
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCovalent design of ionogels: bridging with hydrogels and covalent adaptable networks
Junjia
Zhang
 *a,
Yian
Wang
a,
Yinglu
Liu
ab,
Jérémy
Odent
c and
Yukikazu
Takeoka
*a
*a,
Yian
Wang
a,
Yinglu
Liu
ab,
Jérémy
Odent
c and
Yukikazu
Takeoka
*a
aDepartment of Molecular & Macromolecular Chemistry, Nagoya University, Nagoya 464-8603, Japan. E-mail: ytakeoka1@mac.com; syunnka123456@outlook.com
bState Key Laboratory of Organic–Inorganic Composites, Beijing University of Chemical Technology, Beijing 100029, China
cLaboratory of Polymeric and Composite Materials, Center of Innovation and Research in Materials and Polymers, University of Mons, Place du Parc 20, 7000 Mons, Belgium
First published on 16th April 2025
Abstract
Ionogels are conductive soft matter with ionic liquids as conductive media, exhibiting significant potential as multifunctional materials. Over the past two decades, ionogels have been developed for applications in sensors, actuators, supercapacitors, lithium-ion batteries, adhesives, antifouling coatings, nanotriboelectric generators, thermoelectric devices, etc. To achieve recyclability that is advantageous for various applications, dissociative supramolecular interactions—e.g. electrostatic interactions, hydrogen bonds and π–π stacking—have garnered significant attention in the crosslinking design of ionogels. High-strength ionogels utilizing dissociative supramolecular interactions as a crosslinking mechanism have been synthesized. However, due to the inherently low bond energy and high dynamics of dissociative supramolecular crosslinking, issues such as low thermal stability and insufficient solvent resistance arise, limiting the broader applications of ionogels. To address these challenges, the network structure can be precisely designed, and reversible covalent bonds can be introduced as a crosslinking mechanism to mitigate the trade-off between material durability and dynamic behavior. Several studies provide insights into realizing this approach. For instance, hydrogels, which are also classified as soft materials, can enhance both mechanical strength and deformability by incorporating topological network structures based on organic covalent bonds. Similarly, covalent adaptable networks (CANs), a class of dynamic materials, achieve high thermal stability, solvent resistance, and recyclability by utilizing densely reversible covalent bonds. Hence, we chiefly focus on the critical roles of designing the organic polymer network structures and utilizing reversible covalent bonding to enhance key physical properties of ionogels, including mechanical strength, electrical conductivity, and processability. Last but not least, we discuss the current challenges associated with the design and application of ionogels, while also anticipating potential strategies that leverage superior designs from materials such as hydrogels and CANs to develop innovative ionogels.
 Junjia Zhang | Junjia Zhang received his bachelor's degree in macromolecular materials and engineering from Guangdong University of Technology in 2023. Currently he is a postgraduate student at Nagoya University. His research interest focuses on smart materials e.g. high mechanical strength gels/elastomers, ionic conductive gels/elastomers and 3D printable materials for sensors. |
 Yian Wang | Yian Wang received his bachelor's degree in textile engineering from Soochow University and master's degree in Molecular & Macromolecular Chemistry from Nagoya University. Currently he is a doctoral student at Nagoya University. His research interest focuses on composite hydrogel and synthesis of golden nanoparticles. |
 Yinglu Liu | Yinglu Liu received her Bachelor's degree in Chemical Engineering and Technology from Beijing University of Chemical Technology (BUCT). She is currently a Ph.D. candidate in Chemical Engineering and Technology at BUCT. Her research focuses on optically functional nanodispersions and composite materials for next-generation display technologies. |
 Jérémy Odent | Jérémy Odent received his Ph.D. from the University of Mons (Belgium) in 2014 working on high-impact polylactide-based nanocomposites. He began working as a postdoc at Cornell University (United States) designing novel ionic nanocomposites in 2015, and moved to his current position as Associate Professor at the University of Mons in 2017. Since then, he has developed new capabilities at the Laboratory of Polymeric Materials and Composites in the area of adaptive polymeric materials and additive manufacturing technologies. Research focuses are centered around the development of stimuli-responsive polymeric materials and nanocomposites of desired properties and key functionalities as well as the possibilities sustained by advanced additive manufacturing technologies to meet the ever-increasing demand for complex device platforms. As a means of generating parts with novel functionality, the core technology of this research mainly relies on the generation of smooth structural gradients within the 3D-printed materials. |
 Yukikazu Takeoka | Yukikazu Takeoka received his Ph.D. in the field of electrochemistry from Sophia University (1996). Then he joined Prof. Toyoichi Tanaka's research group at the Massachusetts Institute of Technology with the Uehara Memorial Foundation for Postdoctoral Fellowships and worked on the phase transition of polymer gels and the synthesis of molecular recognition gels. In mid-1998, he moved to Yokohama National University, where he was a research associate. Since 2004, he has been an Associate Professor at Nagoya University. His research focuses on smart materials such as structural colored gels, high mechanical strength gels, densely grafted polymer brushes and photonic band gap materials for optical and biological applications. |
1. Introduction
Ionogels are materials that consist of ionic liquids (ILs) integrated within a supporting network. The network of ionogels can consist of organic polymers,1 organic small molecules,2 inorganic continuous phases,3 inorganic nanomaterials,4 and organic/inorganic composite materials,5 among others. They are distinct from hydrogels, organogels, and aerogels due to their unique solvent characteristics. It is noteworthy that the term “ion gel” has been used in other papers since as early as 2005.6 However, due to its structural similarity to terms such as “hydrogel”, “organogel”, and “aerogel”, this paper adopts the term “ionogel”. Room temperature ILs are ionic compounds that exist in a liquid state at room temperature and consist solely of anions and cations. In contrast to traditional ionic compounds such as salts, ILs remain in liquid state at room temperature, while exhibiting high ionic conductivity and extremely low volatility. Furthermore, ILs possess broad-spectrum bactericidal properties attributable to the positive charges present in their structures. Owing to the approximately 1018 combinations of anions and cations available, ILs exhibit a wide range of adjustable properties, including hydrophilicity, acidity, electrochemical characteristics and flammability.7 Benefiting from the numerous advantages of ILs, ionogels have been developed for applications in sensors,8 actuators,9 supercapacitors,10 lithium-ion batteries,11 adhesives,12 antibacterial materials,13 nanotriboelectric generators,14 thermoelectric devices15etc.The first ionogel was synthesized by Watanabe et al. in 1993, which was formed exclusively through crosslinking via supramolecular interactions.1 Although the term “ionogel” is not used in the paper, judging from its physical properties, it is clearly an ionogel. The interaction between 1-butylpyridinium halide and the ionic groups on the sidechain of poly(1-butyl-4-vinylpyridinium halide) lowers the glass transition temperature (Tg) of the resulting materials. This reduction allows the system to partially decouple the relationship between the number of charge carriers and the mobility of the polymer chain segments, enabling both to increase simultaneously and thereby achieving high conductivity. The first work on an ionogel crosslinked through covalent bonds was published by Noda et al. in 2000.16 This ionogel was synthesized via in situ polymerization, which involved a one-step process of heating a mixture that comprised the monomer 2-hydroxyethyl methacrylate (HEMA), the crosslinker ethylene glycol dimethacrylate (EGDMA), the solvents 1-ethyl-3-methylimidazolium tetrafluoroborate {[EMIm]+[BF4]−} or 1-butylpyridinium tetrafluoroborate {[BP]+[BF4]−}, and the initiator benzoyl peroxide. This method, in contrast to the previous approach of mixing a polymer, organic solvent, and ionic liquid followed by the evaporation of the organic solvent to form an ionogel, reduces the volatilization of organic compounds and yields a precursor with lower viscosity, thereby significantly enhancing processability.17 Subsequently, in 2001 Kimizuka et al. introduced glycolipid molecules into glycophilic lipid ILs {3-(2-methoxyethyl)-1-methyl-1H-imidazol-3-ium bromide and [BMIM]+[PF6]−} to create a double-layer membrane structure via supramolecular self-assembly for gel preparation. They were the first to formally introduce the term ‘ionogels’ to describe gels containing ILs as solvent.2 This designation follows the terminology of materials science, such as aerogel, hydrogel, and organogel. Another organic polymeric ionogel, chemically crosslinked by covalent bonds, was developed by Tiyapiboonchaiya et al. in 2002.18 This ionogel utilized methyl methacrylate (MMA) as the monomer and tetraethylene glycol diacrylate (TEGDA) as the crosslinking agent, with an ionic liquid composed of [EMIm]+ and bis(trifluoromethylsulfonyl)imide ([TFSI]−) prepared as solvents. The incorporation of chemical crosslinking agents enables the use of low Tg polymers, which significantly enhances the ionic conductivity of ionogel electrolytes. Since then, ionogels built from small-molecule gelling reagents, linear organic polymers, or permanently crosslinked networks of conventional organic polymers have been extensively developed.
Although the covalent network structure of ionogels has gradually attracted attention, hydrogel networks have been extensively developed during the last two decades. In contrast to the simpler covalent network designs of most ionogels, various advanced covalent design strategies for polymer networks have emerged, including interpenetrating polymer networks,19 semi-interpenetrating polymer networks,20 homogeneous polymer networks,21 slide-ring polymer networks,22 hyperbranched polymer networks,23 highly entangled polymer networks,24 bottlebrush polymer networks,25 force-induced growth polymer networks26,27 and polymer networks with hidden length28 (see section 2). These strategies enable a range of extreme mechanical properties (e.g. high mechanical strength) while maintaining versatility. To achieve recyclability that is advantageous for various applications, dissociative supramolecular interactions—e.g. electrostatic interactions, hydrogen bonds—have garnered significant attention in the crosslinking design of ionogels. Although high-strength ionogels have been synthesized utilizing dissociative supramolecular interactions as the primary crosslinking mechanism, in contrast to interlocking supramolecular interactions, e.g. rotaxanes and catenanes—namely, two molecules are interconnected through a form of mechanical interlocking. Since the covalent bonds remain intact, these two molecules cannot be separated. Consequently, they are typically considered as a single molecule—, dissociative supramolecular interactions separate into two distinct parts when subjected to heat, leading to the breaking of the reversible crosslinking between polymer chains. Hence, issues such as poor thermal stability and insufficient solvent resistance—stemming from the inherent low bond energy and high dynamics of dissociative supramolecular crosslinking—restrict the broader application of ionogels. (Unless otherwise specified, all supramolecular interactions discussed here refer specifically to dissociative supramolecular interactions.) To address this, the trade-off between material durability and dynamics can be mitigated to some extent by judiciously designing the network structure and employing reversible covalent bonds as the crosslinking mechanism. Covalent adaptable networks (CANs), which are also dynamic materials, attain high thermal stability, solvent resistance, and recyclability by utilizing dense reversible covalent bonds. CANs based on dynamic covalent bonds have demonstrated thermal stability without compromising recyclability. This includes processes e.g. the Diels–Alder reaction,29 transimination reaction,30 dynamic exchange of disulfide bonds,31 boronic transesterification reaction,32 dynamic exchange of acylhydrazone bonds,33 transesterification reaction,34 transcarbamoylation reaction,35 transcarbamoylation reaction of oxime-urethanes,36 transamination reaction,37,38 transalkylation reaction,39,40 dynamic exchange of silyl ethers,41 and dynamic exchange of acetals42–45 (see section 3). Fortunately, ionogels exhibit excellent compatibility with various design concepts of hydrogel network structures and possess the potential for direct transplantation. It is noteworthy that the universality of network design in hydrogels and ionogels must consider the solubility of the corresponding components in ILs. Only when a homogeneous phase is formed do the ionogels with the corresponding network design exhibit properties similar to those of hydrogels. Furthermore, in addition to serving as a plasticizer that enhances chain segment mobility, the IL itself may function as a catalyst, potentially eliminating the need for additional catalysts in CANs to activate reversible covalent bonds.46 Therefore, it is reasonable to hypothesize that ionogels, when crafted with meticulously designed covalent structures, can not only combine the benefits of hydrogels and CANs but may also demonstrate unprecedented properties (Fig. 1).
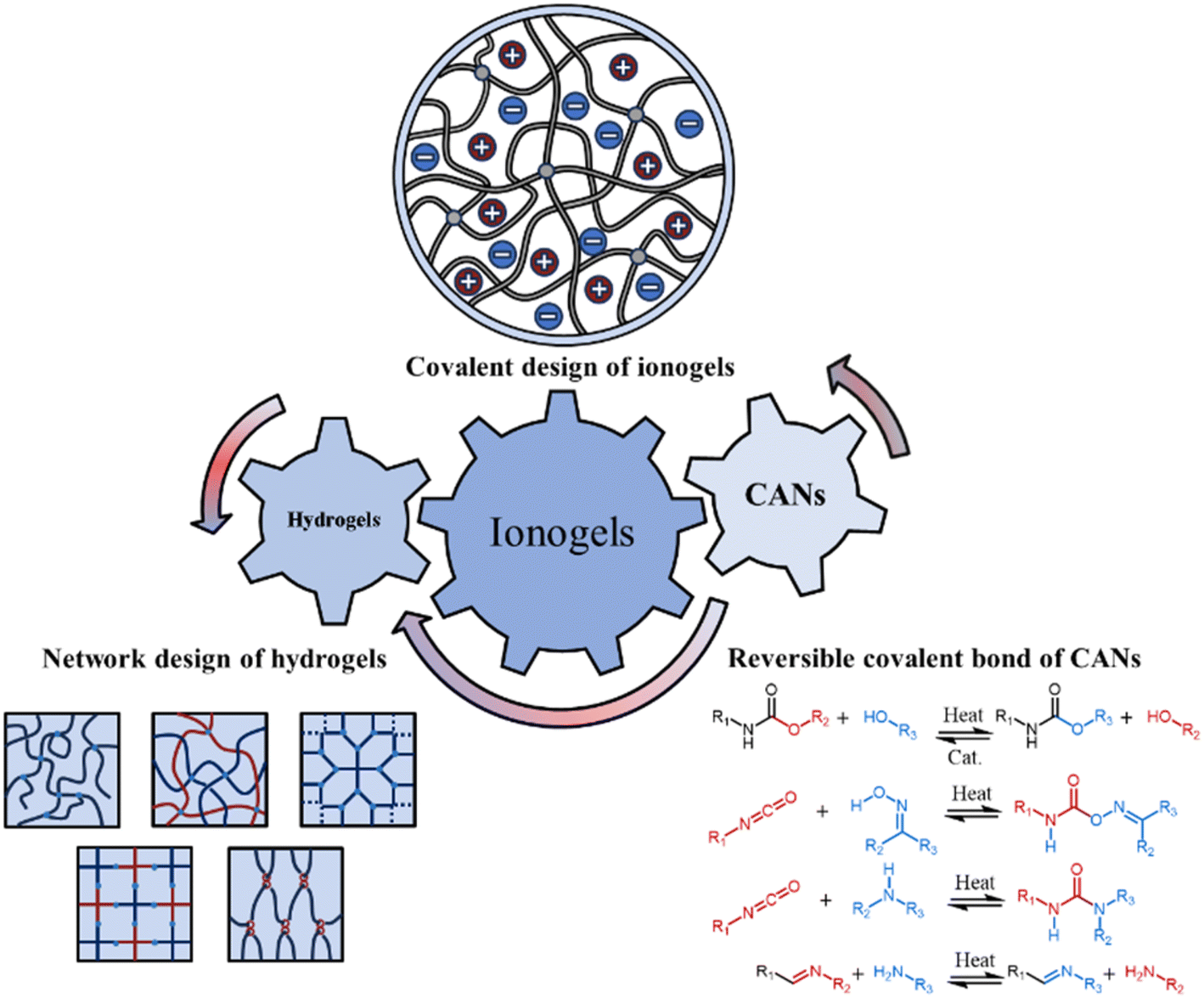 | ||
| Fig. 1 Concept illustration of ionogels with novel properties drawing inspiration from the outstanding covalent designs of hydrogels and covalent adaptable networks (CANs). | ||
It should be noted that both supramolecular crosslinking and covalent crosslinking are crucial mechanisms for the formation of ionogels. The issue of ambiguous interactions with the polymer network, stemming from the diverse range of ILs, has been highlighted in previous reviews.47 Additionally, a specialized review has been published to elucidate the significance of supramolecular interactions involving ILs in modulating the properties of ionogels.48 However, a review detailing the covalent design of ionogels remains absent. Hence, in this illustrative review, we focus on the design of ionogels characterized by organic polymer covalent network structures and reversible covalent bonds, which have potential to provide excellent mechanical properties, electrical properties, and processability. We will draw parallels with hydrogels and CANs to enhance our discussion. Another impetus for comparing ionogels with hydrogels and CANs is the emphasis on multi-scale design principles. Proposed strategies, including in situ phase separation49 and block copolymer-induced phase separation,50 have resulted in abundant ordered sequences at the nanometer scale due to supramolecular interactions. However, material design at the scale of polymer networks (micrometers) and reversible covalent bonds (angstroms) has not yet received adequate attention in the synthesis of ionogels. Furthermore, we will address the challenges and opportunities associated with the manufacturing of new ionogels, inspired by the covalent design concepts of other materials, and propose potential strategies for the future design of innovative ionogels.
2. Unconventional covalent network structures for improving properties of ionogels
Ionogel networks based on organic polymers have emerged as the most extensively studied system, owing to their advantages of strong designability and mild manufacturing conditions. In the organic polymer framework, there are many parameters that can be changed, e.g. crosslinking mechanism, crosslinking density, monomer type, polymerization method etc. Compared with traditional inorganic ionogels, an organic ionogel synthesized through photopolymerization can be produced within minutes and does not necessitate hazardous conditions such as high temperature and high pressure. Conventional covalently crosslinked networks are defined as non-uniform structured network structures composed of linear organic polymers crosslinked exclusively by irreversible covalent bonds. This type of network is generally considered to contain drawbacks like crosslinking points with varying numbers of chains connected to them, polymer chains with different lengths between crosslinking points, dangling chains with one end unconstrained within the network, looped chains with both ends connected at the same point in the network, and entangled structures of polymer chains. Unconventional network structures can be achieved by altering one or more design features typical of conventional networks (Fig. 2). For example, if an additional network or uncrosslinked polymer is prepared within an already formed network structure, an interpenetrating polymer network (IPN) or semi-interpenetrating polymer network (SIPN) can be created (see section 2.1). By transforming linear polymers into bottle-brush or hyperbranched structures, network structures with minimal entanglement between polymers can be constructed (see sections 2.2 and 2.5). If the chain length and number between crosslinking points can be made nearly uniform, a homogeneous polymer network structure can be obtained (see section 2.3). By incorporating supramolecular components such as supramolecular assemblies where cyclic molecules can move along axle molecules into the polymer network, it becomes possible to create a slide-ring network (see section 2.4). In this network, the crosslinking points can move in response to deformation of the polymer network under external forces, thus preventing breakage of the crosslinks. The cyclic molecule cannot detach from the axis until the covalent bond is broken; that is, the energy required to break this type of sliding crosslink is at least equal to or greater than the energy needed to break the corresponding covalent bond. Consequently, although dissociative supramolecular interactions are employed during the synthesis of polyrotaxanes, their use as crosslinking agents can still be considered a covalent design. If the number of entanglements between polymer chains significantly exceeds the number of crosslinking points, a highly entangled polymer network can be formed. The following sections will discuss representative works in which unconventional network designs enhance the physical properties of ionogels, drawing analogies with hydrogels. | ||
| Fig. 2 Schematics of polymer networks, including (a) conventional polymer networks, unconventional polymer networks e.g. (b) interpenetration polymer networks, (c) semi-interpenetration polymer networks, (d) hyperbranched polymer networks, (e) homogeneous polymer networks, (f) slide-ring polymer networks, (g) high entanglement polymer networks, and (h) bottlebrush polymer networks. | ||
2.1. Interpenetrating and semi-interpenetrating polymer networks
An interpenetrating polymer network (IPN) refers to polymer networks composed of different types of polymers that interpenetrate each other. If one of the polymers is not crosslinked, it is called a semi-interpenetrating polymer network (SIPN). By combining polymers or polymer networks with different properties, IPNs and SIPNs can exhibit characteristics that are not present in either individual component, enabling functions unattainable with conventional polymer networks.51 For example, an IPN using a polymer network made of polyacrylamide and another made of polyacrylic acid combines a hydrogen-donating polymer with a polymer having hydrogen-accepting side chains.52 This combination causes the polymer network to contract due to hydrogen bonding even at low temperatures in aqueous environments, namely, above a certain temperature, the hydrogen bonds dissociate, leading to swelling. This volume change is reversible with temperature variations, leading to the potential of temperature-responsive drug carriers capable of releasing an antipyretic agent during fever. An IPN combining a mechanically brittle polymer network with a flexible polymer network is known to form a highly tough polymer gel.53 The brittle polymer network fractures when an external force is applied to the IPN. This behavior dissipates energy, and subsequently, the intact network in the IPN holds the fractured network together, preventing the polymer gel from failing entirely. This mechanism is believed to arise from the presence of sacrificial bonds. The first high-strength IPN hydrogel was developed by Gong et al., utilizing a brittle polymer network made of poly(2-acrylamido-2-methylpropanesulfonic acid) (PAMPS) with high crosslinking density and low polymer concentration as the first network, and a flexible polymer network made of polyacrylamide (PAAm) with low crosslinking density and high concentration as the second network.19 The Young's modulus and fracture strength of this gel are primarily influenced by the first network with a higher crosslink density, while the fracture toughness and fatigue threshold are determined by the second network with a lower crosslinking density. This finding challenges the Lake–Thomas theory, which posits that the modulus and toughness of a gel cannot be simultaneously improved. In this IPN hydrogel system, it is possible to achieve simultaneously high Young's modulus, high fracture strength, and high toughness, owing to the synergistic effects of the network. Thus, it facilitates the decoupling of the mechanical properties of the gel material.The first SIPN ionogel was developed by Zhou et al. through combining methyl methacrylate (MMA), tetraethylene glycol diacrylate (TEGDA), [EMIm]+[TFSI]−/[BMIm]+[PF6]−, and dibenzoyl peroxide (BPO) as components of the second network.54 This mixture was then infiltrated into linear polypyrrole (PPy) that had been deposited on platinum-coated polyvinylidene difluoride (PVDF) sheets for application in actuators. Polypyrrole is a conjugated polymer known for its electrical conductivity. To attain optimal electrical fatigue properties, the surrounding electrolyte must effectively dope and dedope polypyrrole, necessitating that the ionic counterpart possesses a high degree of environmental stability. Consequently, ionic liquids with such stability are considered a suitable choice. The SIPN formed through in situ polymerization of monomers within the pores of PPy and platinum metal not only enhanced interface stability between polymer and platinum metal but also preserved the high conductivity of 3.2 × 10−4 S cm−1 provided by the IL and polypyrrole. Additionally, the SIPN actuator exhibited a rapid response time, capable of bending 180° in just 30 seconds, and boasted a cycle life exceeding 3600 cycles, outperforming traditional carbonate electrolytes which can bend 180° in the same duration but only achieve a cycle life of 2400. Although the authors did not evaluate the mechanical properties of the material, the results suggest that the SIPN structure enhanced the fatigue resistance of the actuator. Similarly, SIPN network ionogels composed of poly(3,4-ethylenedioxythiophene) (PEDOT) and polyurethane (PU) have also been utilized in actuators.55
Although IPN/SIPN hydrogels and ionogels were developed around the same time, the enhancement of the mechanical properties of ionogels through network design was not given significant attention, which may stem from the varying research interests of the associated researchers. Subsequently, as researchers investigated ionogels with improved mechanical properties, the design of IPN/SIPN evolved from solely organic polymer materials to include organic/inorganic hybrid materials, aiming to achieve superior mechanical properties by incorporating inorganic networks. The increased modulus is attributed to both the structural energy and high crosslinking density, described by the equation E = U/b3, where E represents the modulus, U denotes the structural energy, and b signifies the structural size. Due to their extremely small structural dimensions, inorganic materials exhibit a significantly high Young's modulus, as indicated by the aforementioned formula. Consequently, employing an organic/inorganic hybrid strategy can enhance the mechanical strength of IPN ionogels. Jones et al. utilized bicontinuous emulsions of IL [BMIm]+[BF4]− and polyethylene (PE)/poly(ethylene-b-ethylene-alt-propylene) (PE-PEP) as templates to create two different pore size distributions of 6–7 nm and 90–100 nm porous silica.56 Although both the polymer and the IL were removed at the conclusion of the process, an organic–inorganic hybrid interpenetration ionogel (OIHIG) was formed during the synthesis. This study represents the first known example of OIHIG synthesis. Subsequently, Kamio et al. utilized tetraethoxysilane (TEOS) and N,N-dimethylacrylamide (DMAAm) dissolved in [BMIm]+[TFSI]−/[BMIm]+[BF4]− as precursors for the inorganic and organic networks, respectively, to synthesize OIHIG (Fig. 3a).5 When the silica network is formed prior to the poly(N,N-dimethylacrylamide) (PDMAAm) network, the material can develop an organic/inorganic bicontinuous phase, resulting in a fracture energy of 437 kJ cm−3 for the “DN ionogels”. This energy is achieved through the rupture of covalent bonds and the supramolecular interactions at the surface of the inorganic phase. Conversely, when the PDAAm network is established before the silica network, the silica fails to create a continuous inorganic phase due to the steric hindrance imposed by the organic polymer chain. This gel is referred to as “μ-DN ionogels”. It is noteworthy that all ionogels containing 80 wt% ionic liquids exhibit an exceptionally high compressive stress of at least 28 MPa at a compressive strain of 95%.
 | ||
| Fig. 3 Schematics of ionogels with SIPNs. (a) Schematic illustrations of the concept of selective formation of inorganic/organic composite in an IL. When the silica particles are allowed to synthesize prior to the PDMAAm network, a spatially continuous silica particle network is formed to obtain DN ionogels. When the PDMAAm network is synthesized prior to the silica particles, spatially dispersed clusters of silica particle networks are formed to obtain μ-DN ionogels. Adapted with permission.5 Copyright 2017 Wiley. (b) Schematic illustration of preparation process and energy dissipation mechanism of ionogels. After the microspheres were swollen in the mixture of DMAA and ([EMIm]+[TFSI]−), a topological configuration of interpenetrating entanglements of linear networks and covalent-network microspheres was prepared through polymerization (the volume fraction of microspheres is not actual). Elastic covalent networks dissipate energy and passivate cracks through deformation when subjected to external load. The synergistic effect of reversible entanglement interaction and dynamic bonds allows the ionogels to be insensitive to crack propagation in any direction. Adapted with permission.57 Copyright 2022 Wiley. | ||
In addition to achieving high mechanical strength through the selection of different precursors, strategies for forming IPN and SIPN within the pores of dispersed phase have garnered significant attention, owing to their enhanced fatigue resistance. Li et al. prepared SIPN ionogels through in situ photopolymerization of DAAm within PAMPS microspheres that were swollen with [EMIm]+[TFSI]− and DMAAm (Fig. 3b).57 The microspheres acted as the first network, facilitating the deformation of microspheres at front of cracks, while the hydrogen bonds formed between ILs and the main chain of PDMAAm also functioned as an energy dissipation mechanism, leading to passivated cracks. This mechanism of crack passivation allowed stress to be evenly distributed at the front end of the crack, avoiding catastrophic crack propagation. Samples containing 2 wt% PAMPS microspheres and of up to 66 wt% of IL could be easily stretched to 33 times their original length while maintaining a fracture stress of 280 kPa. Additionally, the ionogel exhibited an ultra-high fatigue threshold of 2.12 kJ m−2, with no significant stress attenuation observed after 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles at 30% strain. The cracked samples demonstrated a fracture strain of approximately 3000%, even when the initial crack length was one-fifth of the sample width, highlighting the crack insensitivity of this type of ionogel. By varying the IL content between 50 wt% and 72 wt%, ultrahigh ionic conductivity values ranging from 0.001 to 1.340 S m−1 could be achieved. The combination of these outstanding physical properties renders this type of ionogel an effective flexible resistive sensor. The same authors also developed ionogels through in situ photopolymerization of monomers and crosslinkers within a covalent organic framework (COF) that was swollen with [EMIm]+[TFSI]−, AAm, and chemical crosslinkers.58 The resulting IPN ionogels exhibited negligible hysteresis under cyclic tensile test with tensile amplitude of 1500%. This behavior was attributed to the ability of the PAAm main chain to slide freely within the macropores of the COF as strain increases, leading to an orderly orientation of the polymer accompanied by the formation of high-density hydrogen bonds between the side chain amide groups. Once the external force is removed, the entropic elasticity, driven by the chemical bond crosslinking between PAAm chains, facilitates a reversible return of the polymer to its original position. This strategy is highly scalable and applicable to various types of polymers.59
000 cycles at 30% strain. The cracked samples demonstrated a fracture strain of approximately 3000%, even when the initial crack length was one-fifth of the sample width, highlighting the crack insensitivity of this type of ionogel. By varying the IL content between 50 wt% and 72 wt%, ultrahigh ionic conductivity values ranging from 0.001 to 1.340 S m−1 could be achieved. The combination of these outstanding physical properties renders this type of ionogel an effective flexible resistive sensor. The same authors also developed ionogels through in situ photopolymerization of monomers and crosslinkers within a covalent organic framework (COF) that was swollen with [EMIm]+[TFSI]−, AAm, and chemical crosslinkers.58 The resulting IPN ionogels exhibited negligible hysteresis under cyclic tensile test with tensile amplitude of 1500%. This behavior was attributed to the ability of the PAAm main chain to slide freely within the macropores of the COF as strain increases, leading to an orderly orientation of the polymer accompanied by the formation of high-density hydrogen bonds between the side chain amide groups. Once the external force is removed, the entropic elasticity, driven by the chemical bond crosslinking between PAAm chains, facilitates a reversible return of the polymer to its original position. This strategy is highly scalable and applicable to various types of polymers.59
The preparation method for most IPN/SIPN ionogels based on covalent crosslinking closely resembles that of hydrogels. Specifically, the precursor required to synthesize the second network is immersed in the first network and subsequently polymerized in situ to form the IPN/SIPN structure, a process known as the two-step method. While this method is straightforward, it compromises the molding accuracy of the network and prolongs processing time. It is advisable to consider the preparation process of one-step IPN hydrogels, which utilize two orthogonal polymerization mechanisms to form the gel. Furthermore, conventional covalently crosslinked IPN or SIPN ionogels exhibit limited fatigue resistance, which constrains their applicability. Although methods for in situ polymerization within the pores of dispersed phase have been developed to enhance the fatigue resistance of ionogels, the synthesis of specialized nanomaterials still involves time-consuming procedures. To adopt a simpler approach for improving fatigue resistance, further reference can be made to the covalent design of hydrogels; for instance: (1) increasing the reorganization speed of the sacrificial network to align with the external cyclic loading rate in the working environment;60 (2) alleviating stress concentration by minimizing the occurrence of dangling chains and ring chains within the network;61 (3) reducing the crosslinking density at crack tips through specific triggering mechanisms for crosslinking density to achieve elastic shielding against cracks;62 (4) increasing chain length by introducing monomers with force-induced ring-opening reactions into networks with higher crosslink density;28 and (5) performing force-induced radical polymerization by immersing the IPN gel in a solution containing vinyl monomers and crosslinking agents.27
2.2. Hyperbranched polymer networks
Hyperbranched polymers are highly branched polymers characterized by their three-dimensional structure, typically formed through the polymerization of a central core and branched monomers. Unlike randomly branched polymers, which exhibit indefinite branch functionality and chain lengths, and dendrimers, which have well-defined structures, hyperbranched polymers possess statistically determined branch functionality and chain length. They also exhibit a broad molecular weight distribution due to uncontrolled polymerization. The unique branched architecture results in a high number of terminal groups at the periphery, preventing the molecular chains from becoming entangled. Consequently, hyperbranched polymers demonstrate high solubility, low viscosity, and significant chemical reactivity in solvents that have a strong affinity for the terminal groups. For instance, the first hyperbranched polymer was synthesized by Kim and Webster in 1990 through the Suzuki coupling of hydrophobic 3,5-dibromophenylboronic acid in mixed solvents.63 However, the carboxylate obtained by treating the bromine atoms on the periphery of the polymer with butyllithium and carbon dioxide exhibited high water solubility, in contrast to the polymerized monomer. This observation supports the potential use of hyperbranched polymers for hydrogel synthesis. Jiang et al. recently introduced hyperbranched polycaprolactone (PCL) with carboxyl and acrylate terminal groups into linear acrylamide-co-acrylic acid (AAm-co-AAc) polymers, resulting in hydrogels with exceptional mechanical properties.64 The non-entangled geometry of the hyperbranched crosslinker enhanced the mobility of the polymer chain segments, while the abundant end groups acted as high-functionality crosslinking points, enabling the simultaneous hardening of multiple polymer chains during stretching. This approach achieved a remarkable combination of mechanical strength ranging from 4 to 9 MPa, toughness between 14.1 and 44.1 MJ m−3, and stretchability of 300% to 1100%.To our knowledge, the first study that simultaneously investigated ionic liquids and hyperbranched polymers was conducted by Stebani et al. in 1995, who synthesized protonated ionic liquid crystals.65 The core of these crystals consists of four tertiary amines that have been protonated by hydrochloric acid, while the peripheries are linked to catechol derivatives modified with long carbon chains through an amidation reaction. In the absence of protonation, the hydrogen bonds between the amide groups lead to a high melting point and a lack of liquid crystallinity. However, the tertiary amine at the molecular center exhibits excessive flexibility. Upon protonation, the hydrogen bonds of the amides are disrupted, resulting in a lowered melting point. Furthermore, the formation of quaternary ammonium salts increases the rigidity at the molecular center, thus facilitating the formation of smectic liquid crystals. Although the system is not an ionogel, the interactions among the numerous ionic groups within the hyperbranched compound significantly alter the morphology of the polymer, thereby demonstrating the potential of hyperbranched structures to enhance the properties of ionogels. The synthesis methods of ionogels featuring hyperbranched polymer backbones can be categorized into two main approaches: (1) the formation of hyperbranched structures via in situ polymerization of a mixture of monomers and ionic liquids, and (2) the blending of pre-synthesized precursors containing hyperbranched polymers with ionic liquids, followed by in situ crosslinking. Zhang et al. designed a hyperbranched polyurea ionogel through in situ polymerization of monomers and utilizing [EMIm]+[TFSI]− as the solvent (Fig. 4a).66 The hyperbranched structure enhanced the supramolecular urea hydrogen bond clusters, yielding a tensile strength of 3.5 MPa and an elongation of 495%. This ionogel, exhibiting a toughness of 5.5 MJ m−3, has potential applications in cluster luminescence (cluster luminescence is generated through spatial conjugation and can be enhanced by the proximity of clusters, which could be promoted by hyperbranched structures) flaw detection, soft robotic tentacles, and nanotriboelectric generators.
 | ||
| Fig. 4 Schematics of ionogels with hyperbranched polymer networks. (a) Preparation of hyperbranched polyurea ionogels. Adapted with permission.66 Copyright 2024 Wiley. (b) Synthesis of acrylate terminated hyperbranched polymer (HP-A) and ionogel from acrylate terminated hyperbranched polymer by photopolymerization. Adapted with permission.68 Copyright 2019 Multidisciplinary Digital Publishing Institute. | ||
In addition to enhancing mechanical properties, the introduction of a hyperbranched network structure also has the potential to improve the conductivity of ionogels, because the loose and amorphous network structure of hyperbranched polymers can mitigate the interference of the polymer network on the mobility of ions, which had been proved by Nishimoto et al. by testing the conductivity of hyperbranched lithium salt polymer electrolyte.67 Zhao et al. synthesized a hyperbranched polymer (HP-A) terminated with 8 hydroxyl groups and 8 acrylate groups, utilizing 2-ethyl-2-(hydroxymethyl)-1,3-propanediol (TMP) and 2,2-dimethylol propionic acid (DMPA) as precursors (Fig. 4b).68 They subsequently conducted in situ crosslinking in [BMIm]+[TFSI]−, resulting in an ionogel with a storage modulus (G′) and a loss modulus (G′′) that exhibited negligible variation with frequency, maintaining stable performance at 200 °C. The gel demonstrated conductivity ranging from 10−3 S cm−1 to 10−1 S cm−1. When employed as an electrolyte for Li/LiFePO4 batteries, it achieved a maximum specific capacity of 153.1 mA h−1 g−1 and exhibited a capacity retention of 98.1% after 100 cycles. However, its tensile strain at break was less than 300%, which limits its effectiveness as a flexible sensing device. Extending the distance between the HP-A terminal crosslinking points in the network by introducing [VBMIm]+[BF4]− as a kind of active diluent could sustain high room temperature ionic conductivity (5.8 mS cm−1) and thermal stability (−60 °C to 250 °C).69 This modification allowed for the production of 3D printable ionogels, exhibiting a response time of 200 ms, and demonstrating a tensile strain exceeding 1000%, making them suitable for flexible sensor applications.
Li et al. synthesized methyl methacrylate-terminated hyperbranched polysiloxane through the self-condensation reaction of a silane coupling agent to create gradient dual-mode sensing ionogels.70 The hyperbranched polymer exhibited brief miscibility in a mixture of AAc and [EMIm]+[ES]− and subsequently underwent vertical phase separation over time. This is because silicon-containing polymers have low surface energy and their compatibility with other components is not particularly good. Therefore, after a certain period of time, components containing silicon elements migrate to the surface of the material, which leads to vertical phase separation of direction. Both the hydrogen bonds between AAc and [EMIm]+[ES]− and high crosslinking density provided by hyperbranched section ensured stretchability greater than 1000% and compressibility of 6.5 MPa. Due to the high sensitivity of the gradient structure to pressure, the ionogel was capable of simultaneously sensing both strain and pressure, which was achieved by measuring the change of resistance in different directions. Additionally, this process achieved a maximum electrical conductivity of over 0.35 mS cm−1, with a pressure sensitivity of 19.33 kPa−1 and a gauge factor (GF) = 2.5 which indicated that the gradient ionogel exhibited great sensitivity.
Currently, ionogels containing hyperbranched structures have been utilized in various fields, including flexible sensors,14 solar cells,71 and solid electrolytes.72 However, the synthesis of hyperbranched components in these ionogels primarily relies on condensation reactions, which tend to be relatively slow. The challenges of controlling reaction kinetics and the chemical structure hinder the expansion of their applications. It is advisable to employ click chemistry under orthogonal conditions to enhance synthesis efficiency.73 Additionally, while the interaction between solvents and polymers is well understood, the uncertainty surrounding the interactions between ILs and functional groups limits the effective utilization of the diverse end groups of hyperbranched polymers, necessitating further investigation.74 Lastly, future designs should not only focus on utilizing hyperbranched networks to enhance the performance of ionogels but also consider the development of hyperbranched ILs. This approach could leverage the high ionic strength properties of their peripheral structures to create innovative ionogels.
2.3. Homogeneous polymer networks
If a homogeneous polymer network (HN) with consistent numbers of polymer chains between crosslinking points and uniform molecular weights of the polymer chains can be formed, it would enable evaluation based on analyses using classical theories. This is because the establishment of theories typically necessitates simplified models to substantially reduce computational load; therefore, many researchers are focusing on synthesizing gels with such networks. The proposed methodology involves synthesizing building blocks that serve as repeating units for HNs. Gels with carefully designed HNs can exhibit several advantages, including anti-swelling properties,75 adjustable gelling times,76 adjustable degradation times,77 ultra-high crosslinking ratio,78 high fracture strength, and extended tensile length.79In practice, attempts have been made to prepare HNs by synthesizing linear or star-shaped polymers with uniform molecular weights using techniques such as living polymerization and click reactions, and subsequently linking these polymers. However, while past studies have succeeded in synthesizing such polymer building blocks, the formation of networks has often been unsuccessful.80 This failure is attributed to the fact that the spatial arrangement of the building blocks is not adequately considered during network formation. When the network formation is carried out at low concentrations where the building blocks are sparsely dispersed in space, network formation proceeds via site percolation, resulting in microgels forming clusters in various locations. These clusters eventually merge to form a network structure across the entire space. The resulting network, however, consists of building blocks unevenly connected, leading to an inhomogeneous polymer network with large amount of dangling chains. In many past studies, the polymer networks obtained have ended up in such states, and the homogeneity of the resulting networks has not been verified. Even when the concentration of the polymer building blocks is sufficiently high, if network formation is not conducted under conditions where the polymers do not entangle with each other in a good solvent, the resulting polymer network will retain entangled states of the polymers.
Sakai et al. attempted to prepare polymer gels with a HN structure by uniformly mixing aqueous solutions of 4-arm amino ester- and succinimidyl ester-terminated polyethylene glycol (PEG) at the polymer overlap concentration (C*). At this overlap concentration, the ends of the star-shaped polymers are highly likely to come into contact with one another, thereby increasing the probability of forming a uniform network through chain-end reactions. However, based on the results reported in their paper published in 2008,21 this attempt only yielded a nearly homogeneous network structure. The reason for this was that water was not a sufficiently good solvent for these polymers, namely, the polymer chains of 4-arm PEG were not fully extended. However, this uniformity was sufficient for the resulting hydrogel to exhibit compressive strength that significantly surpasses that of conventional hydrogels. Subsequently, Li et al. attempted network formation using star-shaped polymers made from the same PEG under better solvent conditions and at quasi-dilute concentrations of the star-shaped polymers (which are higher than the typical C*).81 Dynamic light scattering observations before and after network formation under these conditions showed no changes in light scattering behavior. This indicates that no cluster formation or polymer chain entanglement occurred during the network formation process. To verify whether the polymer network obtained by this method truly has a uniform structure, it is necessary to swell the network in a good solvent and observe its structure. A truly homogeneous network can sustain its homogeneity even after undergoing swelling, whereas a less uniform network will have its defects amplified during the swelling process, rendering them observable in SAXS. Okaya et al. synthesized star-shaped polymers via living radical polymerization with terminal groups bearing either amino or carboxyl groups. These were dissolved in a good solvent at concentrations above C* in an equimolar ratio, followed by a condensation reaction to attempt network formation. The resulting network was observed by SAXS measurements to have a uniform structure when prepared at concentrations significantly higher than C*, specifically in the range of 1.2C* to 2C*. Notably, the networks used for these measurements were not in their initial state after preparation but were instead equilibrated and swollen in a good solvent, indicating that a uniform network structure was successfully achieved.
Furthermore, Fujiyabu et al. developed a HN hydrogel with a stretch ratio as high as 30 by modifying 4-arm PEG into 3-arm PEG; this high stretchability was initially attributed to the strain-induced crystallization of PEG.79 However, Masubuchi et al. simulated the stretching and fracture behavior of both 4-arm and 3-arm PEG hydrogels using the phantom network method.82 They found that, under various terminal conversion rates, prepolymer molecular weights, and polymer concentrations, the mechanical performance advantages of 3-arm PEG hydrogels can be attributed to smaller stress distribution heterogeneity and a lower modulus, which are determined by intrinsic structural differences.
Before introducing HN ionogels, it is important to note that, due to the limitations of knowledge and understanding at that time, the existing HN ionogel networks are not homogeneously structured in an absolute sense. However, their relative homogeneity is sufficient to demonstrate excellent mechanical properties. Inspired by the gel-forming properties of HN hydrogels at low polymer concentrations, the first ionogel with a uniform network was developed by Fujii et al. This ionogel was synthesized through the end-coupling of amino and succinimidyl esters terminated 4-arm PEG.83 The innovative design effectively decouples the mechanical and conductive properties of the ionogel, enabling the formation of a self-supporting structure within 60 seconds that exhibits almost no attenuation in conductive performance, even at a polymer weight ratio of only 3–6 wt%. Furthermore, the 4-arm PEG ionogel demonstrates remarkable thermal stability at elevated temperatures, maintaining integrity up to 300 °C. It offers significant advantages over triblock polymer ionogels, which typically operate below 100 °C and rely on physical crosslinking, thus positioning it as a viable candidate for use as a polymer electrolyte.84 Subsequently, a series of applications based on HN ionogels were developed sequentially. Fujii et al. discovered that the high content ratio of [EMIm]+[TFSI]− (94 wt%) and the substantial mechanical strength (18 MPa compressive strength; 83.5% compressive strain) of HN ionogels render them suitable for supporting IL membranes (SILM) in traditional CO2 absorption processes.85 These ionogels exhibit high gas permeability and selectivity, and they can endure high gas pressures (up to 3 MPa) without leaking of IL. The combination of HN with ionic liquids not only leverages the excellent carbon dioxide absorption capacity of ionic liquids but also utilizes the mechanical properties of HN to ensure that the network is maximally swollen by the ionic liquids while maintaining mechanical performance, thereby making it a promising material. Ishii et al. employed a 4-arm PEG with amino and maleimide terminals to react in [EMIm]+[TFSI]− or [EMIm]+[PF6]− in order to avoid the generation of additional small molecules during crosslinking, thereby enhancing the electrochemical stability of HN ionogels when utilized as actuators (Fig. 5a).86 Furthermore, due to their high ionic conductivity of up to 5.60 mS cm−1 at 25 °C, the actuation behavior of this type of HN ionogel could be effectively matched to a rectangular voltage of 2 V. Similar studies on crosslinking reactions that do not yield additional small molecules include Ikeda's preparation of 4-arm PEG ionogels via the catalyst-free reaction of electron-deficient alkynes with azide groups, as well as Matsuura et al.'s synthesis of 4-arm PEG ionogels through the Thiol–Michael addition reaction.87,88 Saruwatari et al. exploited the light-induced [4 + 4] cycloaddition reaction of anthracene and also developed healable HN ionogels using [EMIm]+[TFSI]− and 4-arm PEG.89 The material, which had an IL content ratio of 90 wt%, demonstrated a recovery of 66% of its mechanical properties after irradiation with 365 nm light at 150 °C. Despite the challenging conditions, this remains the only documented example of a repairable HN ionogel (Fig. 5b).
 | ||
| Fig. 5 Schematics of ionogels with HNs. (a) Preparation of HN ionogels through Aza-Michael addition reaction and schematic configuration of polymer actuator. Adapted with permission.86 Copyright 2017 American Chemical Society. (b) A conceptual illustration of photohealing of the 4-arm PEG ionogel. Adapted with permission.89 Copyright 2018 The Royal Society of Chemistry. | ||
Although HN ionogels have been utilized in various applications, current research, to our knowledge, primarily focuses on the modification of 4-arm PEG end groups and the control of reaction kinetics. The inherent hydrophilicity of 4-arm PEG may compromise the stability of HN ionogels. In contrast to the single polymer skeleton employed in HN ionogels, HN hydrogels have successfully integrated multiple monomers and crosslinking agents, resulting in enhanced functionalities such as temperature responsiveness90 and photoinduced polymer chain growing.91 Consequently, future material design should thoroughly consider the incorporation of hydrophobic polymer backbones and meticulously designed crosslinking reactions to improve the durability and functionality of HN ionogels. Another equally important point is that if HN ionogels can be synthesized in an absolute sense, it would significantly advance the fundamental research on the properties of HN ionogels. This is because the modeling process can be greatly simplified through the use of well-defined networks. Additionally, hydrogels and elastomers based on 3-arm PEG and 3-arm poly(4-methyl-ε-caprolactone) (PMCL) have been proved both to demonstrate high stretchability, strength, and low hysteresis which are better than that based on 4-arm PEG.79,92 From a preparatory standpoint, HN ionogels derived from 3-arm polymers are feasible and may represent an innovative direction for the advancement of HN ionogels in the future.
2.4. Slide-ring polymer networks
The concept of slide-ring gel was first proposed by de Gennes in 1999.93 The crosslinking of this gel is based on the electrostatic interactions between cationic compounds with multiple positive charges and anionic polymers. Although de Gennes's model differs significantly from current slide-ring gels, the notion of “slidable crosslinking points” served as a substantial inspiration for Ito et al. in their development of polyrotaxane slide-ring gels. A typical Sliding Ring Gel (SRG) actually synthesized by experimentation has a polyrotaxane (PR) structure incorporated into the crosslinking points that make up the polymer network. The most well-known system of SRGs is the polymer network composed of PRs formed by PEG and α-Cyclodextrin (α-CD).94 A representative synthesis method of polyrotaxane involves first mixing α-cyclodextrin, a cyclic molecule with a hydrophobic cavity, with carboxyl-terminated PEG in water. This process allows the PEG to thread through the cyclodextrin, forming a pseudorotaxane structure. The partial hydrophobicity of the PEG backbone, along with the increase in entropy due to the exclusion of water molecules from the cyclodextrin cavity, serves as significant driving forces in this process. The pseudorotaxane exists as a precipitate in water, which is then collected and subjected to a coupling reaction with bulky adamantylamine in DMF, yielding polyrotaxane. The large end groups of adamantylamine effectively prevent the α-cyclodextrin from detaching from the backbone.95 Dissolving PR composed of PEG and α-CD in a solvent and connecting the α-CDs from different PR molecules results in a polymer network with figure-eight-shaped crosslinking structures. In the resulting polymer network, the sliding of crosslinking points allows chains from non-stretching directions to be supplied to the stretching direction, thereby forming the most uniform network possible. This structure helps to offset stress concentration and significantly reduces the crack sensitivity of the material.96 The resulting polymer network can allow the crosslinking points to move when external forces are applied to deform the network, preventing stress concentration in specific parts of the structure. As a result, this polymer network exhibits flexible and tough mechanical properties. The mechanical properties of SRGs can be modified from linear response to nonlinear response by varying the α-CD coverage ratio from 25% to 2%. This adjustment leads to stretch-induced crystallization of PEG, significantly enhancing the tensile length and breaking strength by over 13 times and exceeding 5 MPa, respectively.97 The presence of slidable crosslinking points contributes to the softness of the slide-ring gel,98 while also ensuring independence of modulus and fracture energy,99 low hysteresis,100 effective damping101 and high axial load sensitivity,102 among other favorable mechanical properties. Notably, multiple α-CDs can form pipe-like crystals which are oriented head-to-tail. As a result, traditional α-CD-based polyrotaxanes are soluble only in specific solvents such as dimethyl sulfoxide (DMSO), dimethylacetamide (DMAc) solution of LiCl and aqueous solution of sodium hydroxide (NaOH). To achieve solubility in water or other molecular liquids, PRs must undergo chemical modification to disrupt the symmetry of the α-CD.22,94,103–105 In addition to the SR network crosslinked through the ‘8’-shaped structure obtained by bonding α-CDs on PRs, PRs modified with acrylate on α-CD are also utilized to polymerize with conventional monomers, thereby enhancing the mechanical properties of gels.22,106,107To further investigate suitable solvents for PRs, Samitsu et al. examined the solubility of PR in chloride-containing ILs.108 This study demonstrated that chloride ions facilitate dissolution by disrupting the hydrogen bonds between α-CDs, resulting in a maximum swelling factor that can reach 20 times higher than the original weight. All chloride imidazolium-containing ILs studied could dissolve at least 1 wt% of PRs after being stirred with PRs at 90 °C for several hours. However, the production of slide-ring ionogels necessitated solvent exchange processes that could take up to 14 days. The solubility and processing time observed in the aforementioned work remained significantly below the anticipated processability. Therefore, it is essential to elucidate the interactions involved in order to identify ILs that are more likely to effectively dissolve PRs. Moriyasu et al. prepared SRGs through the hydroxypropylation modification of α-CD on PRs and compared their swelling properties with those of unmodified SRGs.109 The results indicated that the crystallization of α-CD served as the primary limiting factor for swelling in water. Conversely, when swelling occurred in ILs, the affinity of the IL for PEG increased with the Lewis basic strength of the anion. This suggested that the interaction between the anions in the IL and PEG primarily governed the solubility of the SR ionogels. Notably, the highest conductivity observed for the SR ionogel was 3.18 mS cm−1, which corresponds to 92.6% of the conductivity of pure IL.
Building on the previously discussed SR ionogels, Sugihara et al. incorporated Li+[TFSI]− into the system, facilitating the formation of lithium bonds with the hydroxyl groups of α-CD and the ether bonds of PEG.110 This modification enhanced both the puncture resistance and conductivity of the ionogels. Utilizing small amounts of PRs as crosslinkers in the system can significantly enhance processability while preserving the mechanical properties of ionogels. For instance, Du et al. synthesized a macromolecular crosslinking agent by modifying acrylate on PRs and subsequently prepared a slide-ring ionogel exhibiting a hysteresis of only 7% through copolymerization with ethyl acrylate and polyethylene glycol diacrylate (PEGDA) in [BMIm]+[TFSI]− (Fig. 6a).107 The ionogel demonstrated a low breaking strength of approximately 25 kPa and a moderate breaking strain of about 475%, making it suitable for flexible sensors. The conductivity of the PR ionogel varied slightly, ranging from 3 mS cm−1 to 4.15 mS cm−1 across frequencies of 1–1000 Hz. Additionally, its resistance showed a linear relationship with tensile strain within the range of fracture strain, indicating its high sensitivity and feasibility for sensor applications.
 | ||
| Fig. 6 Schematics of ionogels with SR polymer networks. (a) Molecular design of ionogels containing polyrotaxanes and ionic liquids with high stretchability and low hysteresis. Adapted with permission.107 Copyright 2023 Wiley. (b) Detailed illustration of the preparation process of SR ionogels based on crown ether, mainly including two steps, i.e., mechanically interlocked networks were firstly fabricated, followed by immersion in ionic liquids. Adapted with permission.111 Copyright 2025 Wiley. (c) The ring components generate an entropic force as if they behave like gas molecules confined in a cylinder to counteract the chain sliding through the crosslinks. The high ring density disables the chain sliding under small strains, although the chain has enough space to reach the unreal close packing state under the maximum strain. Adapted with permission.112 Copyright 2015 The Royal Society of Chemistry. | ||
In addition to cyclodextrin-based PR crosslinkers, crown ether-based rotaxane crosslinkers have recently garnered attention in the synthesis of ionogels. Yang et al. modified both ends of the rotaxane with methacrylate and copolymerized it with n-butyl methacrylate to prepare an elastomer.111 After swelling this elastomer in a mixed solution of ethanol and ionic liquid, ethanol was subsequently removed to obtain a series of ionogels (Fig. 6b). The ionogels containing the ionic liquid [EMIm]+[TFSI]− and a crosslinker content of 1 mol% exhibited a combination of outstanding properties, including a fracture strain of 473%, a fracture strength of 9.6 MPa, and a toughness of 25.9 MJ m−3. These properties can be attributed to the energy dissipation and stress deconcentration caused by the slippage of the crown ether from its recognition site.
Currently, several SR ionogels have been preliminarily prepared using PRs as the primary structural component or as crosslinking agents, yielding promising results. However, compared with SR hydrogels, there are still disadvantages that require improvement. At present, the α-CD coverage in nearly all SR ionogels is approximately 25%–28%. In this context, the α-CD associated with this type of PR is thought to behave similarly to gas molecules that are confined to the main chain and cannot move due to excessive pressure (Fig. 6c). Namely, the high elasticity observed is primarily attributed to chain entropy.112 To fully exploit the capability of PR to alleviate stress concentration through sliding and the stretch-induced crystallization of PEG, the coverage of the rings should be sufficiently reduced. For instance, Enoki et al. synthesized PR with α-CD coverage as low as 2.6% using PEG with a molecular weight of 40000 g mol−1, and prepared ionogels with [EMIm]+[TFSI]− as the solvent.113 The ionogel with a PR weight ratio of 40 wt% exhibited an impressive combination of properties, including a fracture strain greater than 1900%, a fracture stress exceeding 6.5 MPa, negligible residual strain under 5000 times 100% tensile cycling, and a conductivity of 2 mS cm−1. Through techniques such as wide-angle X-ray scattering (WAXS), small-angle X-ray scattering (SAXS), and small-angle neutron scattering (SANS), it was determined that the excellent performance arises from the low α-CD coverage, which facilitates the orientation and crystallization of PEG during stretching. Additionally, the processability of SR ionogels is a significant factor that limits the expansion of their applications. Current SR ionogels require pre-gelling with other solvents during the preparation process, followed by immersion in ILs to complete processing. The high time cost, which can extend to weeks, is not conducive to large-scale production. It is recommended to enhance the solubility of precursors in ILs by utilizing low-coverage PRs to facilitate in situ polymerization, or to develop PRs based on host molecules e.g. crown ethers, cucurbiturils, calixarenes, pillararenes, and cyclophanes to obtain well-defined precursors that are readily soluble in ILs.
2.5. Other polymer network structures
In addition to the aforementioned examples, various covalent polymer network structures also exist in hydrogels. These structures have the potential to be incorporated into ionogels, with the aim of enhancing the mechanical properties of ionogels while maintaining high conductivity. One such structure is the bottlebrush polymer network, characterized by low interchain entanglement and high deformability, which is formed by constraining one end of a long chain to the same main chain. This network has been demonstrated to exhibit a Young's modulus of approximately 100 Pa in a solvent-free state. Furthermore, its fracture strain and J-shaped stress–strain curve are around 1000%, and the similarity of its mechanical properties in gels has been validated in hydrogels.114,115 Xu et al. prepared bottlebrush polymer ionogels based on PEG and [EMIm]+[OTF]− using a photopolymerization method116 (Fig. 7a). The ionogel with a PEG to IL weight ratio of 1:1.5 exhibited an electrical conductivity of 0.14 S m−1 and a Young's modulus of 1.08 kPa, which is the lowest Young's modulus reported among all ionogels to date. Additionally, in cyclic tensile tests at 100% elongation, the ionogel demonstrated almost negligible hysteresis and residual strain. The combination of these excellent properties renders it suitable for motion detection of silkworm pupae, electrophysiological regulation of Venus flytraps, and human electrocardiogram monitoring. Its ultra-low Young's modulus prevents interference with the movement of silkworm pupae, while its ultra-low hysteresis facilitates the timely triggering and recording of the closure of Venus flytrap leaves (response time 1–7.17 s). Moreover, its signal-to-noise ratio, comparable to that of commercial electrodes, underscores its potential for biological signal acquisition. A highly entangled polymer network is characterized by a significantly greater number of trapped interchain tangles compared with the number of permanent covalent crosslinks. When subjected to external forces, the stress experienced by a single chain can propagate through these entanglements. The slippage of knots is transmitted to numerous nearby chains, thereby distributing the stress. This mechanism underlies the exceptional performance of the highly entangled polyacrylamide hydrogel developed by Kim et al., which achieved a remarkable combination of a fatigue threshold exceeding 200 J m−2 and nearly negligible hysteresis.24 In addition to the various network structures present in hydrogels, certain toughening methods that compromise the conformational entropy of cyclic molecules—typically employed in organic gels or elastomers—also hold potential for application in ionogels.117–119 Chu et al. synthesized highly entangled ionogels using poly(1,8-octanediol-co-citrate-co-caprolactone) (POCL) as the polymer network and [EMIm]+[TFSI]− as the solvent120 (Fig. 7b). The key to the successful synthesis of these ionogels lay in the utilization of polycaprolactone (PCL) with a molecular weight of 37000 g mol−1, which is significantly higher than the critical entanglement molecular weight (14000 g mol−1) of PCL, leading to the formation of a highly entangled network. The highly entangled structure was characterized by the amorphous peak observed in WAXS. It is reasonable to conclude that chain entanglement hinders the crystallization of polycaprolactone (PCL) segments. Furthermore, the absence of the melting peak in Differential Scanning Calorimetry (DSC) tests further corroborated the presence of the entangled structure. When the content of [EMIm]+[TFSI]− was 50 wt%, the ionogel exhibited a conductivity of approximately 1 × 10−4 mS cm−1, with negligible residual strain and a hysteresis as low as 6.2% during 50 cycles of stretching from 0% to 300%. This remarkable performance was attributed to the high elasticity resulting from chain entanglement and the plasticizing effect of the IL on the polymer chains. Although these network structures are not yet widely utilized in ionogels, it is anticipated that such designs will gain broader acceptance in the future.
 | ||
| Fig. 7 Schematics of ionogels with bottlebrush polymer networks and high entanglement polymer networks. (a) Chemical structures of PEGMA, PEGDMA, ionic liquids [EMIm]+[OTF]−, and crosslinked bottlebrush ionogel. (b) The chemical formula of POCL and a schematic diagram of its highly entangled polymer network. Adapted with permission.120 Copyright 2022 American Chemical Society. | ||
3. Covalent adaptable network design for improving properties of ionogels
The crosslinking mechanism of ionogels can be categorized into supramolecular interactions and covalent bonds. Supramolecular crosslinking generates a relatively ordered structure through the physical interactions between functional groups, leading to a significant decrease in entropy. However, at elevated temperatures, the enhanced mobility of the polymer main chain disrupts this ordered structure, resulting in an increase in the number of conformations. Consequently, the material must endure the decline in mechanical properties caused by the dissociation of supramolecular interactions due to an increase in entropy.121 This temperature instability restricts the potential applications of ionogels. In contrast, covalent bonds exhibit higher bond energy than supramolecular interactions, making covalent crosslinking a prevalent method for enhancing the robustness, self-healing ability and mechanical strength of ionogel networks. The types of covalent bonds that form polymer networks include irreversible and reversible covalent bonds. Networks containing reversible covalent bonds, referred to covalent adaptable networks (CANs), can undergo reactions via two mechanisms: dissociation and association.122 In dissociative CANs, the reversible covalent bond initially breaks into two independent groups, which subsequently reform a new reversible covalent bond. Conversely, in associative CANs, a large number of pendant reactive groups are present, and the reversible covalent bond is influenced by external factors. Upon activation, an exchange reaction can occur with dangling groups.123,124 Ionogels featuring reversible covalent bonds have recently been designated as “i-Canogels” by Zhu and You et al.125 This article will adopt this terminology to distinguish these materials from conventional ionogels, which are based on irreversible covalent bonds. Within this classification, i-Canogels are further divided categorized as Di-Canogels (see section 3.1), which are defined by their dissociation mechanism, (Fig. 8a) and Ai-Canogels (see section 3.2), which are characterized by their association mechanism (Fig. 8b). Although the activation energy (or binding energy) of certain supramolecular interactions is comparable to that of reversible covalent bonds (Fig. 8c), leading to ionogels with supramolecular crosslinks and Di-Canogels exhibiting similar behaviors at elevated temperatures—e.g. the ability to dissolve in corresponding good solvents—the crosslinking methods of these two materials are fundamentally distinct.126 For example, at room temperature, ionogels crosslinked by supramolecular interactions can dissolve in corresponding good solvents without the cleavage of covalent bonds, a phenomenon that is unattainable for Di-Canogels. The subsequent sections will discuss representative studies that enhance the physical properties of i-Canogels through the design of reversible covalent bonds, drawing analogies with conventional CANs. | ||
| Fig. 8 Schematic representation of crosslinker exchange reaction in ionogels. (a) Di-Canogels based on dissociative mechanism and (b) Ai-Canogels based on associative mechanism. (c) Activation energy or binding energy of supramolecular interactions and covalent bonds. Adapted with permission.122 Copyright 2021 American Chemical Society. | ||
3.1. Di-Canogels
Research on dissociable CANs can be traced back to 1946. Stern, Tobolsky, and others discovered that the relaxation activation energy of various vulcanized rubbers was nearly uniform by measuring continuous and intermittent stress relaxation and creep under constant load across a temperature range of 35 °C to 120 °C.127 They proposed that this phenomenon was attributable to the balance, disruption, and rearrangement of disulfide bonds. In 1969, Carven synthesized thermally reversible crosslinked materials by incorporating furan groups into the side chains of linear polymers and subsequently reacting them with maleimide-bearing crosslinking agents.128 Chen et al. developed the first dissociative covalent adaptive network entirely composed of reversible covalent bonds, achieved by introducing a reversible Diels–Alder reaction between furan and maleimide at the terminus of a four-arm monomer.129 Unlike traditional materials that incorporate reversible covalent bonds solely within the crosslinking points while maintaining irreversible chemical bonds at the main chains,130 this innovative material features reversible covalent bonds throughout each molecular chain within its three-dimensional network. Furthermore, the chemical bonds can be entirely regenerated by heating the cross-section, thereby facilitating complete reversible reprocessing of polymer materials for the first time. To date, numerous thermally dissociable or photodissociable reversible covalent bond formation reactions have been developed for CANs. These include the Diels–Alder reaction,128 the reversible radical reaction of alkoxyamines,131 the reversible reaction between triazolinediones and alkenes,132 the reversible hindered urea bonds,133 the reversible oxime–carbamate bond,134 the reversible dimerization of nitro-cinnamate,135 the reversible fragmentation of aromatic pinacol,136 reversible guanidine bond,137 reversible amide dissociation reaction,138 reversible Thiol-Michael addition reaction,139 reversible Aza-Michael addition reaction,140 reversible dimerization of anthracene,141 reversible dimerization of coumarin,142 reversible exchange of disulfide bonds,31 reversible exchange of trithiocarbonates,143 and reversible exchange of diselenide bonds144 among others (Fig. 9). | ||
| Fig. 9 Schematics of reversible covalent bonds based on dissociative mechanism, including Diels–Alder reaction, the reversible radical reaction of alkoxyamines, the reversible reaction between triazolinediones and alkenes, the reversible hindered urea bonds, the reversible oxime–carbamate bond, the reversible dimerization of nitro-cinnamate, the reversible fragmentation of aromatic pinacol, reversible guanidine bond, reversible amide dissociation reaction, reversible Thiol-Michael addition reaction, reversible Aza-Michael addition reaction, reversible dimerization of anthracene, reversible dimerization of coumarin, reversible exchange of disulfide bonds, reversible exchange of trithiocarbonates, and reversible exchange of diselenide bonds. | ||
Di-Canogels are CANs based on the dissociate mechanism swollen by ILs, exhibiting both reversible processability due to dissociated dynamic covalent bonds and conductivity attributed to ILs. Tang et al. synthesized a SIPN Di-Canogel through Diels–Alder reaction, which incorporated 80 wt% IL.145 This material maintained a fracture stress of 660 kPa and a fracture strain of 268%, while also demonstrating a high conductivity of 3.3 mS cm−2. Notably, the cracks in the material could completely heal after being heated at 100 °C for 10 seconds, making it a promising candidate for polymer electrolytes and flexible sensors. The contribution of the DA reaction to the self-healing performance was assessed through thermorheological testing and differential scanning calorimetry (DSC) testing. It was observed that the material's G′′ began to exceed G′ when heated to 110 °C, and the endothermic peak in the DSC at 110 °C corresponded to the dissociation of reversible covalent bonds. Zhang et al. developed a series of light-stimulated, repairable Polyurthane (PU) Di-Canogel nanotriboelectric sensors based on disulfide bond exchange reactions.146 The incorporation of acryloyl morpholine (ACMO) and [EMIm]+[TFSI]− significantly reduced the viscosity of the system, enhancing the material's compatibility with photocuring and facilitating its application in 3D printing technology. By varying the mass ratio of IL from 40% to 80%, the tensile strength, breaking strain, and conductivity of the Di-Canogels were found to range from 0.49 MPa to 7.42 MPa, 546% to 977%, and approximately 0.08 mS cm−1 to 4 mS cm−1, respectively, indicating considerable tunability within these parameters. Notably, these materials can recover 100% of their sensing performance after being irradiated by 365 nm UV light for 10 min at room temperature, thereby satisfying the performance requirements for flexible sensors. In another study, Li et al. developed Di-Canogel containing hindered urea bonds by drying a mixed solution of [DEIM]+[TFSI]− and a PU-urea precursor.147 The dynamic crosslinking of hindered urea bonds and hydrogen bonds, along with the excellent crystallinity of PCL, resulted in an Di-Canogel with high mechanical strength of 1.6 MPa and remarkable fatigue resistance of at least 10000 stretching cycles under the condition of 66 wt% IL content. Additionally, it exhibited a conductivity of 1.2 mS cm−1 and impressive self-healing performance, achieving 95% recovery after heating at 65 °C for 3 hours, thereby supporting its application as a durable flexible sensing device. The authors, in conjunction with references and DSC data, demonstrated that heating to 65 °C enables Di-Canogel to facilitate self-healing through the synergistic promotion of the dissociation of hindered urea bonds, the melting of crystalline segments, and the reorganization of hydrogen bonds.133
Fan et al. utilized a combination of the Diels–Alder reaction and lithium bond crosslinked linear PU networks to develop a Di-Canogel flexible sensor containing 75 wt% of [EMIm]+[TFSI]−and 20 wt% of Li+[TFSI]− (BPU75%–20%, BPU refers to polyurethane that is chemically crosslinked by 4,4′-bismaleimidodiphenylmethane.) (Fig. 10a), which exhibited a tensile strength of 11.3 MPa, a tensile strain of 2396% (Fig. 10b), and a conductivity of approximately 0.032 mS cm−1.148 Notably, the energy loss coefficient of BPU75%–20% under 100% strain was only 0.055 (Fig. 10c). The comparative experiments revealed that a lower energy loss coefficient was achieved through the synergistic effects of chemical crosslinking and lithium bonding. Specifically, the linear PU75% without the addition of lithium salt exhibited a high energy loss coefficient ranging from 0.73 to 0.26, whereas the chemically crosslinked BPU75% demonstrated a significant reduction in the energy loss coefficient, decreasing to values between 0.17 and 0.068.
 | ||
| Fig. 10 Design and properties of Di-Canogels. (a) Schematic illustration of polymer network design for robust and recyclable Di-Canogels and synthesis procedure of the dynamic covalent bonds crosslinked polyurethane chains. (b) Stress–strain curves of PU75%, BPU75%, BPU75%–20%, PU75%–20% Di-Canogels in the whole range to 2400%. (c) Comparison of the energy loss coefficient (η) of different Di-Canogels for a strain of 100% during 10 cycle loading-unloading tests. (d) The stress–strain curves of the BPU75%–20% Di-Canogels at different recyclable cycles by DMF solvent method. Adapted with permission.148 Copyright 2024 Wiley. | ||
As an application, when attached to the sole, Di-Canogel could accurately detect the output signal of step speed during 1000 cycles of monitoring. The presence of reversible covalent bonds enabled BPU75%–20% to be recycled at least 10 times by Dimethylformamide (DMF) solvent method while retaining almost unchanged mechanical properties, and it demonstrated good stability in open atmospheric conditions and in organic solvents (Fig. 10d). In contrast, reference samples that utilized only lithium bonds (PU75%–20%) exhibited negligible tensile strength and less than 400% strain at break, while the fracture strain and fracture stress of BPU-75% crosslinked by dynamic covalent bonds could still be maintained at levels exceeding 1700% and 6 MPa, respectively, highlighting the crucial role played by dynamic covalent bonds in enhancing mechanical properties.
Tan et al. efficiently prepared Di-Canogel fibers by incorporating oxime-carbamate bonds into reticulated PU ionogel via hot melt extrusion (Fig. 11a), thereby addressing the challenges associated with the discontinuity of tube mold spinning production and the kinetics of photopolymerization spinning.149 Given that discontinuous spinning methods or prolonged light exposure treatments can diminish spinning efficiency, it is preferable to develop continuous methods that do not depend on light exposure treatment. This method effectively mitigates processing time constraints. Specifically, the principle underlying this approach is that the dissociation of oxime carbamate groups at elevated temperatures decreases the covalent crosslinking density, enhances the mobility of polymer chains, and consequently reduces the viscosity of the ionogel. Upon cooling, the reorganization of oxime carbamate groups and the rearrangement of polymer chains lead to the formation of a covalently crosslinked network within the ionogel, enabling continuous melt spinning to yield Di-Canogel fibers (Fig. 11b). The resulting Di-Canogel fibers demonstrated a mechanical strength of 0.76 MPa, a stretchability of 784%, an electrical conductivity of 12.1 mS cm−1, and notable solvent resistance. Due to the presence of chemical crosslinking, the Di-Canogel exhibited excellent shape stability in THF. In contrast, the ionogel composed of linear molecules became highly fragile in THF, breaking upon being picked up with tweezers.
 | ||
| Fig. 11 Structure and preparation of Di-Canogel fibers. (a) The chemical structure of Di-Canogels. (b) The manufacture process of Di-Canogel fibers. Adapted with permission.149 Copyright 2024 Wiley. | ||
On the other hand, although preliminary findings have been made regarding the improvement of the mechanical properties and self-healing capabilities of Di-Canogels under specific conditions, researchers aspire to develop a solution that combines high mechanical strength with non-stimuli self-healing at room temperature. The key to achieving this desirable combination lies in reducing the activation energy required for the reversible covalent bond reaction. Consequently, a strategy involving the participation of ortho groups has been developed, based on reactions such as boronic transesterification,150 silyl ether exchange,41 phthalic acid transesterification,151 amide–imide exchange reactions,152–154 and β-amino transesterification reactions.140 This approach has been validated in CANs.
Surprisingly, it was not until Huang et al. synthesized Di-Canogels using the neighboring group participation mechanism that anyone had successfully integrated high strength and room temperature self-healing properties into ionogels. Specifically, about the neighboring group participation mechanism, the authors thoroughly considered factors such as electron sharing or attraction, the relative spatial arrangement of the groups, and the conformation of the groups to design a 4-arm crosslinking unit that synergistically enhanced both the network crosslinking density and the mobility of chain segments.125 Tetrafunctional diaminoglyoxime (AMG) reacts with isophorone diisocyanate (IPDI) to form adjacent urea and oxime–urethane bonds within the 4-arm crosslinking unit. The nucleophilic nitrogen atom in the urea bond can attack the oxime–urethane bond, facilitating the exchange of this bond at the crosslinking unit. The strong electron-withdrawing effect of the imine bond in the oxime–urethane contributes to the instability of the carbamate group, making it prone to cleavage and resulting in the generation of oxime and isocyanate groups. These groups can subsequently react to reform the oxime–urethane bond, highlighting the dynamic nature of the system (Fig. 12a). Di-Canogel, with a 1:1 ratio of poly (butylene glycol adipate) diols (PBGAD) to fluorinated tetraethylene glycol (FTEG) (Fig. 12b), exhibited a tensile strength of 4.55 MPa, a toughness of 13.49 MJ m−3, and recovered at room temperature over 48 hours. After recovery, 92.8% of the mechanical properties were retained compared with the original sample. The neighboring group participation mechanism of the Di-Canogel was demonstrated through dynamic mechanical analysis (DMA), rheological testing, comparison of self-healing efficiency, and ATR-FTIR as well as variable-temperature infrared spectroscopy. In the DMA and rheological tests, the authors found that the i-Canogel containing the intrinsic catalytic structure exhibited significant stress relaxation behavior within the temperature range of 25–55 °C, indicating that the dynamic exchange of oxime–carbamate bonds was catalyzed and enhanced by adjacent urea bonds. In the self-healing tests, the self-healing efficiency of the Di-Canogel with introduced AMG units was significantly higher than that of the control group (62.5%). Furthermore, in the infrared tests, the generation of isocyanate was directly observed during the heating process, further substantiating that the neighboring group participation mechanism promoted self-healing at room temperature.
 | ||
| Fig. 12 (a) Internal catalysis mechanism for the oxime–urethane exchange. (b) The multifunctional integrated crosslinking structure design of Di-Canogel. Adapted with permission.125 Copyright 2024 Proceedings of the National Academy of Science. | ||
While the research on Di-Canogels has yielded promising results, it also presents significant challenges. Currently, numerous reactions have been developed for the synthesis of dissociated CANs; however, only a few, e.g. the Diels–Alder reaction, dynamic exchange of disulfide bond, reversible oxime–urethane bond and reversible reaction of hindered urea bond, have been employed to synthesize Di-Canogels. Among these, the Diels–Alder reaction exhibits kinetics that are difficult to manipulate, and the isocyanate released from the oxime–urethane bond and the hindered urea bond is highly sensitive to humidity. Thus, there is an urgent need to develop new reactions to address these limitations. Furthermore, since Di-Canogels involve two independent reactions—bond cleavage and regeneration—the instantaneous crosslink density of these dynamic crosslinks is influenced by the values of the two equilibrium constants associated with the regeneration and broken crosslinking reactions. Consequently, the maintenance time of crosslink density is significantly affected by external conditions. Although the material demonstrates greater stability to external changes compared with supramolecular crosslinked ionogels, when specific external stimuli exceed a certain threshold, the crosslinking overall polymer chain may be lost, leading to a significant decrease of viscosity. This results in reactive fragments diffusing through flow and potentially failing to encounter their complementary segments due to the cessation of external stimulation. In practical applications, chain rearrangements, glass transitions, or crosslink degradation can hinder complete bond regeneration, causing dissociated covalently adaptive networks to exhibit varying crosslink densities after processing. To address this issue, methods e.g. moderately increasing the volume fraction of the IL and incrementally increasing the number of pendant groups can be employed to reduce the glass transition temperature and enhance the likelihood of collisions among crosslinked fragments.
3.2. Ai-Canogels
Associative CANs were named vitrimers by Leibler et al. in 2011 due to their glass-like processability at elevated temperatures and the formation of stable chemical crosslinks at lower temperatures,34 although their existence was established earlier in conventional polymer materials. In 1954, Osthoff et al. found that the presence of water and carbon dioxide could catalyze the rearrangement of silicon–oxygen bonds in the presence of acids and bases, leading to stress relaxation in polydimethylsiloxane elastomers.155 The bond rearrangement mechanism, when influenced solely by either a base or an acid, is understood to conform to the association mechanism. In the subsequent decades, vitrimers have undergone significant development. Transimination reaction,30 transesterification reaction of boronic ester,32 dynamic exchange of acylhydrazone bonds,33 transesterification reaction,34 transcarbamoylation reaction,35 transcarbamoylation reaction of oxime–urethane,36 transamination reaction,37,38 transalkylation reaction,39,40,156 siloxane equilibration,155 dynamic exchange of silyl ethers,41 and dynamic exchange of acetals,42–45 dynamic exchange of aminals,157 transesterification reaction of phosphate triesters,158 transesterification reaction of carbonate,159 olefin metathesis reaction,160 α-acetyl cinnamate/acetoacetate exchange reaction,161 C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C/CN metathesis reaction,162 transesterification of thioester,163 dynamic exchange of anhydride,164 transesterification of cyanurate,165 SNAr reaction of tetrazine166 and allyl sulfide/sulfur-centered radical exchange reaction167etc. have been utilized for the manufacture of vitrimers (Fig. 13).
C/CN metathesis reaction,162 transesterification of thioester,163 dynamic exchange of anhydride,164 transesterification of cyanurate,165 SNAr reaction of tetrazine166 and allyl sulfide/sulfur-centered radical exchange reaction167etc. have been utilized for the manufacture of vitrimers (Fig. 13).
 | ||
| Fig. 13 Schematics of reversible covalent bonds based on associative mechanism, including transimination reaction, transesterification reaction of boronic ester, dynamic exchange of acylhydrazone bonds, transesterification reaction, transcarbamoylation reaction, transcarbamoylation reaction of oxime–urethane, transamination reaction, transalkylation reaction, siloxane equilibration, dynamic exchange of silyl ethers, and dynamic exchange of acetals, dynamic exchange of aminals, transesterification reaction of phosphate triesters, transesterification reaction of carbonate, olefin metathesis reaction, α-acetyl cinnamate/acetoacetate exchange reaction, CC/CN metathesis reaction, transesterification of thioester, dynamic exchange of anhydride, transesterification of cyanurate, SNAr reaction of tetrazine and allyl sulfide/sulfur-centered radical exchange reaction. | ||
Ai-Canogels are vitrimers swollen by ILs, in which bond breaking and regeneration occur simultaneously, resulting in reactants and products that maintain the same bond structure. Consequently, the reaction equilibrium remains unchanged with variations in temperature, and the crosslinking density remains constant below the thermal decomposition temperature.168 It is essential to consider whether the crosslinking density changes in the excited state, as this serves as a key criterion for distinguishing between Di-Canogels and Ai-Canogels.169 Di-Canogels exhibit a critical threshold; when the external stimulus reaches a sufficient intensity, the reversible dynamic bonds are nearly entirely decomposed into their pre-reaction components. At this point, the network lacks chemical crosslinks, allowing the material to dissolve completely in a suitable solvent. In contrast, regardless of the intensity of external stimulation, Ai-Canogels cannot be dissolved in any good solvent until their network structure is irreversibly destroyed.
The connection between ILs and vitrimers predates the invention of Ai-Canogels. For instance, vitrimers based on N-alkylation reactions were first demonstrated in 2014 to exhibit conductivity attributed to counteranions,170,171 because even when monomers are polymerized to form polymers, their counterions remain mobile within the vitrimers. The cationic functional groups of these vitrimers closely resemble the cations found in triazole ILs, allowing them to be considered as a type of polyIL CAN. Other similar cationic functional groups that can be utilized in vitrimers include quaternary ammonium salts156 and pyridinium salts.172 Although vitrimers with cationic functional groups show promise as polymer electrolytes, the number of charge carriers is directly proportional to the number of ionic groups in the backbone. Increasing the number of carriers can inadvertently alter the crosslinking of the network, compromising both the density and overall water resistance of the material. Therefore, it is essential to design materials whose conductivity is either minimized or orthogonal to other physical properties to fulfill specific application requirements. In Ai-Canogels, the number of charge carriers is dictated by the mass fraction of the IL, while the crosslinking density is determined by the number of dynamic covalent bonds. This approach significantly reduces the correlation between conductive properties and mechanical properties.
Current research has demonstrated the feasibility of preparing Ai-Canogels with favorable mechanical, electrical, and processing properties using various chemical compositions. Xu et al. reported that PU Ai-Canogels synthesized through boronic transesterification exhibited a fracture strain of 390% and a conductivity of 14 mS cm−1.173 Due to the presence of boronic ester bonds as reversible chemical crosslinks and the reduction of crystallinity in the PU soft segments due to ionic liquids, these Ai-Canogels exhibited extremely low energy dissipation during stretching. Specifically, even after being stretched ten times at 50% strain, they demonstrated low hysteresis and negligible residual strain. Moreover, following damage, they could restore their original mechanical properties with just 30 minutes of recovery at room temperature. To address the incompatibility between low film thickness and high mechanical properties in carbon dioxide separation membranes, Fu et al. employed in situ crosslinking of amino-terminated poloxamer (a ABA type triblock copolymer of polyethylene glycol, polypropylene glycol and polyethylene glycol) and benzene-1,3,5-tricarboxaldehyde (BTC) to form imine bonds.174 The resulting Ai-Canogels films could be fabricated into samples with a thickness of only 90 μm while maintaining a tensile strength of 1.66 MPa and a breaking strain exceeding 350%. Notably, this material exhibited a combination of low hysteresis (<2%) and self-healing capabilities at room temperature. Specifically, for holes with 0.045 mm2 area on the Ai-Canogels or for Ai-Canogels cut into 2 pieces, they could self-repair within 48 hours. Li et al. emphasized the influence of ILs [EMIm]+[TFSI]− and [EMIm]+[OTF]− on the rearrangement of the Ai-Canogels network, which is based on alkyl transfer between thioether and sulfonium salt moieties.175 The dilution and screening effects of these ILs reduced the alkyl transfer rate, thereby inhibiting network rearrangements and enhancing the mechanical properties of Ai-Canogels in high-temperature environments compared with their non-dynamic counterparts. This conclusion was based on the stress relaxation experiments of Ai-Canogels, which demonstrated that the relaxation time of the material with added ionic liquids (ILs) at 140 °C was extended by more than tenfold compared with the corresponding vitrimers. The authors subsequently fitted the corresponding relaxation times to the Arrhenius equation, revealing that the calculated activation energy of Ai-Canogels was greater than that of the corresponding vitrimers, thereby further supporting this conclusion. Furthermore, the same authors discovered that, for dioxaborane-based photocurable Ai-Canogels, [EMIm]+[TFSI]− and [EMIm]+[OTF]− did not influence the borate transesterification reaction, but did impact the polymer chain. This fluidity produced a plasticizing effect, thereby improving the recyclability of the material.176 In the study conducted by Bui et al., the transesterification reaction of β-hydroxyester was employed to design Ai-Canogels that could be welded with dielectric elastomers (Fig. 14a).177 This reaction could be completed within one hour at a temperature of 160 °C. To address the challenges associated with manufacturing multi-layer stretchable ionic devices, a straightforward and effective approach was proposed (Fig. 14b). The series of Ai-Canogels discussed demonstrated commendable conductivities, all exceeded at least 10−4 S cm−1.
 | ||
| Fig. 14 Structure and welding process of Ai-Canogels. (a) Composition and synthesis route of Ai-Canogels. (b) The manufacture process of multi layer materials by interface welding. Adapted with permission.177 Copyright 2023 Elsevier. | ||
ILs not only function as solvents but also act as catalysts for dynamic covalent bond reactions, enhancing the processability of materials and imparting unique responsiveness to them. For instance, Debuigne et al. (2021) demonstrated that ILs can serve as activators of vitrimers.46 In their study, two types of imidazolium acetate were incorporated into a vitrimer network created by crosslinking bisphenol A epoxy resin with adipic acid, where it acted as monomer and solvent respectively. The catalytic properties of ILs enabled the vitrimer to maintain thermal stability without compromising mechanical properties after being subjected to four cycles of pressing and recovery. Notably, increasing the IL concentration from 3 mol% to 5 mol% in a stress relaxation experiment significantly decreased the network relaxation time τ*—defined as the duration required to release 63% of the stress—from 85 minutes to 29 minutes at 200 °C. Although this relaxation time remains longer than the values reported for zinc acetate as a catalyst, this research is pioneering in demonstrating that imidazolium ILs can function as activators for vitrimer systems, marking a significant advancement in the field. According to the research by Hirose et al., a possible mechanism involves the hydrogen atom at the 2-position of the imidazolium in the ionic liquid forming a hydrogen bond with the carbonyl group of the ester bond. This interaction enhances the electron-withdrawing ability of the oxygen atom, thereby activating the ester group and accelerating the reaction.178 Subsequently, another pioneering study demonstrated that the IL activators of vitrimers are not confined to imidazolium salts. Shin et al. utilized solvated ILs (SILs) [Li(G3)]+[TFSI]− and [Li(G3)]+[OTF]− in their research.179 Additionally, a series of SILs, including [Mg0.5(G4)]+[TFSI]− and [Zn0.5(G4)]+[TFSI]−, was investigated for their effects on the curing reaction of epoxy resin and the catalysis of β-aminoester transamination reactions. The synthesis of SILs involved coordinating the corresponding metal salts with oligomeric PEG [e.g. triethylene glycol dimethyl ether (G3) or tetraethylene glycol dimethyl ether (G4)]. This transformation converted the originally solid salts into liquids, which significantly reduced the amount of catalyst by enhancing their dispersibility in epoxy resins. In addition, the author selected [TFSI]− and [OTF]− as anions due to their hydrophobic properties, which contributed to the stability of SILs in the presence of environmental moisture. All four SILs were found to significantly accelerate the curing reaction without markedly reducing the Tg. The curing onset time and gel time of the resin without a catalyst were 6.0 minutes and 9.4 minutes, respectively. Notably, the catalytic effect of [Mg0.5(G4)]+[TFSI]− was the most pronounced; at a concentration of 3.46 mol%, the onset time and gel time were reduced to 3.0 minutes and 3.2 minutes, respectively. In the model study of transamination reactions, all SILs catalyzed reactions at a low temperature of 70 °C, achieving equilibrium within 10 minutes, which was significantly more efficient than the typical reaction temperature of 120 °C and an equilibrium time of 60 minutes. The catalysis of ring-opening polymerization of epoxy groups and the catalysis of the transamination reaction by SILs involve the coordination of solvated metal ions with the oxygen atom of the epoxy group and the carbonyl group of the acrylate, respectively. This coordination enhances the electrophilicity of the adjacent carbon atoms by increasing the electron-withdrawing effect of the heteroatoms, thereby accelerating the reactions. Zhao et al. introduced [C2C1MIm]+[TFSI]− into the SIPN network of PU and polyhexafluorobutyl acrylate (PHFBA) to create large-span switchable stiffness-gated bicontinuous phase separation Ai-Canogels (Fig. 15a).180 The presence of [C2C1MIm]+[TFSI]− was crucial for achieving high conductivity within the network, as well as for eliciting both positive and negative piezoresistive responses. This was attributed to the fact that IL-catalyzed transesterification and transcarbamoylation significantly altered the stiffness of Ai-Canogel with temperature, while the IL itself exhibited high conductivity (Fig. 15b). The authors investigated the catalytic effects of ionic liquids composed of a series of cations and [TFSI]− on transesterification and transcarbamoylation reactions. It was found that the Ai-Canogels, which were the subjects of the investigation, exhibited high and similar deformation rates at elevated temperatures, indicating that the catalytic mechanism was likely related to the ion–dipole interaction between the cations and the carbonyl groups of the ester bonds. This interaction enhanced the electron-withdrawing ability of the carbonyl groups, thereby increasing the susceptibility of the sp2 hybridized carbon atom in the ester group and carbamate group to nucleophilic attack. The Ai-Canogel sensor exhibited similar negative piezoresistive sensing behavior (ΔR < 0) to the conventional ionogel sensor in the high-stiffness state at room temperature, which could be attributed to the fact that the deformation of the hard phase structure when compressed was similar to folding, which reduces the ion transmission distance. However, when transitioning to a soft state at 80 °C, the Ai-Canogel sensor showed a transition to positive piezoresistivity (ΔR > 0). This was attributed to the softness of the PU hard phase, which makes it easy to deform when compressed, and the deformation was partially blocked into the continuous ionic conduction path (Fig. 15c and d).
 | ||
| Fig. 15 Design, application and mechanism of Ai-Canogels. (a) Chemical structures of reacted precursor components within Ai-Canogels. (b) Bicontinuous phase structure owing to the orthogonal polymerization-induced phase separation. (c) Schematic illustration of Ai-Canogel iontronic piezoresistive sensor (4 × 4) arrays that had the capability of the bidirectional stiffness-gated piezoresistivity. Scale bar, 1 cm. (d) Three-dimensional finite element analysis of Ai-Canogel solid mechanics, demonstrating the variations of the ion transport pathway during the bidirectional stiffness-gated piezoresistivity process. Adapted with permission.180 Copyright 2024 American Association for the Advancement of Science. | ||
The stability and transparency of ILs also render Ai-Canogels suitable for optical devices. Tang et al. utilized borate ester bonds to crosslink polyvinyl alcohol for the preparation of optical prisms.181 When an emitting light source passed through the prism and excited the fluorescence of the reference sample, the detection instrument captured all expected emission peaks between 600 nm and 660 nm. In contrast to conventional inorganic glass prisms, which can only be recycled through ultra-high temperature melting after damage, the scratches on this prism could self-heal within 12 hours. In cases of severe damage, the material could be recast directly by melting at a reduced temperature compared with that of traditional glass, making it a durable optical system which provides a new solution for durable prism design.
Ai-Canogels exhibit advantages in mechanical properties and recycling performance across various applications; however, they face challenges akin to those of Di-Canogels. Notably, while a significant number of reversible covalent bonds have been developed and utilized in vitrimers, only a limited few reactions—e.g. boronic transesterification, transalkylation, transesterification, transcarbamoylation, and transimination reactions—have been employed in the design of Ai-Canogels. Therefore, it is essential to further explore new reversible covalent bonds to diversify the range of materials. On the other hand, although Ai-Canogels avoid incomplete bond reorganization caused by polymer flow and conformational changes compared with Di-Canogels, they still face performance degradation due to oxidative decomposition and interference from external chemical substances. To explore the future application potential of Ai-Canogels, enhancing the robustness of reversible covalent bonds against undesired external interference may become a primary design focus. Chemical designs that consider commercial availability and reaction simplicity will be beneficial for achieving this goal. Regarding the conflict between self-healing performance and mechanical properties, a proven solution in Di-Canogels is to reduce the activation energy of the bond exchange reaction by the neighbouring groups participation mechanism. Furthermore, Ai-Canogels are highly processable and exhibit swelling in solvents without dissolving, distinguishing them as an independent category from thermoplastic and thermosetting ionogels. However, this unique property has largely remained undeveloped, with the exception of applications in welding. We recommend the careful consideration of the challenges faced by traditional materials at the interface to foster the development of novel applications. For instance, in traditional composite materials, the surfaces of inorganic fillers typically contain hydroxyl groups, which allow for a strong bond with the polymer matrix when treated with silane coupling agents.182 It may be feasible to eliminate the need for supplementary coupling agents by employing a transesterification reaction to enhance the interfacial connection. Last but not least, the influence of ILs on bond exchange reaction kinetics and network rearrangement in Ai-Canogels has yet to be thoroughly investigated. A comprehensive understanding of the underlying mechanisms is essential to shift the design mode from a trial-and-error approach to a more goal-oriented methodology.
4. Summary and future outlook
Over the past two decades, hydrogels, CANs, and ionogels have garnered significant attention as multifunctional materials. However, regarding covalent design, there is a notable discrepancy: while the predominant focus has been on the network design of hydrogels and the reversible covalent bond design of CANs, the covalent design of ionogels has received comparatively less scrutiny. Consequently, this review aims to summarize the research findings related to ionogels, emphasizing support network design and reversible covalent bond design by drawing parallels with the covalent design strategies employed in hydrogels and CANs. Furthermore, it proposes potential avenues for future innovation.Although researchers have proposed various successful designs that achieve high fracture strength, high fracture strain, and high conductivity in ionogels, there remains significant potential for further exploration of covalent designs of ionogels in the future. (1) As of today, the network design of ionogels primarily focuses on organic polymers. In contrast, inorganic polymers with inherent functionalities, such as polysulfide (noted for its recyclability) and polyphosphazene (recognized for its flame-retardant properties), have seldom been incorporated into the supporting skeleton of ionogels. (2) The dynamic nature of the polymer network not only enables the polymer to restore its original structure after damage but also serves as a processing method to create new network configurations. Recent studies have demonstrated that isocyanate, released through the thermal dissociation of hindered urea bonds, can react with pendant carboxyl groups in the network or with residual water, transforming single network elastomers into double network elastomers.183 This strategy, which holds significant potential for development, aligns well with the covalent design principles of ionogels. (3) Currently, the covalent design scope of ionogels is primarily concerned with the design of the supporting network, while the covalent designability of ILs has not been thoroughly explored. Enhancing the physiological compatibility of ILs by designing suitable anions and cations represents a crucial objective in this context. (4) Regarding the recyclability of ionogels, most recycling methods rely on thermally activated dynamic covalent bond exchange. Given the transparency and conductivity of ILs, the development of low-energy-consuming light-activated or electrically activated devices could significantly improve the practicality of recycling strategies through reversible covalent bonds. Amamoto et al. achieved self-healing of damaged materials by incorporating thiuram disulfide units into polyurethane elastomers under the illumination of a tabletop lamp.184 This reaction can be completed using visible light and in an atmospheric environment, aligning with the concept of low energy consumption. In the field of electrochemical recycling, Hansen-Felby et al. successfully achieved the cleavage of disulfide bonds in polydisulfide derivatives with an efficiency exceeding 60% under electrified conditions, utilizing anthraquinone as a redox mediator to recycle these polymers into monomers.185 Given the recent surge in lipoic acid chemistry and the strong interest among scientists in incorporating lipoic acid molecules into ionogels, as a result, it is foreseeable that with further development of dynamic covalent bonds and their integration with ionogels, the recycling of i-Canogels under mild conditions within a short time frame will become feasible. (5) The high cost of ILs and the complexities associated with the synthesis of efficient reversible covalent bonds remain significant barriers to the practical application of i-Canogels. Designs that thoroughly account for real-world production constraints will substantially enhance the application of this type of material.
In summary, this review elucidates the covalent design of ionogels, encompassing the design of support networks and reversible covalent bonds. Given the diverse types of polymer networks and the growing utilization of reversible covalent bonds, it is anticipated that ionogels with novel properties, distinct from existing gels, will continue to emerge in the coming years. This research field remains in its infancy, and we believe that a focus on covalent design will facilitate the accelerated commercial application of ionogels.
Abbreviations
| [BF4]− | Tetrafluoroborate |
| [BMIm]+ | 1-Butyl-3-methylimidazolium |
| [BP]+ | 1-Butylpyridinium |
| [C2C1MIm]+ | 1-Ethyl-2-methyl-3-methylimidazolium |
| [DEIM]+ | 1,2-Dimethyl-3-ethoxyethyl-imidazolium |
| [EMIm]+ | 1-Ethyl-3-methylimidazolium |
| [ES]− | Ethyl sulfate |
| [OTF]− | Trifluoromethanesulfonate |
| [PF6]− | Hexafluorophosphate |
| [TFSI]− | Bis(trifluoromethylsulfonyl)imide |
| [VBMIm]+ | 1-Vinyl-3-butylimidazolium |
| AAc | Acrylic acid |
| AAm | Acrylamide |
| ACMO | Acryloyl morpholine |
| AMG | Diaminoglyoxime |
| BPO | Dibenzoyl peroxide |
| BTC | Benzene-1,3,5-tricarboxaldehyde |
| DMAAm | N,N-Dimethylacrylamide |
| DMAc | Dimethylacetamide |
| DMF | Dimethylformamide |
| DMPA | 2,2-Dimethylol propionic acid |
| DMSO | Dimethyl sulfoxide |
| EGDMA | Ethylene glycol dimethacrylate |
| FTEG | Fluorinated tetraethylene glycol |
| G3 | Triethylene glycol dimethyl ether |
| G4 | Tetraethylene glycol dimethyl ether |
| HEMA | 2-Hydroxyethyl methacrylate |
| IPDI | Isophorone diisocyanate |
| MMA | Methyl methacrylate |
| PAAm | Polyacrylamide |
| PAMPS | Poly(2-acrylamide-2-methyl propane sulfonic acid) |
| PBGAD | Poly(butylene glycol adipate)diols |
| PCL | Polycaprolactone |
| PDMAAm | Poly(N,N-dimethylacrylamide) |
| PE | Polyethylene |
| PEDOT | Poly(3,4-ethylenedioxythiophene) |
| PEG | Polyethylene glycol |
| PEGDA | Polyethylene glycol diacrylate |
| PEGDMA | Polyethylene glycol dimethacrylate |
| PEGMA | Polyethylene glycol methacrylate |
| PE-PEP | Poly(ethylene-b-ethylene-alt-propylene) |
| PHFBA | Polyhexafluorobutyl acrylate |
| PMCL | Poly(4-methyl-ε-caprolactone) |
| POCL | Poly(1,8-octanediol-co-citrate-co-caprolactone) |
| PPy | Polypyrrole |
| PU | Polyurthane |
| PVDF | Polyvinylidene difluoride |
| TBC | Benzene-1,3,5-tricarboxaldehyde |
| TEGDA | Tetraethylene glycol diacrylate |
| TEOS | Tetraethoxysilane |
| TMP | 2-Ethyl-2-(hydroxymethyl)-1,3-propanediol |
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.References
- M. Watanabe, S.-I. Yamada, K. Sanui and N. Ogata, J. Chem. Soc., Chem. Commun., 1993, 929–931 RSC.
- N. Kimizuka and T. Nakashima, Langmuir, 2001, 17, 6759–6761 CrossRef.
- S. Dai, Y. H. Ju, H. J. Gao, J. S. Lin, S. J. Pennycook and C. E. Barnes, Chem. Commun., 2000, 243–244 RSC.
- J.-B. Ducros, N. Buchtová, A. Magrez, O. Chauvet and J. L. Bideau, J. Mater. Chem., 2011, 21, 2508–2511 RSC.
- E. Kamio, T. Yasui, Y. Iida, J. P. Gong and H. Matsuyama, Adv. Mater., 2017, 29, 1704118 CrossRef.
- Md. A. B. H. Susan, T. Kaneko, A. Noda and M. Watanabe, J. Am. Chem. Soc., 2005, 127, 4976–4983 CrossRef PubMed.
- C.-C. Yan, W. Li, Z. Liu, S. Zheng, Y. Hu, Y. Zhou, J. Guo, X. Ou, Q. Li, J. Yu, L. Li, M. Yang, Q. Liu and F. Yan, Adv. Funct. Mater., 2024, 34, 2314408 CrossRef CAS.
- Z. Yu and P. Wu, Adv. Mater., 2021, 33, 2008479 CrossRef CAS.
- Y. Deng, G. Liu, A. Brûlet, G. T. M. Nguyen, D. Dudzinski, F. Vidal, C. Plesse, C. Vancaeyzeele and M.-H. Li, Adv. Funct. Mater., 2024, 2403892 CrossRef CAS.
- X. Ming, L. Shi, H. Zhu and Q. Zhang, Adv. Funct. Mater., 2020, 30, 2005079 CrossRef CAS.
- G. Tan, F. Wu, C. Zhan, J. Wang, D. Mu, J. Lu and K. Amine, Nano Lett., 2016, 16, 1960–1968 CrossRef.
- Z. Yu and P. Wu, Mater. Horiz., 2021, 8, 2057–2064 RSC.
- N. Jiang, X. Chang, D. Hu, L. Chen, Y. Wang, J. Chen and Y. Zhu, Chem. Eng. J., 2021, 424, 130418 CrossRef CAS.
- S. Hao, T. Li, X. Yang and H. Song, ACS Appl. Mater. Interfaces, 2022, 14, 2029–2037 CrossRef CAS PubMed.
- Y. Fang, H. Cheng, H. He, S. Wang, J. Li, S. Yue, L. Zhang, Z. Du and J. Ouyang, Adv. Funct. Mater., 2020, 30, 2004699 CrossRef.
- A. Noda and M. Watanabe, Electrochim. Acta, 2000, 45, 1265–1270 CrossRef.
- J. Fuller, A. C. Breda and R. T. Carlin, J. Electrochem. Soc., 1997, 144, L67–L70 CrossRef.
- C. Tiyapiboonchaiya, D. R. MacFarlane, J. Sun and M. Forsyth, Macromol. Chem. Phys., 2002, 203, 1906–1911 CrossRef.
- J. P. Gong, Y. Katsuyama, T. Kurokawa and Y. Osada, Adv. Mater., 2003, 15, 1155–1158 CrossRef.
- Y.-J. Park, J. Liang, Z. Yang and V. C. Yang, J. Controlled Release, 2001, 75, 37–44 CrossRef PubMed.
- T. Sakai, T. Matsunaga, Y. Yamamoto, C. Ito, R. Yoshida, S. Suzuki, N. Sasaki, M. Shibayama and U. Chung, Macromolecules, 2008, 41, 5379–5384 CrossRef.
- A. Bin Imran, K. Esaki, H. Gotoh, T. Seki, K. Ito, Y. Sakai and Y. Takeoka, Nat. Commun., 2014, 5, 5124 CrossRef PubMed.
- Y. Jiang, D. Zhan, M. Zhang, Y. Zhu, H. Zhong, Y. Wu, Q. Tan, X. Dong, D. Zhang and N. Hadjichristidis, Angew. Chem., Int. Ed., 2023, 62, e202310832 CrossRef.
- J. Kim, G. Zhang, M. Shi and Z. Suo, Science, 2021, 374, 212–216 CrossRef PubMed.
- F. Vashahi, M. R. Martinez, E. Dashtimoghadam, F. Fahimipour, A. N. Keith, E. A. Bersenev, D. A. Ivanov, E. B. Zhulina, P. Popryadukhin, K. Matyjaszewski, M. Vatankhah-Varnosfaderani and S. S. Sheiko, Sci. Adv., 2022, 8, eabm2469 CrossRef PubMed.
- Z. J. Wang, J. Jiang, Q. Mu, S. Maeda, T. Nakajima and J. P. Gong, J. Am. Chem. Soc., 2022, 144, 3154–3161 CrossRef.
- T. Matsuda, R. Kawakami, R. Namba, T. Nakajima and J. P. Gong, Science, 2019, 363, 504–508 CrossRef PubMed.
- Z. Wang, X. Zheng, T. Ouchi, T. B. Kouznetsova, H. K. Beech, S. Av-Ron, T. Matsuda, B. H. Bowser, S. Wang, J. A. Johnson, J. A. Kalow, B. D. Olsen, J. P. Gong, M. Rubinstein and S. L. Craig, Science, 2021, 374, 193–196 CrossRef PubMed.
- L. Li, X. Qin, H. Mei, L. Liu and S. Zheng, Eur. Polym. J., 2021, 160, 110811 CrossRef.
- J. Zhang, Z. Lei, S. Luo, Y. Jin, L. Qiu and W. Zhang, ACS Appl. Nano Mater., 2020, 3, 4845–4850 CrossRef.
- M. Chen, L. Zhou, Y. Wu, X. Zhao and Y. Zhang, ACS Macro Lett., 2019, 8, 255–260 CrossRef CAS PubMed.
- X. Zhang, S. Wang, Z. Jiang, Y. Li and X. Jing, J. Am. Chem. Soc., 2020, 142, 21852–21860 CrossRef CAS PubMed.
- Y. Zhao, Y. Huang, W. Hou, T. Zhang, Y. Zhang, Y. Wang and X. Bai, J. Polym. Sci., 2023, 61, 3266–3275 CrossRef CAS.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed.
- D. J. Fortman, J. P. Brutman, C. J. Cramer, M. A. Hillmyer and W. R. Dichtel, J. Am. Chem. Soc., 2015, 137, 14019–14022 CrossRef CAS.
- W.-X. Liu, C. Zhang, H. Zhang, N. Zhao, Z.-X. Yu and J. Xu, J. Am. Chem. Soc., 2017, 139, 8678–8684 CrossRef CAS.
- W. Denissen, G. Rivero, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. Du Prez, Adv. Funct. Mater., 2015, 25, 2451–2457 CrossRef CAS.
- P. R. Christensen, A. M. Scheuermann, K. E. Loeffler and B. A. Helms, Nat. Chem., 2019, 11, 442–448 CrossRef CAS PubMed.
- M. M. Obadia, B. P. Mudraboyina, A. Serghei, D. Montarnal and E. Drockenmuller, J. Am. Chem. Soc., 2015, 137, 6078–6083 CrossRef CAS PubMed.
- B. Hendriks, J. Waelkens, J. M. Winne and F. E. Du Prez, ACS Macro Lett., 2017, 6, 930–934 CrossRef CAS PubMed.
- Y. Nishimura, J. Chung, H. Muradyan and Z. Guan, J. Am. Chem. Soc., 2017, 139, 14881–14884 CrossRef CAS.
- Q. Li, S. Ma, S. Wang, W. Yuan, X. Xu, B. Wang, K. Huang and J. Zhu, J. Mater. Chem. A, 2019, 7, 18039–18049 RSC.
- Q. Li, S. Ma, S. Wang, Y. Liu, M. A. Taher, B. Wang, K. Huang, X. Xu, Y. Han and J. Zhu, Macromolecules, 2020, 53, 1474–1485 CrossRef CAS.
- S. Yu, S. Wu, C. Zhang, Z. Tang, Y. Luo, B. Guo and L. Zhang, ACS Macro Lett., 2020, 9, 1143–1148 CrossRef CAS PubMed.
- H. Feng, S. Ma, X. Xu, Q. Li, B. Wang, N. Lu, P. Li, S. Wang, Z. Yu and J. Zhu, Green Chem., 2021, 23, 9061–9070 RSC.
- P. G. Falireas, J.-M. Thomassin and A. Debuigne, Eur. Polym. J., 2021, 147, 110296 CrossRef CAS.
- M. Wang, J. Hu and M. D. Dickey, JACS Au, 2022, 2, 2645–2657 CrossRef CAS PubMed.
- Z. Luo, W. Li, J. Yan and J. Sun, Adv. Funct. Mater., 2022, 32, 2203988 CrossRef CAS.
- M. Wang, P. Zhang, M. Shamsi, J. L. Thelen, W. Qian, V. K. Truong, J. Ma, J. Hu and M. D. Dickey, Nat. Mater., 2022, 21, 359–365 CrossRef CAS.
- R. Tamate, K. Hashimoto, T. Horii, M. Hirasawa, X. Li, M. Shibayama and M. Watanabe, Adv. Mater., 2018, 30, 1802792 CrossRef PubMed.
- X. Zhao, X. Chen, H. Yuk, S. Lin, X. Liu and G. Parada, Chem. Rev., 2021, 121, 4309–4372 CrossRef CAS PubMed.
- H. Katono, A. Maruyama, K. Sanui, N. Ogata, T. Okano and Y. Sakurai, J. Controlled Release, 1991, 16, 215–227 CrossRef CAS.
- J. P. Gong, Soft Matter, 2010, 6, 2583–2590 RSC.
- D. Zhou, G. M. Spinks, G. G. Wallace, C. Tiyapiboonchaiya, D. R. MacFarlane, M. Forsyth and J. Sun, Electrochim. Acta, 2003, 48, 2355–2359 CrossRef CAS.
- F. Vidal, C. Plesse, D. Teyssié and C. Chevrot, Synth. Met., 2004, 142, 287–291 CrossRef CAS.
- B. H. Jones and T. P. Lodge, Chem. Mater., 2011, 23, 4824–4831 CrossRef CAS.
- W. Li, L. Li, S. Zheng, Z. Liu, X. Zou, Z. Sun, J. Guo and F. Yan, Adv. Mater., 2022, 34, 2203049 CrossRef CAS.
- W. Li, X. Wang, Z. Liu, X. Zou, Z. Shen, D. Liu, L. Li, Y. Guo and F. Yan, Nat. Mater., 2023, 23, 131–138 CrossRef PubMed.
- Y. Ohya, R. Dohi, F. Seko, Y. Nakazawa, K. Mizuguchi, K. Shinzaki, T. Yasui, H. Ogawa, S. Kato, Y. Yoshizaki, N. Murase and A. Kuzuya, Angew. Chem., Int. Ed., 2024, 63, e202317045 CrossRef CAS PubMed.
- Y. Xiao, Q. Li, X. Yao, R. Bai, W. Hong and C. Yang, Extreme Mech. Lett., 2022, 53, 101679 CrossRef.
- T. Nakajima, Y. Fukuda, T. Kurokawa, T. Sakai, U. Chung and J. P. Gong, ACS Macro Lett., 2013, 2, 518–521 CrossRef CAS.
- B. Liu, T. Yin, J. Zhu, D. Zhao, H. Yu, S. Qu and W. Yang, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2217781120 CrossRef.
- Y. H. Kim and O. W. Webster, J. Am. Chem. Soc., 1990, 112, 4592–4593 CrossRef CAS.
- Y. Jiang, D. Zhan, M. Zhang, Y. Zhu, H. Zhong, Y. Wu, Q. Tan, X. Dong, D. Zhang and N. Hadjichristidis, Angew. Chem., Int. Ed., 2023, 62, e202310832 CrossRef CAS PubMed.
- U. Stebani and G. Lattermann, Adv. Mater., 1995, 7, 578–581 CrossRef CAS.
- Z. Zhang, L. Qian, B. Zhang, C. Ma and G. Zhang, Angew. Chem., Int. Ed., 2024, e202410335 CAS.
- A. Nishimoto, K. Agehara, N. Furuya, T. Watanabe and M. Watanabe, Macromolecules, 1999, 32, 1541–1548 CrossRef CAS.
- K. Zhao, H. Song, X. Duan, Z. Wang, J. Liu and X. Ba, Polymers, 2019, 11, 444 CrossRef.
- Z. Wang, J. Zhang, J. Liu, S. Hao, H. Song and J. Zhang, ACS Appl. Mater. Interfaces, 2021, 13, 5614–5624 CrossRef CAS.
- X. Li, Y. Gao, J. Nie and F. Sun, J. Colloid Interface Sci., 2024, 678, 703–712 CrossRef.
- L.-C. Tseng, M. Kuo and R.-H. Lee, J. Polym. Res., 2016, 23, 157 CrossRef.
- H. Wang, X. Li, Q. Zeng, Z. Li, Y. Liu, J. Guan, Y. Jiang, L. Chen, Y. Cao, R. Li, A. Wang, Z. Wang and L. Zhang, Energy Storage Mater., 2024, 66, 103188 CrossRef.
- B. Song, D. Lu, A. Qin and B. Z. Tang, J. Am. Chem. Soc., 2022, 144, 1672–1680 CrossRef CAS PubMed.
- W. Xu, P. A. Ledin, V. V. Shevchenko and V. V. Tsukruk, ACS Appl. Mater. Interfaces, 2015, 7, 12570–12596 CrossRef CAS.
- H. Kamata, Y. Akagi, Y. Kayasuga-Kariya, U. Chung and T. Sakai, Science, 2014, 343, 873–875 CrossRef CAS PubMed.
- K. Hashimoto, K. Fujii, K. Nishi, T. Sakai, N. Yoshimoto, M. Morita and M. Shibayama, J. Phys. Chem. B, 2015, 119, 4795–4801 CrossRef CAS PubMed.
- X. Li, Y. Tsutsui, T. Matsunaga, M. Shibayama, U. Chung and T. Sakai, Macromolecules, 2011, 44, 3567–3571 CrossRef CAS.
- S. Nakagawa and N. Yoshie, Polym. Chem., 2022, 13, 2074–2107 RSC.
- T. Fujiyabu, N. Sakumichi, T. Katashima, C. Liu, K. Mayumi, U. Chung and T. Sakai, Sci. Adv., 2022, 8, eabk0010 CrossRef CAS.
- M. Malkoch, R. Vestberg, N. Gupta, L. Mespouille, P. Dubois, A. F. Mason, J. L. Hedrick, Q. Liao, C. W. Frank, K. Kingsbury and C. J. Hawker, Chem. Commun., 2006, 2774–2776 RSC.
- X. Li, S. Nakagawa, Y. Tsuji, N. Watanabe and M. Shibayama, Sci. Adv., 2019, 5, eaax8647 CrossRef CAS PubMed.
- Y. Masubuchi, Y. Doi, T. Ishida, N. Sakumichi, T. Sakai, K. Mayumi and T. Uneyama, Macromolecules, 2023, 56, 2217–2223 CrossRef CAS.
- K. Fujii, H. Asai, T. Ueki, T. Sakai, S. Imaizumi, U. Chung, M. Watanabe and M. Shibayama, Soft Matter, 2012, 8, 1756–1759 RSC.
- Y. He, P. G. Boswell, P. Bühlmann and T. P. Lodge, J. Phys. Chem. B, 2007, 111, 4645–4652 CrossRef CAS.
- K. Fujii, T. Makino, K. Hashimoto, T. Sakai, M. Kanakubo and M. Shibayama, Chem. Lett., 2015, 44, 17–19 CrossRef.
- S. Ishii, H. Kokubo, K. Hashimoto, S. Imaizumi and M. Watanabe, Macromolecules, 2017, 50, 2906–2915 CrossRef CAS.
- T. Ikeda, Polym. J., 2020, 52, 1129–1135 CrossRef CAS.
- S. Matsuura, M. Shibata, J. Han and K. Fujii, ACS Appl. Polym. Mater., 2020, 2, 1276–1282 CrossRef CAS.
- A. Saruwatari, R. Tamate, H. Kokubo and M. Watanabe, Chem. Commun., 2018, 54, 13371–13374 RSC.
- G. Gao, M. Hara, T. Seki and Y. Takeoka, Sci. Technol. Adv. Mater., 2024, 25, 2302795 CrossRef.
- M. Chen, Y. Gu, A. Singh, M. Zhong, A. M. Jordan, S. Biswas, L. T. J. Korley, A. C. Balazs and J. A. Johnson, ACS Cent. Sci., 2017, 3, 124–134 CrossRef CAS.
- S. Nakagawa, D. Aoki, Y. Asano and N. Yoshie, Adv. Mater., 2023, 35, 2301124 CrossRef CAS.
- P.-G. de Gennes, Phys. A, 1999, 271, 231–237 CrossRef CAS.
- A. Harada, J. Li and M. Kamachi, Macromolecules, 1993, 26, 5698–5703 CrossRef CAS.
- J. Araki, C. Zhao and K. Ito, Macromolecules, 2005, 38, 7524–7527 CrossRef CAS.
- C. Liu, H. Kadono, H. Yokoyama, K. Mayumi and K. Ito, Polymer, 2019, 181, 121782 CrossRef CAS.
- C. Liu, N. Morimoto, L. Jiang, S. Kawahara, T. Noritomi, H. Yokoyama, K. Mayumi and K. Ito, Science, 2021, 372, 1078–1081 CrossRef CAS.
- K. Ito, Polym. J., 2012, 44, 38–41 CrossRef CAS.
- C. Liu, H. Kadono, K. Mayumi, K. Kato, H. Yokoyama and K. Ito, ACS Macro Lett., 2017, 6, 1409–1413 CrossRef CAS.
- S. Wang, Y. Chen, Y. Sun, Y. Qin, H. Zhang, X. Yu and Y. Liu, Commun. Mater., 2022, 3, 1–9 CrossRef.
- Z. Xu, J. Lu, D. Lu, Y. Li, H. Lei, B. Chen, W. Li, B. Xue, Y. Cao and W. Wang, Nat. Commun., 2024, 15, 4895 CrossRef CAS PubMed.
- T. Aoyama, K. Kato and K. Urayama, ACS Macro Lett., 2022, 11, 362–367 CrossRef CAS.
- Y. Okumura and K. Ito, Adv. Mater., 2001, 13, 485–487 CrossRef CAS.
- J. Araki and K. Ito, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 6312–6323 CrossRef CAS.
- J. Araki and K. Ito, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 532–538 CrossRef CAS.
- H. Gotoh, C. Liu, A. B. Imran, M. Hara, T. Seki, K. Mayumi, K. Ito and Y. Takeoka, Sci. Adv., 2018, 4, eaat7629 CrossRef CAS.
- R. Du, T. Bao, T. Zhu, J. Zhang, X. Huang, Q. Jin, M. Xin, L. Pan, Q. Zhang and X. Jia, Adv. Funct. Mater., 2023, 33, 2212888 CrossRef CAS.
- S. Samitsu, J. Araki, T. Kataoka and K. Ito, J. Polym. Sci., Part B: Polym. Phys., 2006, 44, 1985–1994 CrossRef CAS.
- T. Moriyasu, T. Sakamoto, N. Sugihara, Y. Sasa, Y. Ota, T. Shimomura, Y. Sakai and K. Ito, Polymer, 2013, 54, 1490–1496 CrossRef CAS.
- N. Sugihara, K. Nishimura, H. Nishino, S. Kanehashi, K. Mayumi, Y. Tominaga, T. Shimomura and K. Ito, Electrochim. Acta, 2017, 229, 166–172 CrossRef CAS.
- M. Yang, J. Li, C. Wang, L. Yang, Z. Fan, W. Wang, G. Liu, L. Cheng, S. Qu, Z. Zhang, J. Zou, W. Yu, G. Gu and X. Yan, Angew. Chem., Int. Ed., 2025, e202423847 CAS.
- K. Kato, Y. Okabe, Y. Okazumi and K. Ito, Chem. Commun., 2015, 51, 16180–16183 RSC.
- T. Enoki, K. Hashimoto, T. Oda, K. Ito and K. Mayumi, Macromolecules, 2024, 57, 11498–11506 CrossRef CAS.
- W. F. M. Daniel, J. Burdyńska, M. Vatankhah-Varnoosfaderani, K. Matyjaszewski, J. Paturej, M. Rubinstein, A. V. Dobrynin and S. S. Sheiko, Nat. Mater., 2016, 15, 183–189 CrossRef CAS.
- F. Vashahi, M. R. Martinez, E. Dashtimoghadam, F. Fahimipour, A. N. Keith, E. A. Bersenev, D. A. Ivanov, E. B. Zhulina, P. Popryadukhin, K. Matyjaszewski, M. Vatankhah-Varnosfaderani and S. S. Sheiko, Sci. Adv., 2022, 8, eabm2469 CrossRef CAS PubMed.
- P. Xu, S. Challa, X. Wu, S. Wang, P. Pan and X. Liu, ChemRxiv, 2024, preprint, DOI:10.26434/chemrxiv-2024-rrgfn.
- M. A. Nosiglia, N. D. Colley, M. K. Danielson, M. S. Palmquist, A. O. Delawder, S. L. Tran, G. H. Harlan and J. C. Barnes, J. Am. Chem. Soc., 2022, 144, 9990–9996 CrossRef CAS.
- R. Bai, Z. Zhang, W. Di, X. Yang, J. Zhao, H. Ouyang, G. Liu, X. Zhang, L. Cheng, Y. Cao, W. Yu and X. Yan, J. Am. Chem. Soc., 2023, 145, 9011–9020 CrossRef CAS.
- D. Zhao, H. Wang, Z. Wei, Z. Liu, B. Zheng, Z. Zhang, X. Yan, L. He and T. Li, Angew. Chem., Int. Ed., e202416790 Search PubMed.
- X. Chu, R. Wang, H. Zhao, M. Kuang, J. Yan, B. Wang, H. Ma, M. Cui and X. Zhang, ACS Appl. Mater. Interfaces, 2022, 14, 16631–16640 CrossRef CAS.
- S. Seiffert and J. Sprakel, Chem. Soc. Rev., 2012, 41, 909–930 RSC.
- N. Zheng, Y. Xu, Q. Zhao and T. Xie, Chem. Rev., 2021, 121, 1716–1745 CrossRef CAS PubMed.
- C. J. Kloxin, T. F. Scott, B. J. Adzima and C. N. Bowman, Macromolecules, 2010, 43, 2643–2653 CrossRef CAS PubMed.
- W. Denissen, J. M. Winne and F. E. D. Prez, Chem. Sci., 2015, 7, 30–38 RSC.
- H. Huang, W. Sun, L. Sun, L. Zhang, Y. Wang, Y. Zhang, S. Gu, Z. You and M. Zhu, Proc. Natl. Acad. Sci. U. S. A., 2024, 121, e2404726121 CrossRef CAS.
- S. Samanta, S. Kim, T. Saito and A. P. Sokolov, J. Phys. Chem. B, 2021, 125, 9389–9401 CrossRef CAS PubMed.
- M. D. Stern and A. V. Tobolsky, J. Chem. Phys., 1946, 14, 93–100 CrossRef CAS.
- J. M. Craven, United States, US3435003A, 1969 Search PubMed.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 CrossRef CAS PubMed.
- L. P. Engle and K. B. Wagener, J. Macromol. Sci., Part C, 1993, 33, 239–257 CrossRef.
- Z. P. Zhang, M. Z. Rong, M. Q. Zhang and C. Yuan, Polym. Chem., 2013, 4, 4648–4654 RSC.
- S. Billiet, K. De Bruycker, F. Driessen, H. Goossens, V. Van Speybroeck, J. M. Winne and F. E. Du Prez, Nat. Chem., 2014, 6, 815–821 CrossRef CAS PubMed.
- H. Ying, Y. Zhang and J. Cheng, Nat. Commun., 2014, 5, 3218 CrossRef PubMed.
- X. Meng, Z. Xing, X. Hu, Z. Huang, T. Hu, L. Tan, F. Li and Y. Chen, Angew. Chem., Int. Ed., 2020, 59, 16602–16608 CrossRef CAS.
- B. Jin, H. Song, R. Jiang, J. Song, Q. Zhao and T. Xie, Sci. Adv., 2018, 4, eaao3865 CrossRef.
- Z. P. Zhang, M. Z. Rong and M. Q. Zhang, Adv. Funct. Mater., 2018, 28, 1706050 CrossRef.
- A. L. Koenig, K. M. Allis, J. S. Lehr and M. B. Larsen, Polym. Chem., 2025, 16, 52 RSC.
- M. Hutchby, C. E. Houlden, M. F. Haddow, S. N. G. Tyler, G. C. Lloyd-Jones and K. I. Booker-Milburn, Angew. Chem., Int. Ed., 2012, 51, 548–551 CrossRef CAS PubMed.
- B. Zhang, Z. A. Digby, J. A. Flum, P. Chakma, J. M. Saul, J. L. Sparks and D. Konkolewicz, Macromolecules, 2016, 49, 6871–6878 CrossRef CAS.
- C. Taplan, M. Guerre and F. E. Du Prez, J. Am. Chem. Soc., 2021, 143, 9140–9150 CrossRef CAS PubMed.
- P. Froimowicz, H. Frey and K. Landfester, Macromol. Rapid Commun., 2011, 32, 468–473 CrossRef CAS PubMed.
- M. Nagata and Y. Yamamoto, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2422–2433 CrossRef CAS.
- Y. Amamoto, J. Kamada, H. Otsuka, A. Takahara and K. Matyjaszewski, Angew. Chem., Int. Ed., 2011, 50, 1660–1663 CrossRef CAS.
- S. Ji, W. Cao, Y. Yu and H. Xu, Angew. Chem., Int. Ed., 2014, 53, 6781–6785 CrossRef CAS.
- Z. Tang, X. Lyu, A. Xiao, Z. Shen and X. Fan, Chem. Mater., 2018, 30, 7752–7759 CrossRef CAS.
- M. Zhang, X. Tao, R. Yu, Y. He, X. Li, X. Chen and W. Huang, J. Mater. Chem. A, 2022, 10, 12005–12015 RSC.
- T. Li, Y. Wang, S. Li, X. Liu and J. Sun, Adv. Mater., 2020, 32, 2002706 CrossRef.
- X. Fan, Y. Luo, K. Li, Y. J. Wong, C. Wang, J. C. C. Yeo, G. Yang, J. Li, X. J. Loh, Z. Li and X. Chen, Adv. Mater., 2024, 36, 2407398 CrossRef CAS PubMed.
- H. Tan, L. Sun, H. Huang, L. Zhang, R. E. Neisiany, X. Ma and Z. You, Adv. Mater., 2024, 36, 2310020 CrossRef CAS.
- O. R. Cromwell, J. Chung and Z. Guan, J. Am. Chem. Soc., 2015, 137, 6492–6495 CrossRef CAS.
- M. Delahaye, J. M. Winne and F. E. Du Prez, J. Am. Chem. Soc., 2019, 141, 15277–15287 CrossRef CAS.
- Y. Chen, H. Zhang, S. Majumdar, R. A. T. M. van Benthem, J. P. A. Heuts and R. P. Sijbesma, Macromolecules, 2021, 54, 9703–9711 CrossRef CAS.
- F. Van Lijsebetten, Y. Spiesschaert, J. M. Winne and F. E. Du Prez, J. Am. Chem. Soc., 2021, 143, 15834–15844 CrossRef CAS PubMed.
- H. Zhang, A. van Hertrooij, T. Schnitzer, Y. Chen, S. Majumdar, R. A. T. M. van Benthem, R. P. Sijbesma and J. P. A. Heuts, Macromolecules, 2023, 56, 6452–6460 CrossRef CAS PubMed.
- R. C. Osthoff, A. M. Bueche and W. T. Grubb, J. Am. Chem. Soc., 1954, 76, 4659–4663 CrossRef CAS.
- P. Chakma, Z. A. Digby, M. P. Shulman, L. R. Kuhn, C. N. Morley, J. L. Sparks and D. Konkolewicz, ACS Macro Lett., 2019, 8, 95–100 CrossRef CAS PubMed.
- A. Chao and D. Zhang, Macromolecules, 2019, 52, 495–503 CrossRef CAS.
- S. Majumdar, H. Zhang, M. Soleimani, R. A. T. M. van Benthem, J. P. A. Heuts and R. P. Sijbesma, ACS Macro Lett., 2020, 9, 1753–1758 CrossRef CAS PubMed.
- R. L. Snyder, D. J. Fortman, G. X. De Hoe, M. A. Hillmyer and W. R. Dichtel, Macromolecules, 2018, 51, 389–397 CrossRef CAS.
- Y.-X. Lu, F. Tournilhac, L. Leibler and Z. Guan, J. Am. Chem. Soc., 2012, 134, 8424–8427 CrossRef CAS PubMed.
- H. Feng, S. Wang, J. Y. C. Lim, B. Li, W. Rusli, F. Liu, N. Hadjichristidis, Z. Li and J. Zhu, Angew. Chem., Int. Ed., 2024, 63, e202400955 CrossRef CAS.
- P. Li, X. Jiang, R. Gu, H. Tian and D.-H. Qu, Angew. Chem., Int. Ed., 2024, 63, e202406708 CrossRef CAS.
- B. T. Worrell, M. K. McBride, G. B. Lyon, L. M. Cox, C. Wang, S. Mavila, C.-H. Lim, H. M. Coley, C. B. Musgrave, Y. Ding and C. N. Bowman, Nat. Commun., 2018, 9, 2804 CrossRef.
- M. I. Lawton, K. R. Tillman, H. S. Mohammed, W. Kuang, D. A. Shipp and P. T. Mather, ACS Macro Lett., 2016, 5, 203–207 CrossRef CAS.
- Z. Lei, H. Chen, C. Luo, Y. Rong, Y. Hu, Y. Jin, R. Long, K. Yu and W. Zhang, Nat. Chem., 2022, 14, 1399–1404 CrossRef CAS PubMed.
- T. Santos, D. S. Rivero, Y. Pérez-Pérez, E. Martín-Encinas, J. Pasán, A. H. Daranas and R. Carrillo, Angew. Chem., Int. Ed., 2021, 60, 18783–18791 CrossRef CAS PubMed.
- T. F. Scott, A. D. Schneider, W. D. Cook and C. N. Bowman, Science, 2005, 308, 1615–1617 CrossRef CAS.
- C. J. Kloxin and C. N. Bowman, Chem. Soc. Rev., 2013, 42, 7161–7173 RSC.
- J. M. Winne, L. Leibler and F. E. D. Prez, Polym. Chem., 2019, 10, 6091–6108 RSC.
- B. P. Mudraboyina, M. M. Obadia, I. Allaoua, R. Sood, A. Serghei and E. Drockenmuller, Chem. Mater., 2014, 26, 1720–1726 CrossRef CAS.
- M. M. Obadia, B. P. Mudraboyina, A. Serghei, D. Montarnal and E. Drockenmuller, J. Am. Chem. Soc., 2015, 137, 6078–6083 CrossRef CAS PubMed.
- Y. Oba, T. Kimura, M. Hayashi and K. Yamamoto, Macromolecules, 2022, 55, 1771–1782 CrossRef CAS.
- J. Xu, H. Wang, X. Du, X. Cheng, Z. Du and H. Wang, Chem. Eng. J., 2021, 426, 130724 CrossRef CAS.
- Y. Fu, L. Chen, F. Xu, X. Li, Y. Li and J. Sun, J. Mater. Chem. A, 2022, 10, 4695–4702 RSC.
- F. Li, G. T. M. Nguyen, C. Vancaeyzeele, F. Vidal and C. Plesse, RSC Adv., 2023, 13, 6656–6667 RSC.
- F. Li, G. T. M. Nguyen, C. Vancaeyzeele, F. Vidal and C. Plesse, Gels, 2022, 8, 381 CrossRef CAS PubMed.
- K. Bui, G. T. M. Nguyen, C. Vancaeyzeele, F. Vidal, X. Hu, C. Wan and C. Plesse, Chem. Eng. J., 2023, 474, 145533 CrossRef CAS.
- D. Hirose, S. B. W. Kusuma, S. Nomura, M. Yamaguchi, Y. Yasaka, R. Kakuchi and K. Takahashi, RSC Adv., 2019, 9, 4048–4053 RSC.
- J.-H. Shin, M.-B. Yi, T.-H. Lee and H.-J. Kim, Adv. Funct. Mater., 2022, 32, 2207329 CrossRef CAS.
- Z. Zhao, Z. Cao, Z. Wu, W. Du, X. Meng, H. Chen, Y. Wu, L. Jiang and M. Liu, Sci. Adv., 2024, 10, eadl2737 CrossRef CAS PubMed.
- J. Tang, J. Yang, H. Yang, R. Miao, R. Wen, K. Liu, J. Peng and Y. Fang, J. Mater. Chem. C, 2018, 6, 12493–12497 RSC.
- Y. Liu, D. Wang, C. Liu, Q. Hao, J. Li, J.-X. Wang, X. Chen, P. Zhong, X. Shao and J.-F. Chen, Engineering, 2024, 37, 96–104 CrossRef CAS.
- Z. Fang, H. Mu, Z. Sun, K. Zhang, A. Zhang, J. Chen, N. Zheng, Q. Zhao, X. Yang, F. Liu, J. Wu and T. Xie, Nature, 2024, 631, 783–788 CrossRef CAS PubMed.
- Y. Amamoto, H. Otsuka, A. Takahara and K. Matyjaszewski, Adv. Mater., 2012, 24, 3975–3980 CrossRef CAS.
- M. Hansen-Felby, S. U. Pedersen and K. Daasbjerg, Molecules, 2022, 27, 6292 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |