 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Methylation using dimethylcarbonate catalysed by ionic liquids under continuous flow conditions
Toma N.
Glasnov
a,
John D.
Holbrey
*b,
C. Oliver
Kappe
*a,
Kenneth R.
Seddon
b and
Ting
Yan
b
aChristian Doppler Laboratory for Microwave Chemistry (CDLMC) and Institute of Chemistry, Karl-Franzens University Graz, Heinrichstrasse 28, A-80010 Graz, Austria. E-mail: oliver.kappe@uni–graz.at
bThe QUILL Research Centre, School of Chemistry and Chemical Engineering, The Queen's University of Belfast, Belfast, BT9 5AG, United Kingdom. E-mail: j.holbrey@qub.ac.uk
First published on 20th September 2012
Abstract
The ionic liquid, tributylmethylammonium methylcarbonate, has been employed as a catalytic base for clean N-methylation of indole with dimethylcarbonate. The reaction conditions were optimised under microwave heating to give 100% conversion and 100% selectivity to N-methylindole, and subsequently transferred to a high temperature/high pressure (285 °C/150 bar) continuous flow process using a short (3 min) residence time and 2 mol% of the catalyst to efficiently methylate a variety of different amines, phenols, thiophenols and carboxylic acid substrates. The extremely short residence times, versatility, and high selectivity have significant implications for the synthesis of a wide range of pharmaceutical intermediates, as high product throughputs can be obtained via this scalable continuous flow protocol. It has also been shown that the ionic liquid can be generated in situ from tributylamine, which has the net effect of transforming an ineffective stoichiometric base into a highly efficient catalyst for this broad class of reactions.
Introduction
Dimethylcarbonate (DMC) is a valuable green reagent with versatile and tunable chemical reactivity1,2 and can be used either as a methoxycarbonylating agent (at lower temperatures, by nucleophilic attack at the carbonyl carbon of DMC) or as a methylating agent (at higher temperatures, typically above 120 °C, via nucleophilic attack at the methyl carbon of DMC). Methylation processes are particularly significant for industrial chemistry, and DMC is a good example of a green methylating agent, since it is non-toxic, biodegradable, relatively inert and safe under ambient conditions, and methylation occurs via methyl–oxygen bond cleavage leading to the generation of only CO2 and methanol as by-products:| (CH3O)2CO + Nu–H → Nu–CH3 + CH3OH + CO2 |
DMC has been used as an alternative to dimethylsulfate and methyl halides and triflate – all highly toxic and hazardous chemicals – in the synthesis of surfactants, detergents, and phase-transfer reagents from amines and phosphines,3 and for the methylation of a variety of substrates including carboxylic acids, phenols, thiols, and NH-containing aliphatic and aromatic amines of interest as pharmaceutical intermediates.4
Usually, selective methylation using DMC requires elevated temperatures (>160 °C) and extended reaction times. In order to achieve reasonable reaction rates, or to drive conversion, inorganic2,5 or strong organic bases (such as 1,8-diazbicycloundec-7-ene (DBU) and 1,4-diazabicyclo[2.2.2]octane (DABCO) etc.4,6) have been used, and although nominally catalytic, they are frequently added in stoichiometric, or even excess, quantities. Weaker organic bases such as tributylamine are usually considered to be inactive.6
Microwave heating has been successfully applied to DMC-based methylation reactions in order to access the high temperatures required and to shorten reaction times.7 However, since microwave batch processes can be difficult to scale,8 the transfer to continuous flow processes is desirable, and examples including high-temperature DBU-catalysed methylation of phenols,9,10 microwave-heated continuous flow processes,11 and several recent procedures using scCO2 as solvent12 have been described.
There is significant interest in integrating ionic liquids13 as solvents with the high temperature and pressure chemical process optimisation elements of microwave14,15 and flow16 technologies. Because of the two modes of microwave absorption, ionic liquids are heated particularly efficiently under microwave irradiation15,17,18 and have zero- to low effective vapour pressures.19 These are characteristics which enable rapid heating while maintaining relatively low reactor pressures leading to safer operation, important to optimisations under conditions where a reaction is “rapidly heated to the highest tolerable temperature, held there for the shortest possible time and then quenched”20 as a route to kinetic products and improved conversions. Indeed, there are a number of reports using organic salts (and ionic liquids) to assist in reactions with DMC, either as solvent,21 microwave absorbers,7 or as phase transfer catalysts.22
Fabris et al.23 have used ionic liquids with methylcarbonate ([CH3OCO2]−) anions as basic catalysts for Michael and Henry reactions. This suggested that such ionic liquids,24 readily prepared by reaction of aromatic or tertiary amines with DMC,25 might serve as promoters for methylation reactions with DMC and so provide a congruent system where both the active catalyst and products were generated using the same reagents.
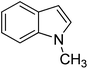
Thus, we set out to evaluate whether basic ionic liquids could be used to promote methylation reactions with DMC. Here, we report on the use of the ionic liquid, tributylmethylammonium methylcarbonate (4), as a catalyst for N-methylation of indole (1) with DMC (Fig. 1), and the optimisation of the reaction under microwave batch conditions to yield 100% conversion and selectivity to N-methylindole (2). We also present a generally applicable continuous flow N-, O,- and S-methylation protocol employing dimethylcarbonate as solvent in its near- or supercritical state, in an extreme high-temperature/high-pressure process window (285 °C/150 bar), which allows a reduction of the required reaction time to approximately 3 min using only 2 mol% of an in situ generated environmentally benign ionic liquid catalyst.
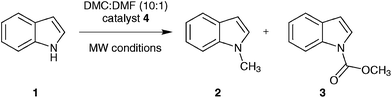 | ||
| Fig. 1 Reaction of indole (1) with DMC forming N-methylindole (2) and N-(methoxycarbonyl)indole (3) catalysed by the basic ionic liquid tributylmethylammonium methylcarbonate (4). | ||
Experimental
All chemicals were purchased from commercial sources and were used without further purification. Analytical HPLC analysis (Shimadzu LC 20 AD) was carried out on a C-18 reversed-phase analytical column (150 mm × 4.6 mm, particle size 5 μm) using mobile phases A (water–ethanenitrile, 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 (v/v) + 0.1% trifluoroethanoic acid) and B (ethanenitrile + 0.1% trifluoroethanoic acid) at a flow rate of 1 cm3 min−1 and linear gradient from solvent 30% B to 100% B in 9 min, hold at 100% solvent B for 1 min. GC/MS (FOCUSGC/DSQ II MS, ThermoFisher) monitoring was based on electron impact ionisation (70 eV) using a HP/5MS column (30 m × 0.250 mm × 0.25 μm). After 1 min at 50 °C the temperature was increased at 25 °C min−1 up to 300 °C and kept at 300 °C for 1 min. The carrier gas was helium and the flow rate was 1.0 cm3 min−1 in constant flow mode. The identity of the peaks was confirmed by computerised comparison with the NIST library. 1H NMR spectra were recorded on a Bruker 300 MHz instrument. Chemical shifts (δ) are expressed in ppm downfield from TMS as internal standard. The letters s, d, t, q, and m are used to indicate singlet, doublet, triplet, quadruplet and multiplet.
:10 (v/v) + 0.1% trifluoroethanoic acid) and B (ethanenitrile + 0.1% trifluoroethanoic acid) at a flow rate of 1 cm3 min−1 and linear gradient from solvent 30% B to 100% B in 9 min, hold at 100% solvent B for 1 min. GC/MS (FOCUSGC/DSQ II MS, ThermoFisher) monitoring was based on electron impact ionisation (70 eV) using a HP/5MS column (30 m × 0.250 mm × 0.25 μm). After 1 min at 50 °C the temperature was increased at 25 °C min−1 up to 300 °C and kept at 300 °C for 1 min. The carrier gas was helium and the flow rate was 1.0 cm3 min−1 in constant flow mode. The identity of the peaks was confirmed by computerised comparison with the NIST library. 1H NMR spectra were recorded on a Bruker 300 MHz instrument. Chemical shifts (δ) are expressed in ppm downfield from TMS as internal standard. The letters s, d, t, q, and m are used to indicate singlet, doublet, triplet, quadruplet and multiplet.
Microwave irradiation experiments were carried out in a Monowave 300 (Anton Paar GmbH, Graz, Austria) in 10 cm3 Pyrex microwave process vials.15,26 Reaction times refer to hold times at the temperature indicated, not to total irradiation times. The temperature was measured using an internal fibre-optic temperature sensor.
The flow chemistry experiments described were performed using an X-Cube Flash flow reactor (ThalesNano Inc.).27 The synthesised compounds were purified using an automated chromatography system (SP1™, Biotage) using cartridges packed with KP-SIL, 60 Å (40–63 μm particle size) and ethyl ethanoate–petroleum ether mixtures as eluent. All products synthesised in this study are known in the literature and were identified and characterised by 1H NMR and MS analysis.
General procedure for the batch microwave synthesis
In a typical experiment, a solution of indole (234 mg, 2 mmol) in DMC–DMF (10:1, 2.2 cm3) and tributylmethylammonium methylcarbonate (5.5–55 mg, 0.01–0.1 eq.) were placed in a 5 cm3 microwave vial equipped with a magnetic stir bar. The sealed reaction vial was placed in the microwave reactor and heated, with stirring, at the indicated temperature over the indicated time. The solution was then cooled to room temperature and the reaction mixture analysed directly by HPLC.
:1) was added tributylamine (7.4 mg, 9.5 μl, 2 mol%) and the reaction mixture was stirred for 30 s. At the same time the X-cube Flash reactor was equipped with a stainless steel reaction coil (4 cm3 volume, ∼3 min residence time at 1.3 cm3 min−1 flow rate). The reaction conditions; temperature (285 °C), flow rate (1.3 cm3 min−1) and pressure (150 bar), were selected and the solvent DMC–DMF (10:1) was pumped through the flow reactor until the instrument had achieved a steady state. Then the freshly prepared reaction mixture was pumped through the flow reactor, collected, and concentrated under vacuum. The product was isolated from the residue via flash chromatography on the Biotage SP1™ instrument. All of the products are known compounds and the obtained analytical data correspond to those reported in the literature.
Results and discussion
Recent work from our laboratories has shown that high-temperature/high-pressure microwave protocols can be translated into scalable continuous flow processes with relative ease.8 Reaction optimisation was performed using controlled microwave heating in a single-mode batch microwave reactor capable of superheating reaction mixtures up to 300 °C and 30 bar pressure (b.p. DMC = 90 °C at ambient pressure) in order to identify desired high-temperature/high-pressure continuous flow conditions.26Initially, we focused our attention on the selective N-methylation of indole (1) as a model reaction (Fig. 1), since this transformation is well studied in the literature.30
The reactions of indole with DMC using DBU as a base has been described by Shieh et al.31 Stoichiometric amounts of base were used and gave improvements to both conversion and rate compared to milder bases such as Na2CO3, or K2CO3 when used at 90 °C. Under microwave irradiation at 160 °C, the reaction times could be substantially reduced and the authors also note that the addition of tetrabutylammonium iodide, presumably as a phase transfer catalyst, enhanced the reaction rate. Subsequently, the same group reported the N-methylation of indoles using DABCO in catalytic amounts.6 DABCO promoted N-methylation using 10 mol% of base (at 90 °C over 5 h), whereas using dimethylaminopyridine or DBU, the N-methoxycarbonylated product was predominantly formed. Using tributylamine, no reaction was observed. The high costs of both DBU and DABCO necessitate their use in catalytic quantities; methylation of phenols with DMC, using DBU in catalyst, has been described at higher temperatures (160–220 °C) operating under continuous flow conditions.10,11
The reaction of indole with DMC, in the presence of tributylmethylammonium methylcarbonate ionic liquid, was investigated here, first under batch conditions using an Anton Parr Monowave 300 microwave reactor, and then in flow using a ThalesNano X flash reactor. Initial results (not shown) in which an excess of the ionic liquid was used as the reaction solvent indicated that the methylation reaction was feasible. Reaction optimisation was subsequently performed using a 10:1 vol/vol mixture of DMC–DMF with sub-stoichiometric quantities of the ionic liquid as a catalyst. The effects of catalyst content, reaction temperature and time were examined, starting from 10 mol% of ionic liquid (relative to indole) and reaction times between 10–30 min.
In a standard microwave experiment, a solution of indole (1) in DMC–DMF (10:1 v/v) together with 10 mol% of tributylmethylammonium methylcarbonate (4) catalyst were heated for 10 min at 90 °C. Analysis by HPLC (215 nm) revealed only 45% indole conversion and formation of N-(methoxycarbonyl)indole (3) as the major product. In subsequent experiments the reaction temperature was gradually increased (results are shown in Table 1). The yield of N-(methoxycarbonyl)indole (3) increased as the temperature increased (entries 1–3, Table 1), with a slight maximum in conversion using a short reaction time at 130 °C (entry 3). At reaction temperatures below 150 °C, no N-methylindole was detected (even when the reaction time was increased to 30 min).
| Entry | Temp./°C | t/min | Catalyst 4/mol% | Conversion/% (HPLC 215 nm) | Selectivity/% | |
|---|---|---|---|---|---|---|
| 2 | 3 | |||||
| 1 | 90 | 10 | 10 | 45 | 0 | 45 |
| 2 | 110 | 10 | 10 | 59 | 0 | 59 |
| 3 | 130 | 10 | 10 | 64 | 0 | 64 |
| 4 | 150 | 10 | 10 | 59 | 2 | 57 |
| 5 | 170 | 10 | 10 | 66 | 15 | 51 |
| 6 | 190 | 10 | 10 | 79 | 47 | 32 |
| 7 | 210 | 10 | 10 | 90 | 79 | 11 |
| 8 | 230 | 10 | 10 | 99 | 98 | 1 |
| 9 | 230 | 20 | 10 | 100 | 100 | 0 |
| 10 | 230 | 20 | 0 | 85 | 6 | 79 |
| 11 | 230 | 20 | 5 | 100 | 100 | 0 |
| 12 | 230 | 20 | 2.5 | 100 | 100 | 0 |
| 13 | 230 | 20 | 2 | 100 | 100 | 0 |
| 14 | 230 | 20 | 1 | 96 | 84 | 12 |
At reaction temperatures at or above 150 °C, formation of N-methylindole (2) in the reaction mixture was observed, and both the overall conversion of indole and selectivity to N-methylindole were significantly improved by increasing the temperature (entries 5–8, Table 1) with 99% conversion of indole and 98% selectivity to N-methylindole (2) observed after 10 min. at 230 °C (∼17 bar). When the reaction time was extended to 20 min, N-methylindole (2) was obtained exclusively (Table 1, entry 9). The results, for 10 min reaction times, are plotted in Fig. 2 and show both the partial optimal production of the N-(methoxycarbonyl)indole at 130 °C, and onset of N-methylindole formation at 150 °C. This is anticipated, as the result of methylcarboxylation via the BAC2 mechanism, whereas at higher temperatures methylation through the BAL2 mechanism dominates.1
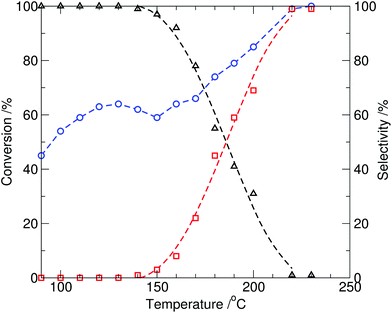 | ||
| Fig. 2 Conversion of indole (1, circle) and relative selectivity to the two observed products, N-methylindole (2, square) and N-(methoxycarbonyl)indole (3, triangle), from the reaction of indole with DMC catalysed by tributylmethylammonium methylcarbonate (10 mol%) as a function of temperature under batch microwave heating (10 min, reaction time). | ||
The effect of the catalyst loading was then investigated. Taking the optimal conditions from the screen at 10 mol% catalyst (230 °C, 20 min reaction time), the amount of ionic liquid added was reduced (Table 1, entries 11–14). It was found that the amount of catalyst 4 required could be reduced to 2 mol%, while retaining complete conversion and high selectivity for N-methylation, within a 20 min reaction time (shown graphically in Fig. 3). By comparison, in the control experiment, reaction of indole with dimethylcarbonate in the absence of 4 (Table 1, entry 10) resulted in only 85% conversion of indole and a reversed product distribution, now strongly favouring the formation of N-(methoxycarbonyl)indole (3). This highlights the catalytic effect of the ionic liquid 4 on the methylation process.
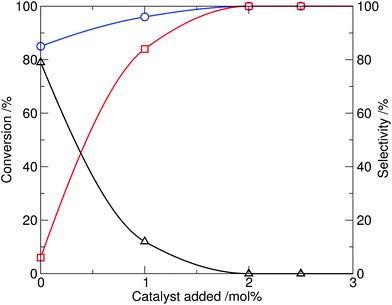 | ||
| Fig. 3 Effect of tributylmethylammonium methylcarbonate catalyst loading on the methylation of indole with DMC, showing conversion of indole (1, circle) and selectivity to N-methylindole (2, square) and N-(methoxycarbonyl)indole (3, triangle) at 230 °C, 20 min reaction time. | ||
Mechanistic investigations have shown that methylation with DMC in the presence of tertiary organic bases (DABCO, DBU, etc.) proceeds through an intermediate formed in situ by reaction of the amine base with DMC, which may be considered to be the actual catalyst. Since tributylmethylammonium methylcarbonate (4) is in fact synthesised by treatment of tributylamine with DMC in MeOH at 160 °C for 10 h,25 it was envisaged that, in the high temperature régime, the methylation reaction might be also catalysed using tributylamine, which would form the ionic liquid 4in situ under the reaction conditions. This would contrast to the inactivity of tributylamine at 90 °C, as reported by Shieh et al.6 Indeed, adding 2 mol% of tributylamine to a 0.9 M solution of indole in DMC–DMF (10:1, v/v) and exposure to 230 °C/20 min microwave heating resulted in full conversion of indole and 100% selectivity to N-methylindole (2) without the need to independently prepare the ionic liquid catalyst 4. Similarly to using the preformed ionic liquid as catalyst, the use of lower amounts of tributylamine resulted in reduced conversion and selectivity (cf. Table 1, entry 14).
Having identified suitable reaction conditions for the selective N-methylation of indole with DMC under batch microwave conditions, the process conditions were then evaluated for transfer to a scalable continuous-flow set-up. For this purpose a high-temperature and high/pressure continuous-flow reactor was employed (X-Cube Flash, ThalesNano Inc.).27 The reactor uses stainless steel coils (i.d. 1 mm) of variable length (4, 8, and 16 cm3 internal volume) that can be directly heated across their full length by electrical resistance heating to temperatures up to 350 °C. The reaction mixture was introduced to the reactor block containing the steel coils and heat exchanger via one or more standard HPLC pumps (flow rate 0.01–10.0 cm3 min−1). The system pressure valve stabilises the set pressure value in the range 50–180 bar. In a typical set-up for indole methylation, the instrument was equipped with a 16 cm3 stainless steel coil, allowing residence times (reaction times) of up to 32 min at flow rates of 0.5 cm3 min−1. Employing the flow reactor system, the batch microwave conditions shown in Table 1, entry 13 (230 °C, 20 min) and a flow rate to 1.2 cm3 min−1 (∼20 min residence time) were used to achieve 100% conversion of indole (1) into N-methylindole (2).
To further intensify the reaction, an even higher temperature/pressure regime (285 °C, 150 bar), potentially reaching the near – or supercritical state for DMC (Tc 275 °C, Pc 45 bar),32 was explored. Using a 4 cm3 stainless steel coil and a flow rate of 1.3 cm3 min−1 (residence time of ∼3 min), the reaction mixture was processed through the continuous-flow instrument at 285 °C and 150 bar pressure. HPLC-analysis of the collected reaction mixture revealed 100% conversion, and subsequent isolation by chromatography gave N-methylindole (2) in 98% yield.33
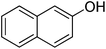
The optimised flow methylation conditions were subsequently evaluated for other substrates (Table 2). Thus, continuous flow methylation experiments with a variety of –NH, –OH, –SH and –CO2H functionalised substrates were carried out without any further optimisation of reaction conditions. Structurally diverse indoles, phenols, thiophenols and carboxylic acids all reacted smoothly at 285 °C and 150 bar pressure to provide the desired methylated products in good to high isolated yields and with excellent selectivity. The degree of conversion was easily determined by monitoring the composition of the reaction mixture by HPLC-UV and/or GC-MS analysis. For all of the examples shown in Table 2, full conversion of the starting material was achieved. Using the conditions described above, typically 250–370 mg of product could be produced within a ∼3 min run, leading to a calculated productivity of ∼100 g per day. This versatility and high selectivity has significant implications for the synthesis of a wide range of pharmaceutical intermediates.
| Entry | Substrate | Product | Yield/%b |
|---|---|---|---|
|
a Reaction conditions: X-Cube Flash, 4 cm3 stainless steel coil, 150 bar, 285 °C, 2.2 cm3 sample of 0.9 M starting material solution in DMC–DMF (10:1), 2 mol% tributylamine, 1.3 cm3 min−1 flow rate.
b Isolated yields obtained after flash chromatography.
|
|||
| 1 |
|
|
98 |
| 2 |
|
|
93 |
| 3 |
|

|
88 |
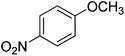
| 4 |
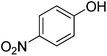
|
|
90 |
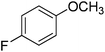
| 5 |
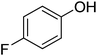
|
|
94 |
| 6 |
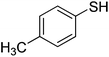
|
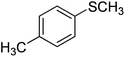
|
87 |
| 7 |

|
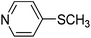
|
52 |
| 8 |
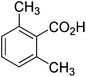
|

|
93 |
| 9 |
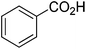
|
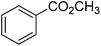
|
86 |
| 10 |
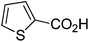
|
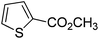
|
81 |
Conclusions
The ionic liquid, tributylmethylammonium methylsulfate, can be used as a base, in catalytic quantities, to promote the archetypal methylation of indole with DMC. The reaction conditions have been optimised, under batch microwave heating, to achieve 100% selectivity and 100% conversion to N-methylindole at 230 °C in 20 min using only 2 mol% loading of the catalyst. These batch optimised conditions were successfully translated into a high-temperature/high-pressure continuous flow protocol for the methylation of a variety of different substrates – amines, phenols, thiophenols and carboxylic acids – using a high temperature/high pressure process window (285 °C/150 bar) and short (3 min) reaction times. This enables high throughput through a scalable continuous flow process, combined with high conversion and selectivities.Importantly, in the high-temperature/pressure reaction window the ionic liquid catalyst can be generated in situ, which has the net effect of transforming an ineffective stoichiometric base (tributylamine) into a highly efficient catalyst for these reactions.
Acknowledgements
This work was supported by the Christian Doppler Research Society and QUILL.Notes and references
- Selected reviews: Y. Ono, Catal. Today, 1997, 35, 15 CrossRef CAS; P. Tundo, Pure Appl. Chem., 2001, 73, 1117 CrossRef; S. Memoli, M. Selva and P. Tundo, Chemosphere, 2001, 43, 115 CrossRef; D. Delledonne, F. Rivetti and U. Romano, Appl. Catal., A, 2001, 221, 241 CrossRef; P. Tundo and M. Selva, Acc. Chem. Res., 2002, 35, 706 CrossRef; S. V. Chankeshwara, Synlett, 2008, 624 CrossRef; M. Selva and A. Perosa, Green Chem., 2008, 10, 457 RSC; F. Arico and P. Tundo, Russ. Chem. Rev., 2010, 79, 479 CrossRef.
- Selected examples: M. Lissel, S. Schmidt and B. Neuman, Synthesis, 1986, 382 CrossRef CAS; M. Lissel, Liebigs Ann. Chem., 1987, 77 CrossRef; Y. Lee and I. Shimizu, Synlett, 1998, 1063 CrossRef; P. Tundo, L. Rossi and A. Loris, J. Org. Chem., 2005, 70, 2219 CrossRef.
- J. H. Werntz, US Pat., 2635100, 1953 Search PubMed; U. Romano, F. Rivetti and N. Di Muzio, US Pat., 4318862, 1981, CA 80141, 1979 Search PubMed; S. Mori, K. Ida and M. Ue, Eur. Pat. Appl., 88107735.8, 1988 Search PubMed; B. Albert and M. Jansen, Z. Anorg. Allg. Chem., 1995, 621, 173 Search PubMed; G. W. Earl, D. E. Weisshaar, D. Paulson, M. Hanson, J. Uilk, D. Wineinger and S. Moeckly, J. Surfactants Deterg., 2005, 8, 325 CrossRef CAS.
- W.-C. Shieh, S. Dell and O. Repic, J. Org. Chem., 2002, 67, 2188 CrossRef CAS; W.-C. Shieh, M. Lozanov, M. Loo, O. Repic and T. J. Blacklock, Tetrahedron Lett., 2003, 44, 4563 CrossRef; S. Ouk, S. Thiebaud and E. Borredon, Synth. Commun., 2005, 35, 3012 CrossRef; R. Juarez, A. Padilla, A. Corma and H. Garcia, Ind. Eng. Chem. Res., 2008, 47, 8043 CrossRef; M. L. Laurilla, N. A. Magnus and M. A. Staszak, Org. Process Res. Dev., 2009, 13, 1199 CrossRef; E. Quaranta, M. Carafa and F. Trani, Appl. Catal., B, 2009, 91, 380 CrossRef; A. Dhakshinamoorthy, A. Sharmila and K. Pitchumani, Chem.–Eur. J., 2010, 16, 1128 CrossRef.
- For example: S. Ouk, S. Thiebaud, E. Borredon, P. Legars and L. Lecomte, Tetrahedron Lett., 2002, 43, 2661 CrossRef CAS; M. Selva, P. Tundo and A. Perosa, J. Org. Chem., 2003, 68, 7374 CrossRef; N. Nagaraju and G. Kuriakose, New J. Chem., 2003, 27, 765 RSC; M. Selva, A. Perosa and M. Fabris, Green Chem., 2008, 10, 1068 RSC.
- W.-C. Shieh, S. Dell, A. Bach, O. Repic and T. J. Blacklock, J. Org. Chem., 2003, 68, 1954 CrossRef CAS.
- W.-C. Shieh, S. Dell and O. Repic, Tetrahedron Lett., 2002, 43, 5607 CrossRef CAS; F. Rajabi and M. R. Saidi, Synth. Commun., 2004, 34, 4179 CrossRef; C. Hou, Y. Chen, W. Chen and W. Li, Carbohydr. Res., 2011, 346, 1178 CrossRef; C. Hou, Y. Chen, W. Chen and W. Li, Carbohydr. Res., 2012, 355, 87 CrossRef.
- T. N. Glasnov and C. O. Kappe, Chem.–Eur. J., 2011, 17, 11956 CrossRef CAS.
- P. Tundo, F. Trotta, G. Moraglio and F. Ligorati, Ind. Eng. Chem. Res., 1988, 27, 1565 CrossRef CAS; A. Bomben, M. Selva, P. Tundo and L. Valli, Ind. Eng. Chem. Res., 1999, 38, 2075 CrossRef; P. Tundo, A. E. Rosamilia and F. Arico, J. Chem. Educ., 2010, 87, 1233 CrossRef.
- U. Tilstam, Org. Process Res. Dev., 2012, 16, 1150 CrossRef CAS.
- W.-S. Shieh, M. Lozanov and O. Repic, Tetrahedron Lett., 2003, 44, 694 Search PubMed.
- P. N. Gooden, R. A. Bourne, A. J. Parrott, H. S. Bevinakatti, D. J. Irvine and M. Poliakoff, Org. Process Res. Dev., 2010, 14, 411 CrossRef CAS; A. J. Parrott, R. A. Bourne, P. N. Gooden, H. S. Bevinakatti, M. Poliakoff and D. J. Irvine, Org. Process Res. Dev., 2010, 14, 1420 CrossRef; R. A. Bourne, R. A. Skilton, A. J. Parrott, D. J. Irvine and M. Poliakoff, Org. Process Res. Dev., 2011, 15, 932 CrossRef; D. J. Jumbam, R. A. Skilton, A. J. Parrott, R. A. Bourne and M. Poliakoff, J. Flow Chem., 2012, 24 CrossRef.
- For example, see: Ionic Liquids in Synthesis, ed. P. Wasserscheid and T. Welton, Wiley-VCH, Weinheim, 2nd edn, 2008 Search PubMed.
- C. O. Kappe, Angew. Chem., Int. Ed., 2004, 43, 6250 CrossRef CAS.
- D. Obermayer and C. O. Kappe, Org. Biomol. Chem., 2010, 8, 114 CAS.
- N. M. Kashid, A. Renken and L. Kiwi-Minsker, Chem. Eng. Sci., 2011, 66, 1480 CrossRef.
- È. Boros, K. R. Seddon and C. R. Strauss, Chim. Oggi – Chem. Today, 2008, 26 (Nov–Dec), 28 Search PubMed.
- See for example, microwave dissolution of cellulose in ionic liquids; R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974 CrossRef CAS.
- L. P. N. Rebelo, J. N. Canongia Lopes, J. M. S. S. Esperança and E. Filipe, J. Phys. Chem. B, 2005, 109, 6040 CrossRef CAS.
- C. Strauss and D. W. Rooney, Green Chem., 2010, 12, 1340 RSC.
- A. Loris, A. Perosa, M. Selva and P. Tundo, J. Org. Chem., 2004, 69, 3953 CrossRef CAS; Z. L. Shen, X. Z. Jiang, W. M. Mo, B. X. Hu and N. Sun, Green Chem., 2005, 7, 97 RSC; J.-Q. Nie, H.-W. Chen, Q.-H. Song, B. Liao and Q.-X. Guo, Energy Fuels, 2010, 24, 5722 CrossRef; J.-G. Xie, J. Quan, S.-B. Li, Y. Zheng and L.-M. Zhu, Synth. Commun., 2011, 41, 871 CrossRef; J. Xie, C. Wu, B. W. Christopher, J. Quan and L. Zhu, Phosphorus, Sulfur Silicon Relat. Elem., 2011, 186, 31 CrossRef.
- S. Ouk, S. Thiebaud, E. Borredon and P. Le Gars, Appl. Catal., A, 2003, 241, 227 CrossRef CAS; A. B. Shivarkar, S. P. Gupte and R. V. Chaudhari, J. Mol. Catal. A: Chem., 2005, 226, 49 CrossRef.
- M. Fabris, V. Lucchini, M. Noè, A. Perosa and M. Selva, Chem.–Eur. J., 2009, 15, 12273 CrossRef CAS; M. Fabris, M. Noè, A. Perosa, M. Selva and R. Ballini, J. Org. Chem., 2012, 77, 1805 CrossRef.
- D. R. MacFarlane, J. M. Pringle, K. M. Johansson, S. A. Forsyth and M. Forsyth, Chem. Commun., 2006, 1905 RSC.
- J. D. Holbrey, R. D. Rogers, S. S. Shukla and C. D. Wilfred, Green Chem., 2010, 12, 407 RSC.
- B. Gutmann, D. Obermayer, B. Reichart, B. Prekodravac, M. Irfan, J. M. Kremsner and C. O. Kappe, Chem.–Eur. J., 2010, 16, 12182 CrossRef CAS.
- For a detailed description of this reactor see: T. Razzaq, T. N. Glasnov and C. O. Kappe, Chem. Eng. Technol., 2009, 32, 1702 CrossRef CAS.
- S. J. Park, J. R. Price and M. H. Todd, J. Org. Chem., 2012, 77, 949 CrossRef CAS.
- L. J. Goossen, C. Linder, N. Rodriguez, P. P. Lange and A. Fromm, Chem. Commun., 2009, 46, 7173 RSC.
- X. Jiang, A. Tiwari, M. Thompson, Z. Chen, T. P. Cleary and T. B. K. Lee, Org. Process Res. Dev., 2001, 5, 604 CrossRef CAS; S.-Y. Zhao, H.-Q. Zhang, D.-Q. Zhang and Z.-Y. Shao, Synth. Commun., 2012, 128 CrossRef.
- W.-C. Shieh, S. Dell and O. Repic, Org. Lett., 2001, 3, 4279 CrossRef CAS.
- S. Camy, J.-S. Pic, E. Badens and J.-S. Condoret, J. Supercrit. Fluids, 2003, 25, 19 CrossRef CAS; P. Pinero, J. Garcia, M. Sokolova and M. J. Cocero, J. Chem. Thermodyn., 2007, 39, 536 CrossRef.
- Although some decomposition of DMC has been observed at the high-temperature conditions (see D. Breuch and H. Löwe, Green Process Synth., 2012, 1, 261), in our hands, possibly due to the very short exposure time (∼3 min), this has not been a major issue.
| This journal is © The Royal Society of Chemistry 2012 |