Sunlight induced atom transfer radical polymerization by using dimanganese decacarbonyl
Mustafa
Ciftci
a,
Mehmet Atilla
Tasdelen
*ab and
Yusuf
Yagci
*ac
aDepartment of Chemistry, Faculty of Science and Letters, Istanbul Technical University, Maslak, TR-34469 Istanbul, Turkey. E-mail: yusuf@itu.edu.tr; Fax: +90 212 285 63 86; Tel: +90 212 285 32 41
bDepartment of Polymer Engineering, Faculty of Engineering, Yalova University, TR-77100 Yalova, Turkey. E-mail: tasdelen@yalova.edu.tr; Fax: +90 226 811 54 01; Tel: +90 226 811 54 12
cKing Abdulaziz University, Center of Excellence for Advanced Materials Research (CEAMR) and Chemistry Department, Faculty of Science, P. O. Box 80203, Jeddah 21589, Saudi Arabia
First published on 30th August 2013
Abstract
A new photoredox catalyst system for atom transfer radical polymerization is developed on the basis of visible light photocatalysis using dimanganese decacarbonyl (Mn2(CO)10) that initiates and controls the polymerization at ambient temperature. The polymerization was performed by a Mn2(CO)10–alkyl halide system with visible- or sunlight in the presence of parts per million (ppm) copper catalysts. The photogenerated ˙Mn(CO)5 radicals are not only able to abstract halogen atoms from alkyl halides to generate carbon centered radicals but also reduce the CuIIBr2 to CuIBr directly, which was used as an activator in the Atom Transfer Radical Polymerization (ATRP) of vinyl monomers such as methyl methacrylate, methyl acrylate and styrene. The method was also used to synthesize graft copolymers from commercially available poly(vinyl chloride) without additional modification.
Introduction
Sunlight covers a broad energy band including infrared, visible, and ultra-violet light, X-rays and gamma rays.1 Since ancient times, it has been used to create hardened and insoluble wood, bamboo, and cotton as building and clothing materials. It would appear that this process involving the action of light became of great importance in the production of thermosetting plastics. Light-induced reactions are advantageous as they are effective, mild and easy processes.2,3 Light can also be used as an external stimulus to switch the reactions “on” and “off” and can trigger the reactions locally at specific positions and spaces.4–6 Light-induced polymerization has been widely employed in conventional radical polymerization as a key technique in various applications, such as coatings, adhesives, medicine, optics and microelectronics.7,8 The use of light in controlled radical polymerization also brings several distinct advantages, including temporal and spatial control over chain growth, easy preparation of well-defined polymers at room temperature and minimization of possible higher temperature side reactions.9,10 Although extensive efforts have been made in this area, it is still crucial to explore an effective light-induced controlled radical polymerization (CRP) method in synthetic polymer chemistry. The commonly used light-induced CRP methods are adapted from thermal counterparts including iniferter,11 nitroxide-mediated radical polymerization (NMRP),12–15 atom transfer radical polymerization (ATRP),16–27 cobalt-mediated radical polymerization (CMRP),28–30 organoiodine-mediated radical polymerization (OMIP),31–35 organotellurium-mediated radical polymerization (TERP)36,37 and reversible addition–fragmentation chain transfer polymerization (RAFT),38–42etc. Among them, light-induced ATRP has been studied extensively because of the broad range of monomers and mild polymerization conditions. The light-induced ATRP is based on photoredox reactions of copper catalysts under various radiation sources with or without various photoinitiators.16–25 Many UV and visible light free radical photoinitiators were reported to be powerful promoters for light-induced ATRP. Recently, Hawker et al. reported the light-induced ATRP of methacrylates regulated by visible light using an iridium based photoredox catalyst.21Dimanganese decacarbonyl (Mn2(CO)10) has a weak Mn–Mn linkage (20–40 kcal mol−1) and visible light photolysis provides the ˙Mn(CO)5 metalloradical with good quantum efficiency.43–45 It abstracts halides from a variety of organohalogen compounds, generating the corresponding carbon centered radicals.46–52 A visible light-induced degenerative iodine transfer polymerization using Mn2(CO)10 in conjunction with alkyl iodides (R–I) was recently developed by Kamigaito and coworkers.31–33 Since the reverse reaction between R˙ radical and I–Mn(CO)5 regenerating R–X and Mn(CO)5˙ is less likely due to the strength of the Mn–I bond, the contribution of the reversible termination mechanism must be quite limited. This system was applicable not only to unconjugated monomers, vinyl acetate, but also to conjugated monomers such as acrylate and styrene derivatives with the use of appropriate initiators (Scheme 1).31–34
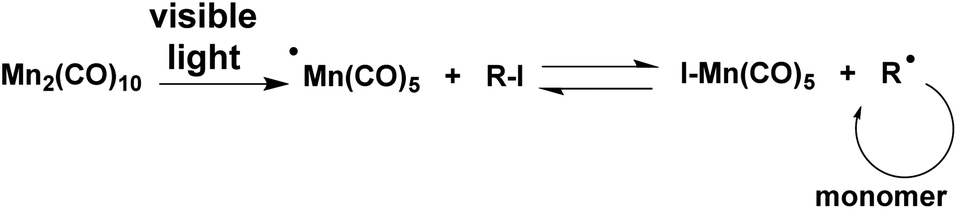 | ||
| Scheme 1 Proposed mechanism of light-induced degenerative iodine transfer polymerization with a Mn2(CO)10–alkyl iodide system. | ||
In the frame of our continuous interest in developing ultraviolet- and visible-light-induced CRP, we herein report a new light-induced ATRP of methyl methacrylate under the sun by using Mn2(CO)10 as the light absorbing component. The polymerization activators, Cu(I)–ligand, can be continuously generated by both Mn(CO)5˙ and R˙ radicals during the polymerization. The mechanism of the light-induced ATRP was studied using a photoreactor emitting light nominally at 400–500 nm at room temperature.
Experimental
Materials
Methyl methacrylate (MMA, 99%; Aldrich), methyl acrylate (MA, 99%; Aldrich) and styrene (St, 99%, Aldrich) were passed through a basic alumina column to remove the inhibitor. N,N,N′,N′′,N′-Pentamethyldiethylenetriamine (PMDETA, 99%; Aldrich) was used as a ligand and was distilled prior to use. Dimanganese decacarbonyl (Mn2(CO)10, 98%; Aldrich) was purified by sublimation and stored in a refrigerator in the dark. Ethyl 2-bromopropionate (EtBP, 98%; Aldrich), methanol (CH3OH, 99.9%; Merck), and copper(II) bromide (CuIIBr2, 99%; Acros) were used as received.Polymerizations
A series of visible light induced ATRP reactions, using MMA, were carried out under different experimental conditions at room temperature. An example detailing a typical procedure is as follows: MMA (2 mL, 18.6 mmol), PMDETA (4 μL, 1.9 × 10−2 mmol), CuIIBr2 (4.2 mg, 1.9 × 10−2 mmol), EtBP (12.2 μL, 9.4 × 10−2 mmol), Mn2(CO)10 (1.8 mg, 4.7 × 10−3 mmol) and methanol (0.1 mL, 2.5 mmol) were put into a Schlenk tube (i.d = 9 mm) equipped with a magnetic stirring bar and the reaction mixture was degassed by three freeze–pump–thaw cycles and left in a vacuum. The mixture was irradiated by a Ker-Vis blue photoreactor equipped with six lamps (Philips TL-D 18 W) emitting light nominally at 400–500 nm at room temperature. The light intensity was 45 mW cm−2 as measured by a Delta Ohm model HD-9021 radiometer. The sunlight ATRP was carried out under sunny weather in April in Istanbul (Turkey); an absolute irradiance measurement leads to an estimated incident energy <60 mW cm−2 in the 400–500 nm range. After the given time the resulting polymers were precipitated in ten-fold excess methanol and then dried under reduced pressure. Conversion of the monomer was determined gravimetrically.Characterization
Gel permeation chromatography (GPC) measurements were obtained from a Viscotek GPCmax autosampler system consisting of a pump, a Viscotek UV detector, and a Viscotek differential refractive index (RI) detector. Three ViscoGEL GPC columns (G2000HHR, G3000HHR, and G4000HHR; 7.8 mm internal diameter, 300 mm length) were used in series. The effective molecular weight ranges were 456–42![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800, 1050–107000, and 10200–2890000, respectively. THF was used as an eluent at a flow rate of 1.0 mL min−1 at 30 °C. Both detectors were calibrated with polystyrene standards having narrow-molecular-weight distribution. Data were analyzed using Viscotek OmniSEC Omni-01 software. The resulting molecular weight distributions were reassessed by universal calibration using Mark–Houwink parameters for poly(methyl methacrylate) (K = 7.56 × 10−5 dL g−1, α = 0.731)53 and for polystyrene (K = 14.1 × 10−5 dL g−1 and α = 0.70).54
800, 1050–107000, and 10200–2890000, respectively. THF was used as an eluent at a flow rate of 1.0 mL min−1 at 30 °C. Both detectors were calibrated with polystyrene standards having narrow-molecular-weight distribution. Data were analyzed using Viscotek OmniSEC Omni-01 software. The resulting molecular weight distributions were reassessed by universal calibration using Mark–Houwink parameters for poly(methyl methacrylate) (K = 7.56 × 10−5 dL g−1, α = 0.731)53 and for polystyrene (K = 14.1 × 10−5 dL g−1 and α = 0.70).54
Results and discussion
The visible-light-mediated photoredox catalysis is an attractive tool to initiate organic reactions because of its low cost, easy availability and almost infinitely available source of energy (e.g. sunlight, household fluorescence or LED bulbs, Xe lamp).55–57 For example, Stephenson et al. reported a visible-light-mediated Appel-type reaction, which converts alcohols into halides in good yields, with exceptional functional group tolerance.58,59 Recently, the radical polymerization of methacrylates was also controlled by visible light using an iridium-based photoredox catalyst.21,25 In order to take advantage of the visible-light-mediated photoredox process, first, optical properties of reaction mixture, dimanganese decacarbonyl, (Mn2(CO)10) and their combination in the visible range were investigated by UV-vis spectroscopy. As can be seen in Fig. 1, only Mn2(CO)10 strongly absorbs visible light whereas the other components of the reaction mixture were completely transparent. Thus, all irradiation was performed at ambient temperature under visible light irradiation using either a photoreactor equipped with six lamps emitting light nominally at 400–500 nm or sunlight. | ||
| Fig. 1 UV-Vis spectra of Mn2(CO)10, reaction mixture and both of them in methanol. The concentration of all components was 2.3 × 10−3 M. | ||
Initially, the polymerization of methyl methacrylate (MMA) was examined by using the CuIIBr2–PMDETA as the catalyst, ethyl 2-bromopropionate (EtBP) as the initiator and Mn2(CO)10 as the photosensitive compound.
The reactants typically consisted of [MMA]0/[EtBP]0/[CuIIBr2]0/[PMDETA]0/[Mn2(CO)10]0 = 200/1/0.2/0.2/0.2 with a small amount of methanol (0.1 mL, 2.5 mmol) to facilitate better dissolution of the CuII complex. Several control experiments were carried out with removal of essential components one-by-one, and the results are shown in Table 1 (entries 1 and 2). Notably, when the polymerizations were conducted in the absence of either light, or Mn2(CO)10 or CuIIBr2, the experiments failed to produce any polymer at the end of irradiation and each component was required for successful controlled radical polymerization. Irradiation of the mixture without CuIIBr2 resulted in free radical polymerization (entry 3, Table 1), most likely due to photogenerated radicals from alkyl halides via classical halogen abstraction reaction.44 A series of polymerization reactions were carried out by changing the molar ratio of Mn2(CO)10 (entries 4–6, Table 1). Encouragingly, initial results employing 0.2 mol% Mn2(CO)10 did afford polymer; however, the reaction displayed little to no control, with a molecular weight distribution value of 1.82. The irreversible radical termination occurred at high Mn2(CO)10 concentration and to gain control over this system the radical concentrations needed to be lowered, which could be achieved by simply reducing the catalyst loading. Compared with the reference conditions of entry 4, reducing the concentration of the Mn2(CO)10 significantly decreased the yield of PMMA and molecular weight distribution of resulting polymers as well (Mn,GPC = 8100, Mw/Mn = 1.16). Furthermore, the experimental molecular weight of the polymer was in good agreement with the theoretical value using 0.05 mol% Mn2(CO)10. Decreasing the catalyst amount to the ppm level resulted in slightly slower polymerization while preserving good control over the molar mass and narrow molecular weight distribution (entry 7, Table 1). Since the light-induced ATRP system only requires photons to drive the reaction, the sunlight can be utilized as an inexpensive, abundant, clean and renewable energy source. Therefore, sunlight was used to drive the polymerization of MMA in both high and low catalyst concentrations (entries 8 and 9, Table 1). When the sunlight was used as the light source, the polymerization proceeded at a relatively slower rate (entry 6, Table 1) than that in the reference process (entry 8, Table 1). Apparently, the low catalyst loadings with sunlight resulted in a polymer with a higher molar mass than that theoretically calculated and a molecular weight distribution of ∼1.43, thus indicating loss of control (entry 9, Table 1). The applicability of this method was extended to other vinyl monomers such as methyl acrylate and styrene (entries 10 and 11 Table 1). In both cases, the control over the polymerization is good, with low polydispersity values and good correlation between experimental and theoretical molecular weights.
| No. | [M]0/[RX]0/[MtX]0/[L]0/[Mn2(CO)10]0 | Mon. | Conv. (%) | M n,theo (g mol−1) | M n,GPC (g mol−1) | M w/Mnb |
|---|---|---|---|---|---|---|
| a M n,theo = [monomer]0/([RX]0 × Mw,monomer × conversion). b Molecular weight (Mn,GPC) and distribution (Mw/Mn) were determined by gel permeation chromatography. c Polymerization was performed under dark, time = 180 min. d Polymerization was performed under visible light irradiation, time = 180 min, light intensity = 45 mW cm−2. e Polymerization was performed with sunlight, time = 180 min, light intensity = 60 mW cm−2. | ||||||
| 1c | 200/1/0.2/0.2/0.2 | MMA | — | — | — | — |
| 2d | 200/1/0.2/0.2/0.0 | MMA | — | — | — | — |
| 3d | 200/1/0.0/0.2/0.2 | MMA | 93 | 18600 |
74800 |
2.65 |
| 4d | 200/1/0.2/0.2/0.2 | MMA | 82 | 16400 |
34500 |
1.82 |
| 5d | 200/1/0.2/0.2/0.1 | MMA | 54 | 8500 | 16500 |
1.48 |
| 6d | 200/1/0.2/0.2/0.05 | MMA | 35 | 7000 | 8100 | 1.16 |
| 7d | 200/1/0.01/0.01/0.05 | MMA | 29 | 5800 | 6200 | 1.21 |
| 8e | 200/1/0.2/0.2/0.05 | MMA | 28 | 6200 | 5600 | 1.28 |
| 9e | 200/1/0.01/0.01/0.05 | MMA | 23 | 4600 | 5500 | 1.43 |
| 10d | 200/1/0.2/0.2/0.05 | MA | 21 | 3600 | 4100 | 1.19 |
| 11d | 200/1/0.2/0.2/0.05 | St | 17 | 3500 | 3800 | 1.23 |
Overall, the control experiment results support the fact that the polymerization proceeds through the photocatalytic initiation mechanism that we have proposed in Scheme 2. The primary photochemical reaction involves the homolysis of Mn2(CO)10, which affords the metal-centered ˙Mn(CO)5 radicals. This radical was not only able to abstract halogen atoms from alkyl halides to generate carbon centered radicals but also reduced the CuIIBr2 to CuIBr directly, which was used as an activator in the ATRP. Subsequently, polymerization was started by the activation of an R–X initiator by the CuIX/L activator.
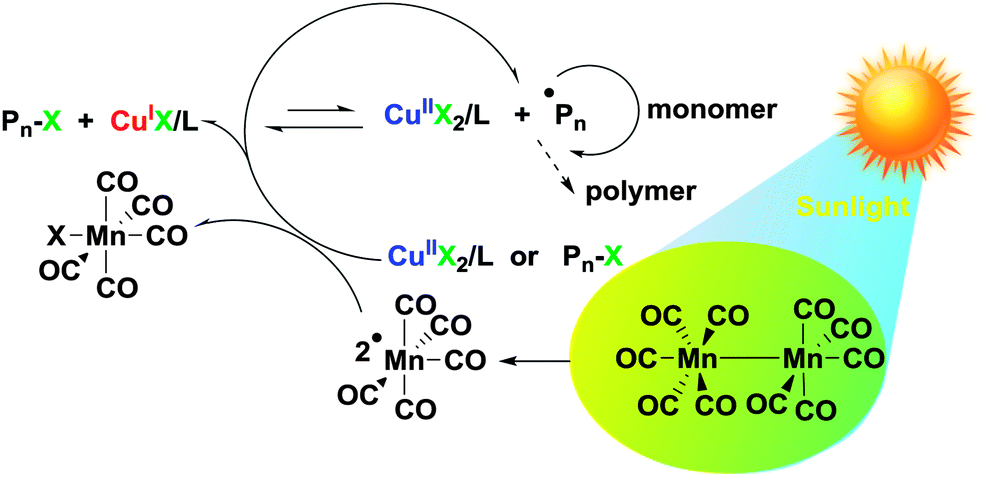 | ||
| Scheme 2 Mechanistic scheme for sunlight induced ATRP using Mn2(CO)10. | ||
In order to gain more insight into the initiation mechanism, the change in the optical absorption spectrum of the polymerization solution was recorded as a function of irradiation time (Fig. 2). The photoredox reaction of Mn2(CO)10 with Pn–X or CuIIX2/L was confirmed experimentally by UV-Vis-spectroscopy as the absorption spectra of Mn2(CO)10 significantly decreased. Although, the maximum absorption spectrum at 345 nm decreased gradually, it was still observable throughout the polymerization period. As formation of CuI ions cannot be observed directly in solution, it was detected indirectly through its catalytic activity in the polymerization.
 | ||
| Fig. 2 Typical UV-vis spectral changes of the initiating system on visible light irradiation, λ = 400–500 nm under nitrogen. | ||
To further investigate the exact role of Mn2(CO)10, visible light irradiation of the reaction mixture without alkyl halide was monitored by UV-Vis spectroscopy. In this case, a new weak band centered at 455 nm appeared (Fig. 3). This absorption corresponding to the ligand-to-metal charge-transfer transition of a copper metal was responsible for the photoreduction of CuII to CuI.60,61 The result implies that photogenerated ˙Mn(CO)5 radicals may directly reduce the CuII to CuI under visible light irradiation.
 | ||
| Fig. 3 Typical UV spectral changes of the initiating system in the absence of alkyl halide under visible light irradiation, λ = 400–500 nm under nitrogen. | ||
Kinetic studies of the visible- and sunlight induced polymerization revealed that the reaction proceeded with first-order kinetics, indicating a constant concentration of growing radicals during polymerization. The linear relationship between monomer consumption, ln([M]0/[M]) and the polymerization time indicated that the termination reaction was suppressed (Fig. 4a). Evolution of the molar mass and molecular weight distribution (Mw/Mn) (Fig. 4b) shows that during visible- and sunlight ATRP of MMA with [MMA]0/[RX]0/[MtX]0/[L]0/[Mn2(CO)10]0 = 200/1/0.2/0.2/0.05 ratio, the polydispersity indexes in both cases were slightly broader (1.13–1.33), and the obtained molar masses were in good agreement with the theoretical values, indicating high initiation efficiency.
 | ||
| Fig. 4 Visible light- or sunlight-induced ATRP of methyl methacrylate ([MMA]0/[EtBP]0/[CuIIBr2]0/[PMDETA]0/[Mn2(CO)10]0 = 200/1/0.2/0.2/0.05): (a) kinetic plot and (b) molecular weights and distributions of the resulting polymers as a function of degree of conversion. | ||
Low catalyst concentrations and mild reaction conditions are desirable to reduce the environmental impact of ATRP. Therefore, Cu catalyst concentrations were reduced to 100 ppm and instead of previous conditions sunlight was used as the irradiation source.
Similar to the previous case, the rate of polymerization was higher using the visible light than that when using the sunlight. As shown in Fig. 5a, a linear dependence was again observed in the kinetic plots, indicating a constant concentration of growing radicals during polymerization. Evolution of the molar mass and molecular weight distribution (Fig. 5b) shows that the experimental molecular weight values were close to the theoretical ones in both cases. In addition, the molecular weight distributions were relatively high, ranging from 1.41 to 1.48 and reasonable control was observed under sunlight irradiation.
 | ||
| Fig. 5 Visible light- or sunlight-induced ATRP of methyl methacrylate with low copper catalyst loading ([MMA]0/[EtBP]0/[CuIIBr2]0/[PMDETA]0/[Mn2(CO)10]0 = 200/1/0.01/0.01/0.05): (a) kinetic plot and (b) molecular weights and distributions of the resulting polymers as a function of degree of conversion. | ||
The effect of visible light irradiation on the polymerization of MMA was further studied by employing a periodic light on–off process (Fig. 6). The reaction mixture was exposed to visible light for a two-hour period to afford approximately 30% conversion. Thereafter, the light source was periodically turned-off and the polymerization proceeded at a much lower rate during this period, indicating a negligible concentration of the active radical present under dark conditions.
 | ||
| Fig. 6 Effect of visible light during the polymerization of MMA at room temperature: [MMA]0/[EtBP]0/[CuIIBr2]0/[PMDETA]0/[Mn2(CO)10]0 = 200/1/0.2/0.2/0.05. | ||
Exposure to visible light for a second two-hour period “woke up” the polymerization, which proceeded with the same kinetic character as that observed in the former light-on process. This indicates that light stimulus not only controlled the initiation steps, but also efficiently regulated the chain growth during the polymerization process.
The “living” nature of the system was further confirmed by treating the macroinitiator made by this technique with the same monomer for chain extension using visible light-induced ATRP conditions. For example, a macroinitiator (Mn = 6200 g mol−1, Mw/Mn = 1.21) prepared by visible light-induced ATRP was employed in chain extension reaction. The GPC trace of the final polymer was clearly shifted to higher molecular weight (conversion = 45%, Mn = 9800 g mol−1, Mw/Mn = 1.26) compared to the precursor macroinitiator and there was no detectable amount of the unreacted initial block (Fig. 7).
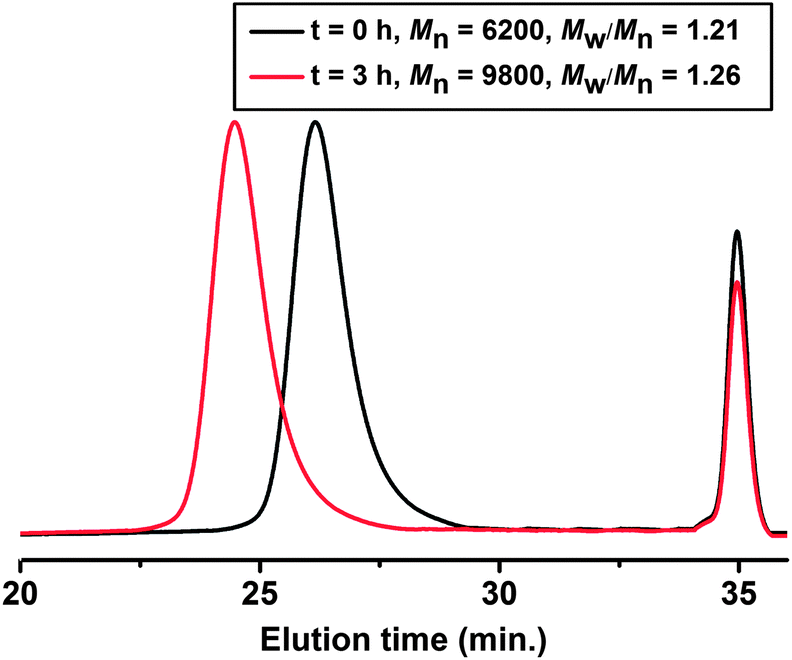 | ||
| Fig. 7 GPC traces of the PMMA–Br macroinitiator before and after chain extension via visible light-induced ATRP using Mn2(CO)10. Experimental conditions: [MMA]0/[PMMA-Br]0/[CuIIBr2]0/[PMDETA]0/[Mn2(CO)10]0 = 200/1/0.01/0.01/0.05 ratio. | ||
One of the key advantages of Mn2(CO)10 chemistry over ATRP is its ability to abstract halogen atoms from primary alkyl halides. Hence, when using alkyl halides, primary carbon-centered radicals were generally easier to form than secondary radicals, while tertiary radicals were even more difficult to prepare and this usually required iodide precursors.62 From this point, chlorine atoms of commercially available poly(vinyl chloride) (PVC, Mn = 63 000, Mw/Mn = 2.20) can act as initiation sites for the direct grafting of MMA by visible light induced ATRP.63,64 The graft copolymerization was carried out for 24 h under similar conditions. The GPC results illustrate an efficient grafting reaction of the PVC and formation of a graft copolymer (conversion = 30%, Mn = 135000 g mol−1, Mw/Mn = 1.91). Their GPC traces show a monomodal molecular weight distribution and a significant shift of the peak value toward higher molecular weights (Fig. 8). This suggests that graft copolymerization occurred without detectable free homopolymer formation.
 | ||
| Fig. 8 GPC traces of PVC and PVC-g-PMMA. Experimental conditions: [MMA]0/[PVC]0/[CuIIBr2]0/[PMDETA]0/[Mn2(CO)10]0 = 200/1/0.2/0.2/0.3 ratio. | ||
Conclusions
In the present article, new initiating systems based on dimanganese decacarbonyl chemistry are proposed for the light-induced ATRP of methyl methacrylate. The advantages of this method are its highly responsive nature, facile reaction setup, use of only ppm level of catalyst, and commercially available and inexpensive catalysts. The growth of the polymer chain can be manipulated by either varying the Mn2(CO)10 concentration or adjusting light intensity, which changes the concentration of the CuI catalyst. Moreover, a simple modification of commercially available PVC by visible light-induced ATRP of MMA forms a corresponding graft copolymer. The present sunlight induced ATRP can be used as an easy, convenient and inexpensive process as an alternative to traditional thermal or photochemical controlled radical polymerization.Notes and references
- S. Protti and M. Fagnoni, Photochem. Photobiol. Sci., 2009, 8, 1499–1516 CAS.
- M. A. Tehfe, J. Lalevee, D. Gigmes and J. P. Fouassier, Macromolecules, 2010, 43, 1364–1370 CrossRef CAS.
- J. Lalevee and J. P. Fouassier, Polym. Chem., 2011, 2, 1107–1113 RSC.
- M. A. Tasdelen and Y. Yagci, Angew. Chem., Int. Ed., 2013, 52, 5930–5938 CrossRef CAS PubMed.
- M. A. Tasdelen and Y. Yagci, Tetrahedron Lett., 2010, 51, 6945–6947 CrossRef CAS PubMed.
- M. A. Tasdelen, G. Yilmaz, B. Iskin and Y. Yagci, Macromolecules, 2012, 45, 56–61 CrossRef CAS.
- Y. Yagci, S. Jockusch and N. J. Turro, Macromolecules, 2010, 43, 6245–6260 CrossRef CAS.
- M. A. Tasdelen and Y. Yagci, Aust. J. Chem., 2011, 64, 982–991 CrossRef CAS.
- S. Yamago and Y. Nakamura, Polymer, 2013, 54, 981–994 CrossRef CAS PubMed.
- M. A. Tasdelen, M. Ciftci, M. Uygun and Y. Yagci, in Progress in Controlled Radical Polymerization: Mechanisms and Techniques, ed. K. Matyjaszewski, B. S. Sumerlin and N. V. Tsarevsky, 2012, vol. 1100, pp. 59–72 Search PubMed.
- T. Otsu, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 2121–2136 CrossRef CAS.
- Y. Guillaneuf, D. Bertin, D. Gigmes, D. L. Versace, J. Lalevée and J. P. Fouassier, Macromolecules, 2010, 43, 2204–2212 CrossRef CAS.
- S. Hu, J. H. Malpert, X. Yang and D. C. Neckers, Polymer, 2000, 41, 445–452 CrossRef CAS.
- E. Yoshida, Colloid Polym. Sci., 2009, 287, 767–772 CAS.
- X. Liu, X. Zhang, X. Zhang, G. Wu, J. Yang, L. Pang, Z. Zeng and Y. Chen, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 2659–2665 CrossRef CAS.
- M. A. Tasdelen, M. Uygun and Y. Yagci, Macromol. Chem. Phys., 2010, 211, 2271–2275 CrossRef CAS.
- M. A. Tasdelen, M. Uygun and Y. Yagci, Macromol. Rapid Commun., 2011, 32, 58–62 CrossRef CAS PubMed.
- M. A. Tasdelen, M. Uygun and Y. Yagci, Macromol. Chem. Phys., 2011, 212, 2036–2042 CrossRef CAS.
- M. A. Tasdelen, M. Ciftci and Y. Yagci, Macromol. Chem. Phys., 2012, 213, 1391–1396 CrossRef CAS.
- G. Zhang, I. Y. Song, K. H. Ahn, T. Park and W. Choi, Macromolecules, 2011, 44, 7594–7599 CrossRef CAS.
- B. P. Fors and C. J. Hawker, Angew. Chem., Int. Ed., 2012, 51, 8850–8853 CrossRef CAS PubMed.
- N. Vargas Alfredo, N. Espinosa Jalapa, S. Lopez Morales, A. D. Ryabov, R. Le Lagadec and L. Alexandrov, Macromolecules, 2012, 45, 8135–8146 CrossRef.
- J. Mosnáček and M. Ilčíková, Macromolecules, 2012, 45, 5859–5865 CrossRef.
- D. Konkolewicz, K. Schröder, J. Buback, S. Bernhard and K. Matyjaszewski, ACS Macro Lett., 2012, 1, 1219–1223 CrossRef CAS.
- J. E. Poelma, B. P. Fors, G. F. Meyers, J. W. Kramer and C. J. Hawker, Angew. Chem., Int. Ed., 2013, 52, 6844–6848 CrossRef CAS PubMed.
- O. S. Taskin, G. Yilmaz, M. A. Tasdelen and Y. Yagci, Polym. Int., 2013 DOI:10.1002/pi.4573.
- J. Yan, B. Li, F. Zhou and W. Liu, ACS Macro Lett., 2013, 2, 592–596 CrossRef CAS.
- C. Detrembleur, D. L. Versace, Y. Piette, M. Hurtgen, C. Jérôme, J. Lalevée and A. Debuigne, Polym. Chem., 2012, 3, 1856–1866 RSC.
- A. Debuigne, M. Schoumacher, N. Willet, R. Riva, X. Zhu, S. Rütten, C. Jérôme and C. Detrembleur, Chem. Commun., 2011, 47, 12703–12705 RSC.
- Y. G. Zhao, M. M. Yu and X. F. Fu, Chem. Commun., 2013, 49, 5186–5188 RSC.
- K. Koumura, K. Satoh and M. Kamigaito, Macromolecules, 2008, 41, 7359–7367 CrossRef CAS.
- K. Koumura, K. Satoh and M. Kamigaito, Macromolecules, 2009, 42, 2497–2504 CrossRef CAS.
- K. Koumura, K. Satoh and M. Kamigaito, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1343–1353 CrossRef CAS.
- A. D. Asandei, O. I. Adebolu and C. P. Simpson, J. Am. Chem. Soc., 2012, 134, 6080–6083 CrossRef CAS PubMed.
- A. Ohtsuki, A. Goto and H. Kaji, Macromolecules, 2012, 46, 96–102 CrossRef.
- E. Mishima, T. Tamura and S. Yamago, Macromolecules, 2012, 45, 2989–2994 CrossRef CAS.
- Y. Nakamura, T. Arima, S. Tomita and S. Yamago, J. Am. Chem. Soc., 2012, 134, 5536–5539 CrossRef CAS PubMed.
- J. F. Quinn, L. Barner, C. Barner-Kowollik, E. Rizzardo and T. P. Davis, Macromolecules, 2002, 35, 7620–7627 CrossRef CAS.
- M. A. Tasdelen, Y. Y. Durmaz, B. Karagoz, N. Bicak and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3387–3395 CrossRef CAS.
- Y. Z. You, C. Y. Hong, R. K. Bai, C. Y. Pan and J. Wang, Macromol. Chem. Phys., 2002, 203, 477–483 CrossRef CAS.
- M. K. Ham, J. HoYouk, Y. K. Kwon and Y. J. Kwark, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2389–2397 CrossRef CAS.
- G. H. Liu, H. Shi, Y. R. Cui, J. Y. Tong, Y. Zhao, D. J. Wang and Y. L. Cai, Polym. Chem., 2013, 4, 1176–1182 RSC.
- C. H. Bamford, P. A. Crowe and R. P. Wayne, Proc. R. Soc. London, Ser. A, 1965, 284, 455–468 CrossRef CAS.
- C. H. Bamford and R. Denyer, Nature, 1968, 217, 59–60 CrossRef CAS.
- M.-A. Tehfe, J. Lalevee, D. Gigmes and J. P. Fouassier, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1830–1837 CrossRef CAS.
- B. C. Gilbert, R. J. Harrison, C. I. Lindsay, P. T. McGrail, A. F. Parsons, R. Southward and D. J. Irvine, Macromolecules, 2003, 36, 9020–9023 CrossRef CAS.
- G. Acik, M. U. Kahveci and Y. Yagci, Macromolecules, 2010, 43, 9198–9201 CrossRef CAS.
- M. U. Kahveci, G. Acik and Y. Yagci, Macromol. Rapid Commun., 2012, 33, 309–313 CrossRef CAS PubMed.
- B. Iskin, G. Yilmaz and Y. Yagci, Macromol. Chem. Phys., 2013, 214, 94–98 CrossRef CAS.
- Y. Yagci and Y. Hepuzer, Macromolecules, 1999, 32, 6367–6370 CrossRef CAS.
- M. Niwa, N. Katsurada, T. Matsumoto and M. Okamoto, J. Macromol. Sci., Part A: Pure Appl.Chem., 1988, 25, 445–466 CrossRef.
- M. Niwa, N. Higashi and M. Okamoto, J. Macromol. Sci., Part A: Pure Appl.Chem., 1988, 25, 1515–1525 CrossRef.
- Y. J. Chen, J. B. Li, N. Hadjichristidis and J. W. Mays, Polym. Bull., 1993, 30, 575–578 CrossRef CAS.
- C. Strazielle, H. Benoit and O. Vogl, Eur. Polym. J., 1978, 14, 331–334 CrossRef CAS.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- J. Lalevee, M. A. Tehfe, F. Morlet-Savary, B. Graff, F. Dumur, D. Gigmes, N. Blanchard and J. P. Fouassier, Chimia, 2012, 66, 439–441 CAS.
- Y. M. Xi, H. Yi and A. W. Lei, Org. Biomol. Chem., 2013, 11, 2387–2403 CAS.
- J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- C. H. Dai, J. M. R. Narayanam and C. R. J. Stephenson, Nature Chem., 2011, 3, 140–145 CrossRef CAS PubMed.
- A. S. Mereshchenko, S. K. Pal, K. E. Karabaeva, P. Z. El-Khoury and A. N. Tarnovsky, J. Phys. Chem. A, 2012, 116, 2791–2799 CrossRef CAS PubMed.
- B. G. Jeliazkova and M. A. Doicheva, Polyhedron, 1996, 15, 1277–1282 CrossRef CAS.
- B. C. Gilbert and A. F. Parsons, J. Chem. Soc., Perkin Trans. 2, 2002, 367–387 RSC.
- A. D. Asandei and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 3392–3418 CrossRef CAS.
- N. Bicak and M. Ozlem, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3457–3462 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |