarizes the chemical strategies in covalent assembling of multi-enzyme complexes.23 The cell-free production of ethanol by enzyme cascades was reviewed in 2014 by Khattak et al. and gives example of the technical use of enzyme cascade in technical productions.24Scope of this article
This review summarizes the different techniques for immobilization of enzymatic cascades and focuses on classifying the different co-immobilization techniques while giving examples of sophisticated approaches. Another excellent review, highlighting the current state of the art, the principles of enzymatic fuel cells, unsolved problems and possible strategies for addressing them was published in 2011 by Osman et al.25Immobilization strategies for co-immobilization of enzymes
Principles of random co-immobilization of enzymatic cascades
Random co-immobilization is mainly achieved by crosslinking the members of the cascade to solid supports or the entrapment of the biocatalysts into polymer films. It leads to a statistical distribution of the enzyme, depending on the ratio of applied enzyme masses, and the density of functional groups on the support material. This method is often used for biosensing application. A simple and fast opportunity for attaching enzymes to solid surfaces is the use of chemical linking agents as for instance classical aldehyde-amino-crosslinking by glutaraldehyde26 or 1-ethyl-3-(4-diaminopropyl) carbodiimide (EDC) to connect functional amino-sidechains from the protein surface to an activated support27 by the activation of carboxylic acids for a nucleophilic attack by an amine. The most commonly employed activation is the conversion of the carboxylic acid to a so-called active ester e.g., the N-hydroxysuccinimide or pentafluorophenol ester. These approaches are fast and simple and enzymes do not need to be extensively modified before immobilization. However, as the distribution of functional groups is random, a specific control of localization and orientation is not possible.Random co-immobilization on surfaces
Two or more enzymes can be co-immobilized to functionalized surfaces in a statistically distributed manner. For some control of the immobilization pattern, either the ratio of the applied enzymes can be tuned or the distribution of different functional groups for enzyme attachment at the surface can be engineered. Deng et al. published an elegant approach for the co-immobilization of proteins on a patterned surface that was generated by chemical vapour deposition (CVD).28 By copolymerization of a controlled ratio of different monomer types, a statistical distribution of alkyne and pentafluorophenyl groups was generated on a surface. Thus, two different proteins were immobilized via azide-alkyne-cycloaddition and activated ester–amine reaction, providing two orthogonal reaction types. Although in this case proteins for cell adhesion were immobilized instead of an enzyme cascade, this approach can be a versatile tool for the realization of a cascade of biocatalysts. However, this approach suffers from one disadvantage: the enzyme groups are statistically distributed, depending on the ratio of monomer. An exact patterning and thereby an ensured maximum distance between the individual enzymes is not possible using this approach.Random co-immobilization by encapsulation
Zhu et al. immobilized GOx and HRP in an electropolymerized pyrrole film that was deposited on an electrode coated with singe-wall carbon nanotubes (SWCT) in order to generate a glucose biosensor.29 By determination of the amperiometric response of the bioelectrode, a signal was recorded that seemed to be proportional to an applied glucose concentration of 3 × 10−5 to 2.43 × 10−3 M. Furthermore, the results indicated a 6.8-fold greater sensitivity, when the enzymes were co-immobilized, compared to a sensor with separately immobilized biocatalysts in different polymer layers. Comparable systems for glucose detection by co-encapsulation of HRP and GOx were also investigated by other groups. In 2002 Wei et al. published the successful incorporation of both enzymes into mesoporous sol–gel materials.30 Ji et al. achieved a co-encapsulation of the cascade by diluting the enzyme into a polyurethane based solution and subsequent co-axial electrospinning, creating a hollow nanofibre membrane that is able to serve as glucose detection strip.31 Eguilaz et al. immobilized a different enzyme mixture, cholesterol oxidase (ChOx) and HRP to composites consisting of coated multiwall carbon nanotubes and magnetic nanoparticles, thus creating a biosensor for the detection of cholesterin.32Random co-immobilization by supportless crosslinking
Another prominent example of random co-immobilization of enzymatic cascades is the immobilization by interconnecting enzymes into so-called combined crosslinked enzyme aggregates (combi-CLEAs). This approach combines two advantages: the co-localization of enzymes and the absence of carriers that lead to a dilution of enzyme activity.33 For instance Mateo et al. developed a combi-CLEA, that consisted of a S-selective oxynitrilase derived from Manihot esculenta and a nonselective nitrilase derived from Pseudomonas fluorescens for the enantioselective conversion of benzaldehyde to (S)-mandelic acid.34,35 By doing so, another advantage of co-immobilized enzyme cascades was exploited: the in situ conversion of the enantioselective product produced by the oxynitrilase is directly converted by the nitrilase, whereby the equilibrium of the first reaction step is driven towards the product.33 Thus, even unfavorable kinetics can be disabled by the co-immobilization of enzyme cascades.Principles of site-specific co-immobilization of enzymatic cascades
This section will describe approaches by which enzymes are not only brought in statistically controlled close proximity by co-encapsulation or co-crosslinking, but also in defined patterns or shapes and with defined spacing between them. For the immobilization of an enzyme cascade in an organized pattern, additional modification steps are necessary. However, due to an enhanced performance of the cascade it is often worth the effort. There are different ways of generating enzyme patterns. For a specific attachment of a defined enzyme to a specific binding site, orthogonal binding mechanisms are required. That means, that binding occurs exclusively between target enzyme and target binding site without any unspecific attachment. Therefore the pattern has to be defined by the distribution of different functional groups on the respective surface. When more than one enzyme type has to be immobilized, several different orthogonal binding mechanisms are required. Here a promising yet somewhat limited approach are methods based on so-called “click chemistry”. These “bio-orthogonal” reactions occur only between the functionalized material surface and specifically introduced residues on the protein surface, enabling the generation of protein patterns.36 One prominent example is the copper-catalysed 1,3-dipolar cycloaddition of azide and alkyne groups or Huisgen-reaction. It occurs at mild reaction conditions without the formation of unwanted byproducts. One group is introduced at the protein surface, while the other group is attached to the desired surface before the coupling step. However, as these groups are often introduced into a protein unspecifically by crosslinking chemistry, the immobilization still takes place in a random orientation. An elegant, but sophisticated way to circumvent this limitation and to generate site-specifically labelled enzymes is the incorporation of unnatural amino acids containing the respective groups, for instance p-azido-phenylalanine, by means of an expanded genetic code.37 Another way of perfectly controlling the orientation of immobilized proteins is the use of genetically encoded tags that are attached to the desired enzyme by genetic engineering leading to the expression of a fusion protein. Many reviews deal with the description of commercially available tagging systems, as for example the paper published by Terpe in 2003 that summarizes molecular basics and the development of such systems.38 An overview of the most prominent and promising examples used for site-directed immobilization is given in Table 1, demonstrating the binding partners and revealing selected examples of sophisticated immobilization approaches. Fig. 1 shows the immobilization of an enzyme by a Histidine-Tag using dip-pen-nanolithography.| Tag name | Binding partner | Selected immobilization example |
|---|---|---|
| Poly-histidine-tag | Transition metals (Cu(II), Co(II), Zn(II), Ni(II) complexed to nitrilotriacetic acid (NTA) or iminodiacetic acid (IDA) | 39, 40 |
| Poly-arginine-tag | Cation exchange material | 41 |
| Biotin | Avidin/streptavidin | 42 |
| Strep-tag | Strep-tactin | 43 |
| FLAG-tag | Anti-flag monoclonal antibody | 44 |
| Cellulose-binding domain | Cellulose | 45, 46 |
| Ribosomal protein L2 (“Si-tag“) | Silica | 47 |
| SNAP-tag | benzylguanine | 48 |
| Halo-tag | Halo-tag-ligand | 49 |
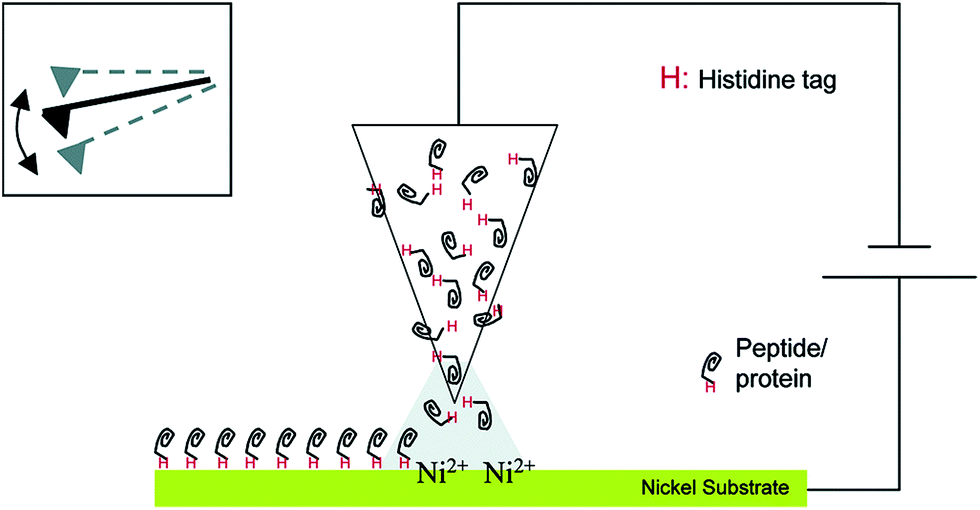 | ||
| Fig. 1 Site-specific immobilization of polyhistidine-tagged proteins to a Ni-substrate by dip-pen-nanolithography. Reprinted (adapted) with permission from.39 Copyright (2015) American Chemical Society. | ||
Site-specific co-immobilization to protein scaffolds
For the generation of distinctive patterns, specifically structured scaffolds are necessary. One potential approach to this is the application of protein scaffolds with specific domains to which modified proteins can bind orthogonally. In 2012 You et al. published a general approach for a self-assembling multi-enzyme-complex basing on a protein scaffold.13 The orthogonal binding mechanisms are mediated by the specific interactions between cohesin and dockerin domains (see Fig. 2). They are derived from a scaffold protein of the cellulosome, which constitutes the cellulose complex by binding different enzymes, carrying dockerin domains. Three enzymes from the glycolysis/gluconeogenesis pathway, triosephosphate isomerase (TIM), aldolase (ALD) and fructose 1,6-bisphosphatase (FBP), were genetically modified with specific dockerin domains and bound by self-assembly to a synthetic trifunctional scaffoldin carrying the appropriate cohesin domains. The authors found that the overall performance of the cascade in co-immobilized form was enhanced up to 21.1-fold compared to soluble enzymes, especially at low substrate concentrations.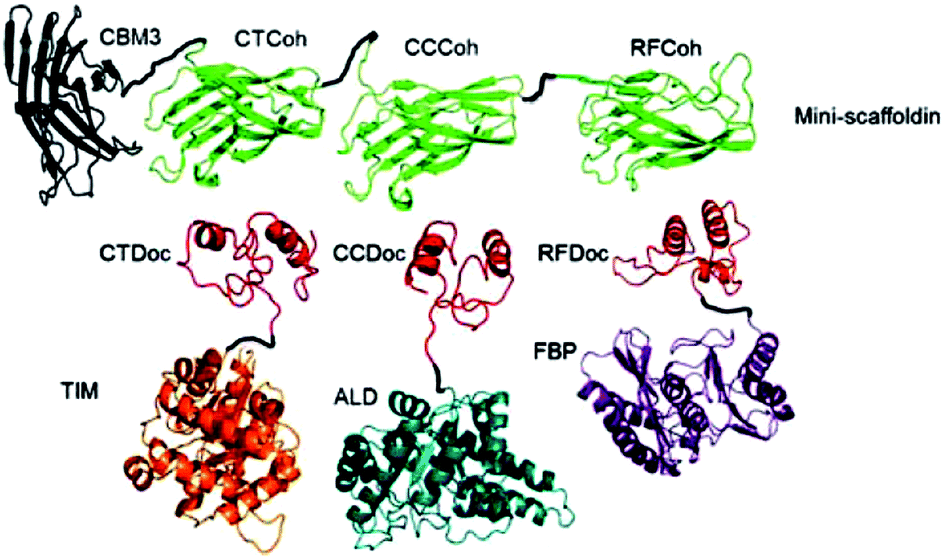 | ||
| Fig. 2 Co-immobilization of three enzymes, triosephosphate isomerase (TIM), aldolase (ALD) and fructose 1,6-bisphosphatase (FBP) fused to three different dockerin-domaines (TIM-CTDoc, ALD-CCDoc, FBP-RFDoc) and coupled in a site-directed fashion to a protein scaffold (CBM3) carrying three cohesion domains (CTCoh, CCCoh, RFCof) for specific binding of the respective dockerin domain. Reprinted from ref. 13 with permission of John Wiley and Sons. | ||
Site-specific co-immobilization to DNA scaffolds
DNA-macromolecules can also be used for creating specific patterns of immobilized enzymes, because of their capability for self-assembly, their physical and chemical stability and their backbone stiffness. Already in 1994, Niemeyer et al. coupled proteins to oligonucleotide strands and immobilized them to a complementary single strand of DNA, leading to macroscopic protein arrays.50 A more sophisticated method is the use of specifically designed DNA-macromolecules that self-assemble by base hybridization into defined 2- and 3-dimensional shapes. This technology, called DNA-origami was originally invented by Rothemund in 2006.51 Underlying molecular principles and general considerations in the process of generating suitable scaffold structures were excellently reviewed by Feldkamp et al. in 2006.52 Many approaches that couple enzymes to DNA microstructures use the biotin–streptavidin binding system for immobilization. However, due to the tetrameric structure of streptavidin and avidin the stoichiometry of the DNA–protein-conjugates is difficult to control. Thus, if the stoichiometry is important for the respective approach other binding mechanisms can be used.53 Most prominent example for the use of such DNA–protein-conjugates is the protein–microarray, where DNA-labeled proteins are site-specifically immobilized to a matrix of complementary DNA-strands attached to a surface. For the generation of soluble biocatalytically active nanostructures, DNA-labeled proteins can be attached to a complementary single strand of DNA, leading to so-called linear protein–DNA-assemblies. One early example was published by Niemeyer et al. in 2002. Here, a bienzymatic assembly of NAD(P)H-FMN oxidoreductase and luciferase were site-specifically immobilized to an complementary single strand DNA via the biotin–streptavidin binding system. The results clearly show, that the spatial orientation of the enzymes is of importance for the performance of the enzyme cascade.54 The same effect could be shown for the enzyme cascade described above, consisting of GOx and HRP.55 If two- or even three-dimensional scaffolds for protein attachment are required, the above described DNA-origami structures engineered by rational strand design can be applied. So far, only simpler approaches with model proteins are reported. For instance, in 2007 Duckworth et al. decorated a DNA tetrahydron56 site-specifically with GFP-molecules, using a click chemistry approach.57 In 2012 Fu et al. were able to immobilize HRP and GOx site-specifically on DNA-origami tiles. Different distances between the enzymes, ranging from 10 to 65 nm were created (see Fig. 3) and further enzymes were immobilized, acting as bridges between the cascade enzymes. Enhanced activity could be observed for enzyme pairs who were in close proximity. However, activity decreased when enzymes were closer than 20 nm suggesting Brownian diffusion of intermediates are responsible for the variation in enzyme activity. The use of further noncatalytic proteins, connecting the hydration shells of the cascade enzymes also led to an enhanced cascade activity.58 As DNA proved to be a suitable scaffold for the site-specific immobilization of enzymes, it is likely to become established as a versatile tool for the immobilization of enzyme cascades exploiting the substrate channeling effect.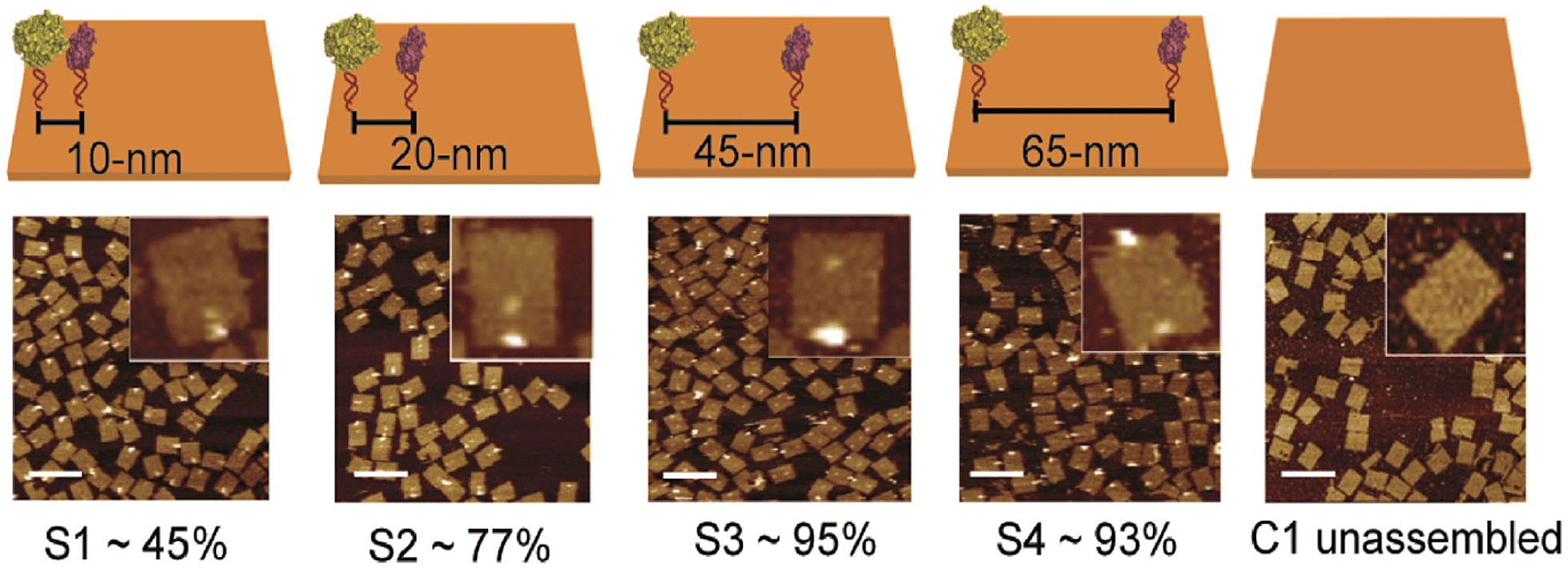 | ||
| Fig. 3 Site-specific immobilization of HRP and GOx in defined distances ranging from 10 to 65 nm. Close proximity of the enzymes leads to an enhanced performance of the cascade due to substrate channeling. Reprinted with permission from.58 Copyright (2015) American Chemical Society. | ||
Site-specific co-immobilization in nanocontainers
An elegant way of the structured immobilization of a three-enzyme-cascade was published by van Dongen et al. in 2009 (see Fig. 4). The approach is based on porous polymersomes composed of isocyanopeptides and styrene block copolymers.17 In order to obtain a structured co-immobilization, mimicking the compartmentalization in living cells, three enzymes were immobilized to different locations of the polymersome: CalB was embedded in the bilayer membrane, GOx was encapsulated in the lumen and HRP was attached to the polymersome surface by means of click chemistry. A specific labelling with metal-ions and subsequent mass spectroscopic analysis revealed the desired distribution of enzymes. Those nanocontainers were shown to be able to convert glucose-acetate and generate a detectable signal of the dye 2,2-azinobis(3-ethylbenzothiazoline-6-sulfuric acid) (ABTS) upon its oxidation from colourless to coloured form. Furthermore it could be demonstrated that the containers can be easily separated from the reaction solution by filtration (Fig. 5). | ||
| Fig. 4 Site-specific immobilization of an enzyme cascade in polymersome nanocontainers: Candida antarctica Lipase B (CalB) is embedded in the polymersome membrane, glucose oxidase (GOx) is entrapped in the inner lumen of the container and horseradish peroxidase (HRP) is attached to the outer polymersome surface by a Click chemistry approach. Reprinted from ref. 17 with permission of John Wiley and Sons. | ||
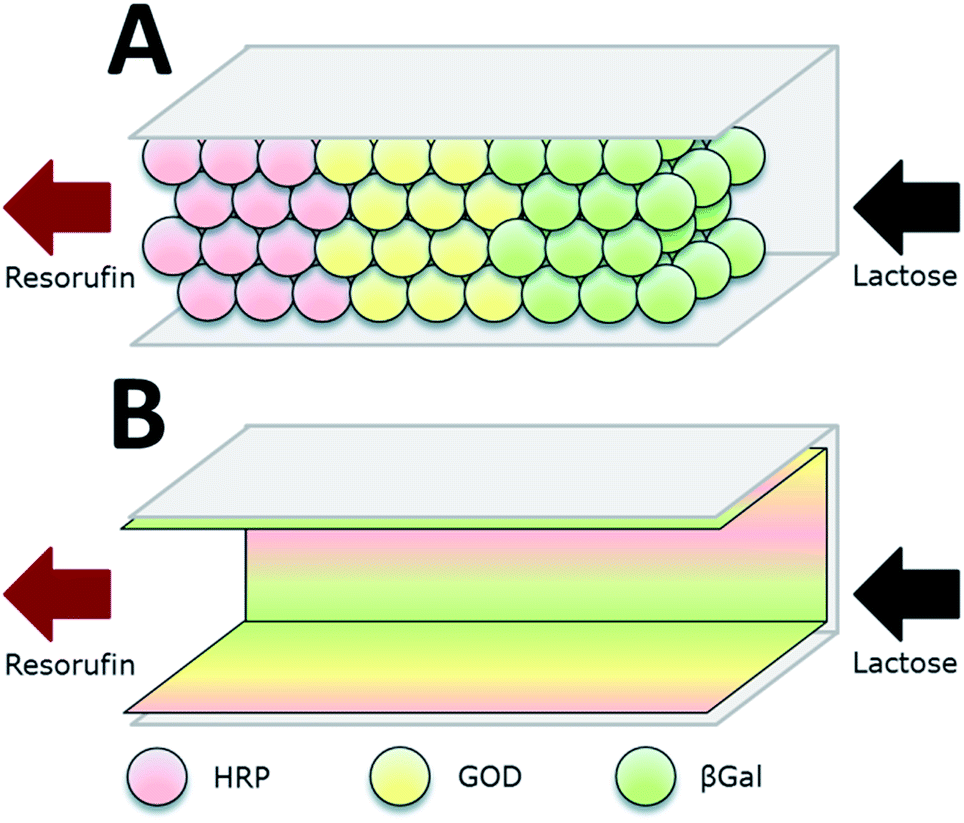 | ||
| Fig. 5 Co-immobilization of β-galactosidase (βGal, green), glucose oxidase (GOx, here GOD, yellow) and horseradish peroxidase (HRP, red) in a microfluidic channel by two approaches: (A) immobilization to microbeads that are subsequently packed in the channel. (B) direct attachment to the inner surface of the microchannels. The packed bed reactor (A) proved to be the more efficient approach. Reprinted from ref. 18 with permission of The Royal Society of Chemistry. | ||
Site-specific co-immobilization in microfluidic devices
Microfluidic devices exploit compartmentalization and efficient control of product and reactant streams, thereby aiming on mimicking the microcompartments of living cells. Microfluidic scaffolds are often combined with immobilized enzymes e.g. for sensor applications, analytics and the small-scale production of several agents. This topic was reviewed by Asanomi et al. in 2011 summarizing recent advantages in the development of microfluidic reactors using immobilized enzymes. The authors focussed on the materials and production of such devices and the advantages of microfluidics in general. Moreover commonly used immobilization techniques were highlighted.59 In this section, only a few of the most relevant examples will be discussed. The described enzymatic system for glucose detection was used by Boehm et al. in 2013 who designed a flow microreactor for synthetic enzyme reactions in vitro (see Fig. 3). A model enzyme cascade, consisting of β-Galactosidase (βGal), GOx and HRP for the conversion of galactose and a fluorescent dye was implemented. The reactor along with its structures was produced by moulding polydimethylsiloxane (PDMS) on a fabricated master and closing it with a glass slide. In order to investigate two different compartmentalization mechanisms, enzymes were immobilized to microbeads and packed subsequently into a microfluidic channel or attached directly to the inner surface of the microchannels. By streaming the channels with substrate solution and readout of the product formation, different kinetic parameters of the reaction were determined and the packed bed reactor with enzymes immobilized to microbeads was shown to be 1.5-fold more efficient than the reactor device with enzymatically active microchannels.18 The same enzyme system was implemented by Fornera et al. in 2012, who introduced a flow-through microfluidic device containing the enzymes immobilized to a dendronized polymer in a predetermined pattern that was obtained by a valve system.19 The system can be applied for the determination of lactose in different lactose-containing solutions by measuring the resulting concentration of fluorescent resorufin, generated by the enzyme cascade. Another sophisticated microfluidic system for the realization of complex enzymatic cascades was currently published by Huebner et al. in 2015. In the introduced system, reaction environments are realized by aqueous plugs separated by immiscible fluidic plugs that are pumped through the reaction cascade of enzymes. The applied biocatalysts are immobilized to magnetic microparticles, that allow the fast and easy separation from the product stream and can be resuspended in the reaction solution by applying alternating electromagnetic fields. (DOI: 10.1002/elsc.201400171).Summarization of co-immobilization techniques
A concluding overview over all discussed techniques, including their advantages and disadvantages is given in Table 2.| Technique | Immobilization chemistry | Support | Advantages | Disadvantages | Example |
|---|---|---|---|---|---|
| Random co-immobilization | |||||
| Crosslinking to solid surfaces | Crosslinking agents, click chemistry approaches | Solid supports: surfaces, particles, fibres etc. | Fast and easy, co-localization of enzymes | Only statistical distribution, no site-specificity | 28 |
| Encapsulation | Encapsulation in polymers | Polymers, surfaces coated with polymers | Less enzyme inactivation, fast and easy, co-localization | Diffusional limitations, as substrate has to enter support, no side-specificity | 29–32 |
| Supportless crosslinking | Crosslinking agents, click chemistry approaches | — | High specific activities, no dilution by support | Only statistical distribution, no site specificity | 35 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||
| Site-specific co-immobilization | |||||
| Immobilization to protein scaffolds | Protein tags, click chemistry approaches | Scaffold protein | High control of localization, exploitation of substrate channeling | Protein tagging necessary (genetic engineering), time-consuming and elaborate | 13 |
| Immobilization to DNA scaffolds | Protein tags, click chemistry approaches | Single stranded scaffold DNA, DNA-origami structures | High control of localization, exploitation of substrate channeling | Protein tagging necessary, time-consuming and elaborate | 54, 57 |
| Immobilization in nanocontainers | Encapsulation, embedding, crosslinking, click chemistry approaches | Porous polymersomes | High control of localization by using different immobilization, compartments, product is soluble but separable by filtration | Time-consuming and elaborate | 17 |
| Immobilization in microfluidic devices | Crosslinking agents, protein tags, click chemistry | Surfaces in microfluidic devices | Compartmentalization comparable to living cells, high level of control of fluidic etc. | Elaborate, special equipment needed | 18, 19 |
Summary and outlook
As enzymes provide some important advantages over traditional chemical syntheses, they have been established as green and cost-saving alternative in many fields. The use of enzyme cascades broadens the potential applications due to complex reaction opportunities, while obtaining the high reaction specificity of enzyme. Immobilization of enzyme cascades allows additional advantages. The catalytic complexes can be easy separated from the product, unfavourable kinetics can be circumvented and by co-localization the performance of the cascade can be enhanced by substrate channelling.In this review an overview over different immobilization techniques has been given. The focus was on the random or site-specific immobilization of enzyme cascades leading to highly active multi-enzyme complexes with enhanced stability and activity. A great variety of techniques and different supports with sophisticated features exists nowadays in order to provide an optimal solution for the realization of enzyme cascades in many fields of application.
Notes and references
- M. T. R. Gomes, M. L. Oliva, M. T. P. Lopes and C. E. Salas, Curr. Protein Pept. Sci., 2011, 12, 417–436 CrossRef CAS.
- K. R. Jegannathan and P. H. Nielsen, J. Cleaner Prod., 2013, 42, 228–240 CrossRef CAS PubMed.
- J. D. Bloom, M. M. Meyer, P. Meinhold, C. R. Otey, D. MacMillan and F. H. Arnold, Curr. Opin. Chem. Biol., 2005, 15, 447–452 CAS.
- J. L. Harris and C. S. Craik, Curr. Opin. Chem. Biol., 1998, 2, 127–132 CrossRef CAS.
- R. A. Sheldon, Adv. Synth. Catal., 2007, 349, 1289–1307 CrossRef CAS PubMed.
- R. A. Sheldon, R. Schoevaart and L. M. Van Langen, Biocatal. Biotransform., 2005, 23, 141–147 CrossRef CAS PubMed.
- D. Avnir, S. Braun, O. Lev and M. Ottolenghi, Chem. Mater., 1994, 6, 1605–1614 CrossRef CAS.
- S. Cosnier, Biosens. Bioelectron., 1999, 14, 443–456 CrossRef CAS.
- C. Mateo, J. M. Palomo, G. Fernandez-Lorente, J. M. Guisan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2007, 40, 1451–1463 CrossRef CAS PubMed.
- E. Ricca, B. Brucher and J. H. Schrittwieser, Adv. Synth. Catal., 2011, 353, 2239–2262 CrossRef CAS PubMed.
- H. O. Spivey and J. Ovadi, Methods, 1999, 19, 306–321 CrossRef CAS PubMed.
- Y. H. P. Zhang, Biotechnol. Adv., 2011, 29, 715–725 CrossRef CAS PubMed.
- C. You, S. Myung and Y. H. P. Zhang, Angew. Chem., Int. Ed., 2012, 51, 8787–8790 CrossRef CAS PubMed.
- J. S. Martin del Campo, J. Rollin, S. Myung, Y. Chun, S. Chandrayan, R. Patino, M. W. W. Adams and Y. H. P. Zhang, Angew. Chem., Int. Ed., 2013, 52, 4587–4590 CrossRef CAS PubMed.
- E. A. Carr, J. Lab. Clin. Med., 1946, 31, 1267–1269 Search PubMed.
- E. H. Yoo and S. Y. Lee, Sensors, 2010, 10, 4558–4576 CrossRef PubMed.
- S. F. M. van Dongen, M. Nallani, J. L. L. M. Cornelissen, R. J. M. Nolte and J. C. M. van Hest, Chem.–Eur. J., 2009, 15, 1107–1114 CrossRef CAS PubMed.
- C. R. Boehm, P. S. Freemont and O. Ces, Lab Chip, 2013, 13, 3426–3432 RSC.
- S. Fornera, P. Kuhn, D. Lombardi, A. D. Schluter, P. S. Dittrich and P. Walde, ChemPlusChem, 2012, 77, 98–101 CrossRef CAS PubMed.
- S. D. Minteer, B. Y. Liaw and M. J. COoney, Curr. Opin. Biotechnol., 2007, 18, 228–234 CrossRef CAS PubMed.
- G. T. R. Palmore, H. Bertschy, S. H. Bergens and G. M. Whitesides, J. Electroanal. Chem., 1998, 443, 155–161 CrossRef CAS.
- L. Betancor and H. R. Luckarift, Biotechnol. Genet. Eng. Rev., 2010, 27, 95–114 CrossRef CAS PubMed.
- S. Schoffelen and J. C. M. van Hest, Curr. Opin. Struct. Biol., 2013, 23, 613–621 CrossRef CAS PubMed.
- W. A. Khattak, M. W. Ullah, M. Ul-Islam, S. Khan, M. Kim, Y. Kim and J. K. Park, Appl. Microbiol. Biotechnol., 2014, 98, 9561–9578 CrossRef CAS PubMed.
- M. H. Osman, A. A. Shah and F. C. Walsh, Biosens. Bioelectron., 2011, 26, 3087–3102 CrossRef CAS PubMed.
- I. Migneault, C. Dartiguenave, M. J. Bertrand and K. C. Waldron, BioTechniques, 2004, 37, 790–802 CAS.
- S. S. Wong and L. J. C. Wong, Enzyme Microb. Technol., 1992, 14, 866–874 CrossRef CAS.
- X. P. Deng, T. W. Eyster, Y. Elkasabi and J. Lahann, Macromol. Rapid Commun., 2012, 33, 640–645 CrossRef CAS PubMed.
- L. D. Zhu, R. L. Yang, J. G. Zhai and C. Y. Tian, Biosens. Bioelectron., 2007, 23, 528–535 CrossRef CAS PubMed.
- Y. Wei, H. Dong, J. Xu and Q. Feng, ChemPhysChem, 2002, 3, 802–808 CrossRef CAS.
- X. Ji, Z. Su, P. Wang, G. Ma and S. Zhang, Analyst, 2014, 139, 6467–6473 RSC.
- M. Eguilaz, R. Villalonga, P. Yanez-Sedeno and J. M. Pingarron, Anal. Chem., 2011, 83, 7807–7814 CrossRef CAS PubMed.
- R. A. Sheldon, Biochem. Soc. Trans., 2007, 35, 1583–1587 CrossRef CAS PubMed.
- A. Chmura, S. Rustler, M. Paravidino, F. van Rantwijk, A. Stolz and R. A. Sheldon, Tetrahedron: Asymmetry, 2013, 24, 1225–1232 CrossRef CAS PubMed.
- C. Mateo, A. Chmura, S. Rustler, F. van Rantwijk, A. Stolz and R. A. Sheldon, Tetrahedron: Asymmetry, 2006, 17, 320–323 CrossRef CAS PubMed.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS PubMed.
- J. W. Chin, S. W. Santoro, A. B. Martin, D. S. King, L. Wang and P. G. Schultz, J. Am. Chem. Soc., 2002, 124, 9026–9027 CrossRef CAS PubMed.
- K. Terpe, Appl. Microbiol. Biotechnol., 2003, 60, 523–533 CrossRef CAS PubMed.
- G. Agarwal, R. R. Naik and M. O. Stone, J. Am. Chem. Soc., 2003, 125, 7408–7412 CrossRef CAS PubMed.
- M. Miyazaki, J. Kaneno, S. Yamaori, T. Honda, M. P. P. Briones, M. Uehara, K. Arima, K. Kanno, K. Yamashita, Y. Yamaguchi, H. Nakamura, H. Yonezawa, M. Fujii and H. Maeda, Protein Pept. Lett., 2005, 12, 207–210 CrossRef CAS.
- J. Jeong, T. H. Ha and B. H. Chung, Anal. Chim. Acta, 2006, 569, 203–209 CrossRef CAS PubMed.
- A. Waldbaur, B. Waterkotte, K. Schmitz and B. E. Rapp, Small, 2012, 8, 1570–1578 CrossRef CAS PubMed.
- T. G. Schmidt and A. Skerra, J. Chromatogr. A, 1994, 676, 337–345 CrossRef CAS.
- T. Kinoshita, H. Sato, A. Okada, E. Ohuchi, K. Imai, Y. Okada and M. Seiki, J. Biol. Chem., 1998, 273, 16098–16103 CrossRef CAS PubMed.
- A. Fishman, I. Levy, U. Cogan and O. Shoseyov, J. Mol. Catal. B: Enzym., 2002, 18, 121–131 CrossRef CAS.
- E. Ong, N. R. Gilkes, R. C. Miller, R. A. J. Warren and D. G. Kilburn, Enzyme Microb. Technol., 1991, 13, 59–65 CrossRef CAS.
- K. Taniguchi, K. Nomura, Y. Hata, T. Nishimura, Y. Ksami and A. Kuroda, Biotechnol. Bioeng., 2007, 96, 1023–1029 CrossRef CAS PubMed.
- S. Engin, D. Fichtner, D. Wedlich and L. Fruk, Curr. Pharm. Des., 2013, 19, 5443–5448 CrossRef CAS.
- Y. Zhang, M. K. So, A. M. Loening, H. Q. Yao, S. S. Gambhir and J. H. Rao, Angew. Chem., Int. Ed., 2006, 45, 4936–4940 CrossRef CAS PubMed.
- C. M. Niemeyer, T. Sano, C. L. Smith and C. R. Cantor, Nucleic Acids Res., 1994, 22, 5530–5539 CrossRef CAS PubMed.
- P. W. K. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS PubMed.
- U. Feldkamp and C. M. Niemeyer, Angew. Chem., Int. Ed., 2006, 45, 1856–1876 CrossRef CAS PubMed.
- B. Sacca and C. M. Niemeyer, Chem. Soc. Rev., 2011, 40, 5910–5921 RSC.
- C. M. Niemeyer, J. Koehler and C. Wuerdemann, ChemBioChem, 2002, 3, 242–245 CrossRef CAS.
- J. Muller and C. M. Niemeyer, Biochem. Biophys. Res. Commun., 2008, 377, 62–67 CrossRef PubMed.
- R. P. Goodman, I. A. Schaap, C. F. Tardin, C. M. Erben, R. M. Berry, C. F. Schmidt and A. J. Turberfield, Science, 2005, 310, 1661–1665 CrossRef CAS PubMed.
- B. P. Duckworth, Y. Chen, J. W. Wollack, Y. Sham, J. D. Mueller, T. A. Taton and M. D. Distefano, Angew. Chem., 2007, 46, 8819–8822 CrossRef CAS PubMed.
- J. Fu, M. Liu, Y. Liu, N. W. Woodbury and H. Yan, J. Am. Chem. Soc., 2012, 134, 5516–5519 CrossRef CAS PubMed.
- Y. Asanomi, H. Yamaguchi, M. Miyazaki and H. Maeda, Molecules, 2011, 16, 6041–6059 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |