
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cyclic thioacetal carbonates for dual-stimuli degradable poly(vinyl ether)s with cleavable thioacetal and carbonate bonds evenly distributed in the main chains by cationic degenerative chain-transfer copolymerization†
Mineto
Uchiyama
 *,
Kaoru
Matoba
and
Masami
Kamigaito
*
*,
Kaoru
Matoba
and
Masami
Kamigaito
*
Department of Molecular and Macromolecular Chemistry, Graduate School of Engineering, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8603, Japan. E-mail: uchiyama@chembio.nagoya-u.ac.jp; kamigait@chembio.nagoya-u.ac.jp
First published on 17th April 2025
Abstract
We report the synthesis of dual-stimuli degradable poly(vinyl ether)s with cleavable thioacetal and carbonate bonds evenly distributed in the main chains using cationic degenerative chain-transfer (DT) copolymerization of vinyl ethers with macrocyclic thioacetal carbonates (CTAC). The 22- and 26-membered cyclic thioacetal carbonates (22-CTAC and 26-CTAC) were initially synthesized by a cationic thiol–ene reaction between divinyl ether with a carbonate bond and dithiol with or without a carbonate bond under dilution conditions. These compounds were subjected to cationic copolymerization with vinyl ethers using the HCl-adduct of isobutyl vinyl ether as an initiator and ZnCl2 as a catalyst and were consumed much faster than the vinyl ethers by ring-opening reactions despite the large ring to introduce thioacetal and carbonate bonds in the main chains of the products. The in-chain thioacetal bonds subsequently served as dormant bonds for the cationic DT polymerization of vinyl ethers and enabled the synthesis of poly(vinyl ether)s with controlled total and segmental molecular weights between the thioacetal and carbonate bonds. The orthogonal degradations were successful when acid and base catalysts were used for the thioacetal and carbonate bonds, respectively; this resulted in low-molecular-weight products with controlled molecular weights. Furthermore, multiblock copolymers were synthesized by the one-time addition of the second monomer (B) to the cationic DT polymerization of the first monomer (A) and 22-CTAC and were selectively degraded into ABA and BAB triblock copolymers with acid and base catalysts, respectively.
Introduction
The development of degradable polymer materials is urgently needed since the environmental issues caused by plastic waste have become more severe. In addition to microbial degradation in the natural environment, chemical degradation induced by external stimuli is also useful for developing sustainable polymer materials. Recently, interest in the synthesis of novel polymers with specific bonds that can be cleaved by chemical stimuli has increased.1–5 These polymers can be chemically degraded and further recycled. Alternatively, chemically degraded low-molecular-weight products can undergo biodegradation naturally, although a rational design of the polymers is needed. Furthermore, more recently, multi-stimuli degradable polymers have attracted increased interest because of their ability to degrade in response to different conditions.6–8Among various synthetic polymers, vinyl polymers have the most significant challenges for degradation because their main chains are composed of stable carbon–carbon single bonds. Several strategies have been developed to increase the degradability or recyclability of vinyl polymers.9–22 One effective direct approach is the use of heteroatom-containing cyclic monomers that can copolymerize with common vinyl monomers. These cyclic monomers can undergo ring-opening reactions to enable the incorporation of more cleavable carbon–heteroatom bonds in the main chain of the resulting copolymers.23–51 Alternatively, recent publications have revealed that even acyclic compounds with heteroatoms can be used for the incorporation of heteroatoms in the main chains via copolymerization with vinyl monomers, as reported in the direct polymerization of carbon–heteroatom double bonds such as C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S and CO in thiocarbonyl and carbonyl copmpounds52–57 and isomerization radical polymerizations of specific vinyl ethers accompanying 1,5-shift of atoms and groups.58–60 In these copolymerization approaches, however, the controlled incorporation of heteroatoms in the main chain is generally difficult except for alternating copolymerization because the comonomers are randomly copolymerized according to the monomer reactivity ratios. Controlled incorporation of carbon–heteroatom bonds is more desirable in terms of regulating the molecular weights of the degraded vinyl oligomer products; these degraded products may further undergo biodegradation.61–63
S and CO in thiocarbonyl and carbonyl copmpounds52–57 and isomerization radical polymerizations of specific vinyl ethers accompanying 1,5-shift of atoms and groups.58–60 In these copolymerization approaches, however, the controlled incorporation of heteroatoms in the main chain is generally difficult except for alternating copolymerization because the comonomers are randomly copolymerized according to the monomer reactivity ratios. Controlled incorporation of carbon–heteroatom bonds is more desirable in terms of regulating the molecular weights of the degraded vinyl oligomer products; these degraded products may further undergo biodegradation.61–63
One of the most useful methods to control the molecular weight of the vinyl polymers is “living”/controlled polymerization or reversible deactivation polymerization; here, the dormant polymer terminal functions as a reversibly cleavable bond during the polymerization to enable continuous controlled chain growth. However, dormant bonds are usually located at the polymer terminal. Recently, we developed controlled cationic polymerizations that proceeded via reversible activation of the C–S bond via degenerative chain transfer (DT) or reversible addition–fragmentation chain transfer (RAFT) of the cationic propagating species to the dormant thioester and thioacetal terminals.64–80 We also found that thioacetal bonds could be constructed as main-chain linkages of polythioacetals via cationic thiol–ene polyaddition of dithiol and divinyl ethers72,81 or cationic ring-opening polymerization of cyclic thioacetal (CTA).80 These bonds could be used as in-chain dormant bonds for the subsequent cationic DT polymerization of vinyl ethers, resulting in copolymers with sulfur atoms in the main chain. In particular, a 7-membered cyclic thioacetal (7-CTA) efficiently undergoes cationic copolymerization with vinyl ethers through a ring-opening reaction. Here, 7-CTA is consumed much faster than vinyl ethers and is able to produce mostly polythioacetals in the initial stage. The resulting thioacetal bonds in the main chain serve as in-chain dormant species for the cationic DT polymerization of the remaining vinyl ethers. Thus, vinyl ethers are schematically inserted into the main-chain thioacetal bonds to form the copolymers, in which both the whole and segmental molecular weights of the poly(vinyl ether)s are controlled via the DT mechanism. Subsequent treatment of the resulting copolymers with silver nitrate or organic acid solution cleaves the main-chain thioacetals to finally result in poly(vinyl ethers)s with low and controlled molecular weights. Thus, the thioacetal bond in CTA has multiple functions, i.e., a polymerizable group of the cyclic monomer, an in-chain dormant bond for the cationic polymerization of vinyl ethers, and a degradable bond in the resulting vinyl polymers.
In this study, we investigated the synthesis of dual-stimuli degradable poly(vinyl ether)s via the incorporation of another degradable bond in CTA and subsequent cationic DT copolymerization with vinyl ethers (Scheme 1). Herein, in addition to the acid-cleavable thioacetal bond, carbonate was chosen as the other degradable bond, and could be cleaved under basic conditions, thus enabling dual-stimuli degradation. Novel macrocyclic thioacetals with carbonate bonds were synthesized by a cationic thiol–ene reaction between divinyl ether with a carbonate bond and dithiol with or without a carbonate bond under dilution conditions. The obtained macrocyclic thioacetal carbonate was used as a comonomer and a precursor for the in-chain dormant species in the cationic copolymerization with vinyl ethers. In this case, the carbonate bond could be introduced into the main chains along with the thioacetal bond via the ring-opening reaction of the macrocyclic thioacetal carbonate. The cationic DT polymerization of vinyl ethers based on the in-chain thioacetal bonds enabled the formation of poly(vinyl ether)s with controlled segment lengths, which were linked with both thioacetal and carbonate bonds. The orthogonal degradation of the obtained copolymers was investigated using acid and base catalysts for the cleavage of thioacetal and carbonate bonds, respectively. Furthermore, the one-pot synthesis of multiblock copolymers with dual degradability was also investigated.
 | ||
| Scheme 1 Synthesis and selective degradation of poly(vinyl ether)s with periodically distributed thioacetal and carbonate bonds in main chains by cationic degenerative chain-transfer copolymerization of vinyl ethers and cyclic thioacetal carbonate. | ||
Results and discussion
Synthesis of macrocyclic thioacetal carbonate
Macrocyclic thioacetal carbonates were synthesized by a cationic thiol–ene reaction between a divinyl ether with a carbonate group and a dithiol with or without a carbonate group using benzenesulfonic acid (BSA) as an acid catalyst under dilution conditions (Scheme 2).81 | ||
| Scheme 2 Synthesis of cyclic thioacetal carbonate by cationic thiol–ene reaction between divinyl ether with carbonate linkage and dithiol. | ||
The synthesis of a 22-membered cyclic thioacetal carbonate (22-CTAC) with one carbonate and two thioacetal bonds is first described (Scheme S1†). A symmetrical divinyl ether with a carbonate group, i.e., bis[4-(vinyloxy)butyl]carbonate (DVE-1), was prepared by the reaction between 4-hydroxybutyl vinyl ether and 1,1′-carbonyldiimdazole. A dithiol, bis(2-mercaptoethyl)ether (DT-1), was commercially available. Then, the cationic thiol–ene reaction between DVE-1 and DT-1 was examined with equimolar amounts under dilution conditions ([DVE-1]0/[DT-1]0 = 25/25 mM) in the presence of a catalytic amount of BSA ([BSA]0 = 0.10 mM) in CH2Cl2 at −40 °C. Both thiol and vinyl ether were quantitatively consumed. The peak-top molecular weight (Mp) of the size-exclusion chromatography (SEC) curve of the main product was 340; this result indicated the formation of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 adduct (Fig. 1A). After purification by silica column chromatography, the SEC curve became monomodal. In addition, the 1H NMR spectrum showed characteristic peaks; these peaks were attributed to thioacetal (b) and carbonate (e) groups and were present at a 2:1 ratio (Fig. 1B). In addition, the MALDI-TOF-MS spectrum also confirmed that the main product was the desired 1:1 adduct (Fig. S3†). Thus, a 22-membered cyclic thioacetal carbonate (22-CTAC) with two thioacetal bonds and one carbonate bond was successfully synthesized.
:1 adduct (Fig. 1A). After purification by silica column chromatography, the SEC curve became monomodal. In addition, the 1H NMR spectrum showed characteristic peaks; these peaks were attributed to thioacetal (b) and carbonate (e) groups and were present at a 2:1 ratio (Fig. 1B). In addition, the MALDI-TOF-MS spectrum also confirmed that the main product was the desired 1:1 adduct (Fig. S3†). Thus, a 22-membered cyclic thioacetal carbonate (22-CTAC) with two thioacetal bonds and one carbonate bond was successfully synthesized.
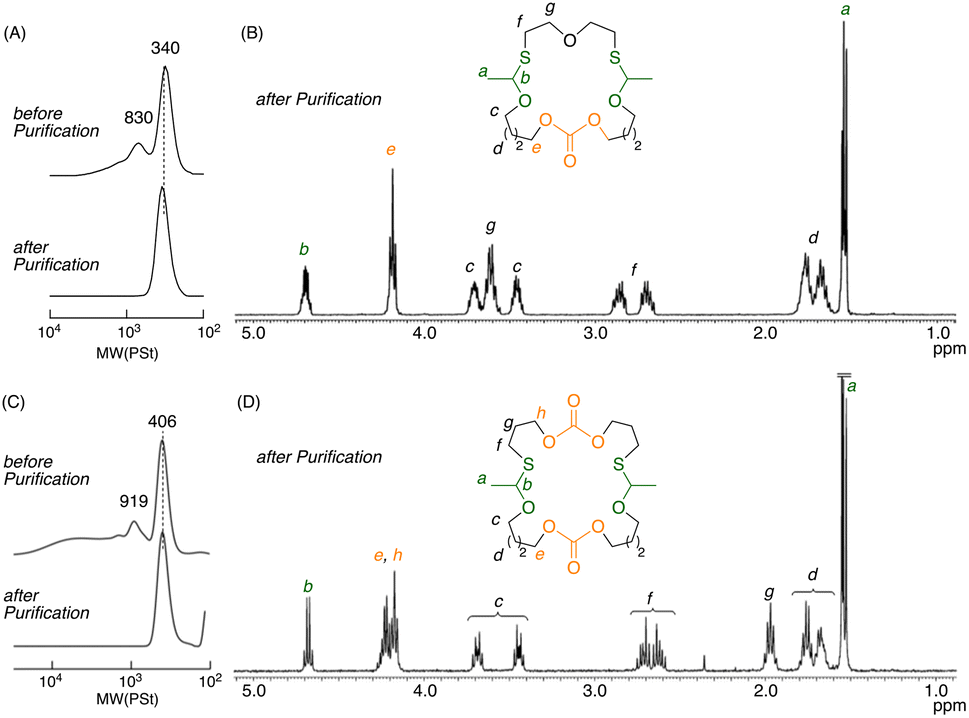 | ||
| Fig. 1 SEC curves (A and C) and 1H NMR spectra (CDCl3, r.t.) (B and D) of 22-CTAC (for A and B) and 26-CTAC (C and D) obtained by cationic thiol–ene reaction of DVE-1 and dithiols under dilute conditions: [DVE-1]0/[DT-1]0/[BSA]0 = 25/25/0.10 mM or [DVE-1]0/[DT-2]0/[BSA]0 = 50/50/0.20 mM in CH2Cl2 at −40 °C. | ||
Another cyclic thioacetal carbonate (26-CTAC) with a 26-membered ring linked by two carbonate and two thioacetal bonds was also prepared by a similar cationic thiol–ene reaction involving an equimolar mixture of DVE-1 and bis(3-mecaptopropyl)carbonate (DT-2); DT-2 was prepared from bis(3-choloropropyl)carbonate and potassium thioacetate followed by deacetylation (Scheme S2†). In this case, both divinyl ether (DVE-1) and dithiol (DT-2) possess a carbonate bond. Although slightly higher amounts of high-molecular-weight products were observed in the SEC curve of the products, the main product with an Mp of 406 was purified by column chromatography (Fig. 1C). The 1H NMR spectrum showed two kinds of carbonate peaks (e and h) and one thioacetal peak (b) with a peak area ratio of (e + h):b = 4:1 (Fig. 1D); these results indicated that this molecule had two different carbonate bonds and two identical thioacetal bonds. The formation of macrocyclic 26-membered cyclic thioacetal carbonate was also supported by the MALDI-TOF-MS spectrum (Fig. S8†).
22-Membered cyclic thioacetal carbonate for cationic DT copolymerization with ethyl vinyl ether
Cationic copolymerization of ethyl vinyl ether (EVE) and 22-CTAC was investigated with the HCl adduct of isobutyl vinyl ether (1) as an initiator and ZnCl2 as a catalyst in a similar way to the copolymerization of EVE and 7-CTA.80Fig. 2 shows representative results for the copolymerization in the mixture of CH2Cl2, toluene, and Et2O (80/10/10) at −40 °C, where the feed ratios of the monomers (EVE + 22-CTAC) to the initiator (1) and EVE to 22-CTAC were 200 + 10 and 20, respectively ([EVE]0/[22-CTAC]0/[1]0 = 4000/200/20 mM). The cyclic thioacetal carbonate was consumed more rapidly than EVE, irrespective of the large ring, and both monomers were quantitatively consumed (Fig. 2A). In the SEC curves, two peaks were present, and both shifted to the high-molecular-weight regions along with the consumption of EVE (Fig. 2B). As the polymerization proceeded, the lower-molecular-weight peak was attributed to the cyclic oligomers formed via the intramolecular DT reaction and decreased.80 The number-average molecular weight (Mn) of the main peak linearly increased with monomer conversion and was in good agreement with the theoretical values, assuming that one copolymer chain was generated from one molecule of 1 (Fig. 2B). However, the dispersities (Đ) were relatively broad during the polymerization. This was attributed to the shuffling of poly(EVE) segments among the polymer chains because the growing carbocationic species could attack the in-chain dormant thioacetal bonds to induce reversible main-chain cleavage via the DT mechanism. These results were similar to those obtained with 7-CTA, as reported previously.80 Thus, despite the larger ring size, the 22-membered macrocyclic thioacetal carbonate, i.e., 22-CTAC, successfully underwent cationic DT copolymerization with EVE. | ||
| Fig. 2 Time-conversion curve (A), Mn values (B), and SEC curves (C) of the polymers obtained in cationic DT copolymerization of EVE and 22-CTAC: [EVE]0/[22-CTAC]0/[1]0/[ZnCl2]0 = 4000/200/20/4.0 mM in CH2Cl2/n-hexane/Et2O (20/10/10) at −40 °C. | ||
Then, the final product was purified to remove the lower-molecular-weight oligomers using preparative SEC to produce the isolated main product (Mn = 17900, Đ = 2.20), and this product was then analyzed by 1H NMR spectroscopy. The characteristic peaks derived from the ring-opened structure of 22-CTAC were the methine proton of the thioacetal at 4.7 ppm (e), the methylene proton next to the sulfur atom at 2.7 ppm (f), and the methylene proton (l) next to the carbonate group at 4.1 ppm; these peaks were observed in addition to the large peaks of the poly(EVE) segments (Fig. 3A). These results indicated that the thioacetal and carbonate bonds derived from 22-CTAC were incorporated into the poly(EVE) main chains via a ring-opening reaction.
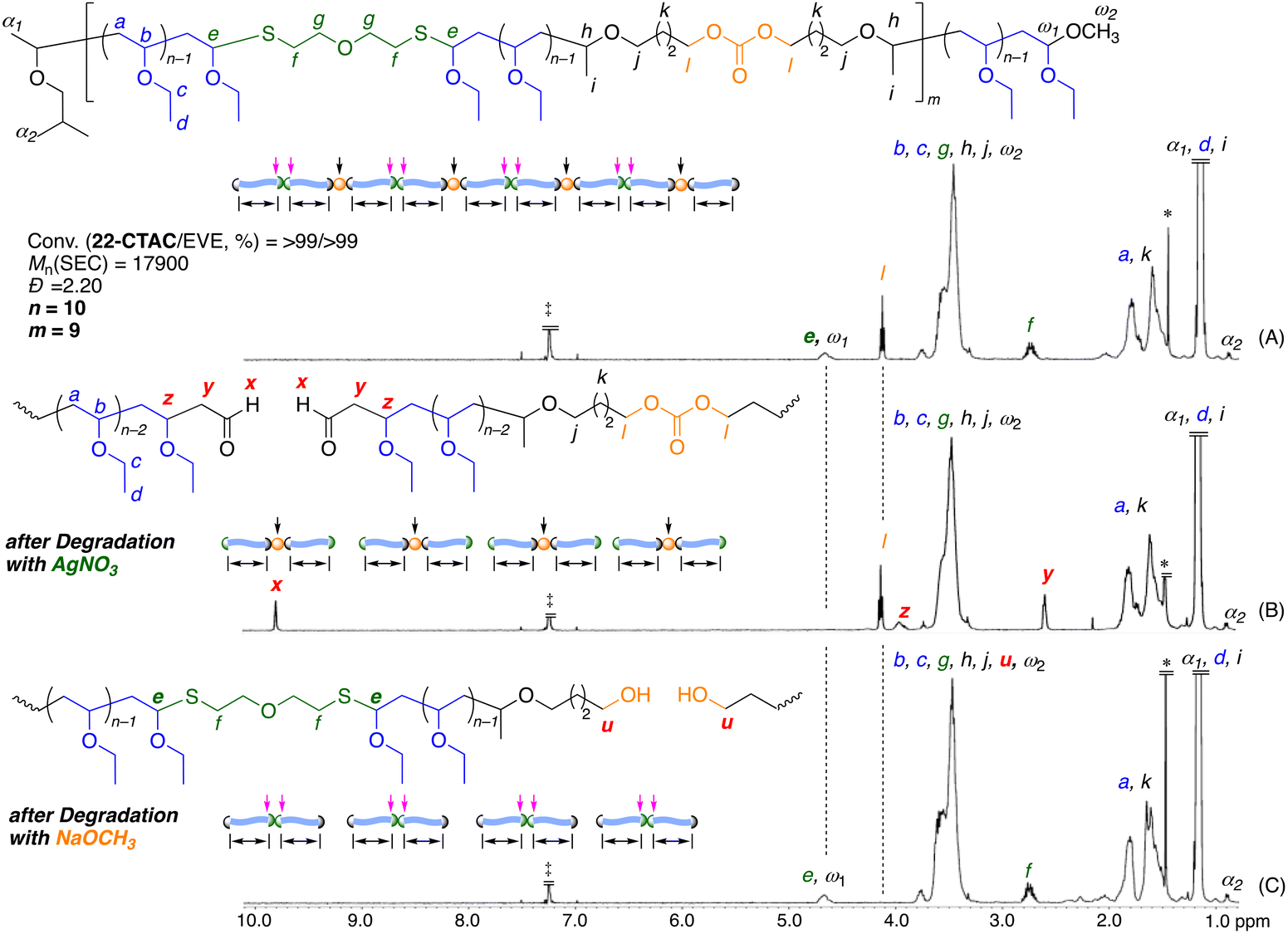 | ||
| Fig. 3 1H NMR spectra (CDCl3, 55 °C) of the obtained copolymer of EVE and 22-CTAC (A) and the polymers after degradation using an AgNO3 solution (B) and using a NaOCH3 solution (C): [thioacetal unit]0/[AgNO3]0 = 5.0/50 mM in THF/H2O at 20 °C or [carbonate unit]0/[NaOCH3]0 = 5.0/50 mM in CH3OH at 60 °C. *: H2O, ‡: CHCl3. | ||
Furthermore, additional small peaks were present and attributed to the terminal protons, such as the methyl proton (α2) of the isobutyl group at the α-chain end derived from the initiator at 0.9 ppm and the methine proton (ω1) of the acetal at the ω-chain end generated via termination reaction with methanol at 4.6 ppm; this peak overlapped with the methine proton of the main-chain thioacetal (e). The average number (2m) of the incorporated thioacetal bonds calculated from the integral ratio of f to α2 was 18. Additionally, the average number (m) of the incorporated carbonate bonds calculated from the integral ratio of l to α2 was 9; this value was in agreement with half the number of thioacetal bonds. These numbers were close to the theoretical values calculated from the feed ratio of 22-CTAC to 1 ([22-CTAC]0/[1]0 = 10); these results indicated that 22-CTAC was quantitatively incorporated into the polymer main chains. Moreover, the number-average degree (n) of poly(EVE) segments per thioacetal bond was calculated from the integral ratio of EVE units to the sum of in-chain thioacetal and terminal acetal bonds and was determined to be 10. These results indicated that the obtained copolymer was composed of 19 (= 2m + 1) poly(EVE) segments with 10 (= n) EVE units on average and contained 18 (= 2m) thioacetal bonds and 9 (= m) carbonate bonds in the main chain.
Degradation of the copolymer
The thioacetal and carbonate bonds in the polymer main chains could be selectively cleaved by different stimuli or catalysts. The selective degradation of the obtained copolymers was investigated using different catalysts. The degradation via hydrolysis of the thioacetal bonds was initially examined using an AgNO3 solution in a mixture of THF and H2O at room temperature.80,82–85 After 3 h, the SEC curve of the products shifted to the low-molecular-weight region (Fig. 4A). The Mn value decreased to 2200 and was in agreement with the calculated value (2000), assuming that one thioacetal bond formed one poly(EVE) segment. In addition, the dispersity of the degraded products decreased (Đ = 1.27). The 1H NMR spectrum of the resulting products revealed that the peak (e) derived from the thioacetal bond at 4.6 ppm completely disappeared and that a new peak (x) attributed to the aldehyde terminal was formed via hydrolysis and appeared at 9.8 ppm (Fig. 3B). Furthermore, the peaks derived from carbonate bonds remained unchanged after treatment with the AgNO3 solution. These results indicated that the thioacetal bonds were evenly distributed in the main chain of the obtained copolymers and that they were selectively cleaved by AgNO3. A similar selective cleavage of the thioacetal bonds was also examined using a common organic acid such as p-toluenesulfonic acid. The degradation was slower, but the selective degradation was supported by the SEC and 1H NMR analyses (Fig. S9 and S10†). | ||
| Fig. 4 SEC curves of the products before and after degradation with AgNO3 (A) and NaOCH3 (B), and SEC curves of the products obtained by dual degradation with the sequential use of NaOCH3 and AgNO3 (C): [thioacetal unit]0/[AgNO3]0 = 5.0/50 mM in THF/H2O at 20 °C. [Carbonate]0/[NaOCH3]0 = 5.0/50 mM in CH3OH at 60 °C. | ||
The selective degradation of the carbonate bonds was then investigated via methanolysis using a NaOCH3 solution in CH3OH at 60 °C. The SEC curve of the products obtained after 48 h shifted to the low-molecular-weight region (Mn = 2500) and became narrow (Đ = 1.23) (Fig. 4B), as in the case of AgNO3. More interestingly, peak (l) of the methylene proton adjacent to the carbonate groups completely disappeared, whereas peak (e) attributed to the thioacetal bonds remained intact (Fig. 3C). These results indicated that selective complementary degradation occurred when the base catalyst was used in place of the acid catalyst. The degradations were thus orthogonal. Additionally, kinetic comparison under these specific conditions revealed that degradation of carbonate bonds with NaOCH3 proceeded much slower than that of thioacetal bonds with AgNO3. The degradation progress with NaOCH3 was examined by SEC and 1H NMR (Fig. S11 and S12†). As shown in Fig. S12,† the Mn values of the products were measured by SEC and decreased with increasing degradation of the carbonate moiety, as measured by 1H NMR (Fig. S11†). The plots were effectively fit with the theoretical curves, assuming that the carbonate bonds were evenly incorporated into the main chains of the copolymers.
Based on these results, stepwise degradation was investigated using a base catalyst followed by an acid catalyst. The first degradation using the NaOCH3 solution resulted in a low-molecular-weight polymer with a narrow dispersity (Mn = 2600, Đ = 1.24) (Fig. 4C). The recovered polymer with thioacetal bonds was subsequently subjected to a second degradation using the AgNO3 solution. The SEC curve further shifted to the low-molecular-weight region; this resulted in a product with Mn = 1400, which was reduced by approximately half of the first degraded products. These results indicated that the thioacetal bonds were located around the middle of the first degradation products and that dual-stimuli degradable poly(EVE) with thioacetal and carbonate bonds, which were evenly distributed in the main chain, was obtained by the cationic DT copolymerization of EVE and 22-CTAC.
Tunable molecular weights before and after degradation
In this copolymerization, the overall molecular weights of the products before degradation could be controlled by the feed ratio of total monomers to initiator, whereas the molecular weights of the degraded products could be controlled by the feed ratio of vinyl ether to cyclic thioacetal.80Here, the control of the total molecular weight of the copolymer was investigated by changing the feed ratio of the total monomers to the initiator (([EVE]0 + [22-CTAC]0)/[1]0 = 100 + 5, 200 + 10, and 400 + 20), while the feed ratio of vinyl ether to cyclic thioacetal carbonate was fixed ([EVE]0/[22-CTAC]0 = 20). This enabled the synthesis of polymers with different overall molecular weights while maintaining the same molecular weights of the degraded products. In all cases, 22-CTAC was consumed much faster than EVE (Fig. S13†), resulting in copolymers (Fig. S14†). The Mn values of the products varied with the feed ratio of total monomers to initiator (Mn = 10000–40000) and effectively agreed with the theoretical values, assuming that one initiator generates one polymer chain (Fig. 5A).
 | ||
| Fig. 5 Effect of varying ([EVE]0 + [22-CTAC]0)/[1]0 with constant [EVE]0/[22-CTAC]0 on Mn values (A) and SEC curves (B and C) of the polymers obtained by cationic DT copolymerizations of EVE and 22-CTAC before and after degradation: [EVE]0/[22-CTAC]0/[1]0/[ZnCl2]0 = 4000/200/10, 20, 40/4.0 mM in CH2Cl2/n-hexane/Et2O (20/10/10) at −40 °C. [Thioacetal unit]0/[AgNO3]0 = 5.0/50 mM in THF/H2O at 20 °C. [Carbonate]0/[NaOCH3]0 = 5.0/50 mM in CH3OH at 60 °C. | ||
The selective degradation of a series of copolymers with different total molecular weights was examined similarly using AgNO3 and NaOCH3 solutions. In all cases, the SEC curves shifted to the low-molecular-weight region and became narrower (Đ = 1.2–1.3) (Fig. 5B and C). Despite the different original molecular weights and different stimuli used, the Mp and Mn after degradation were nearly the same (Mn ∼ 2000) and agreed well with theoretical values. Thus, this method enabled the synthesis of a series of dual-stimuli degradable polymers; these polymers had overall different molecular weights tunable by the varied feed ratios of monomers and initiators but could be degraded into the same molecular weights determined by the fixed feed ratio of vinyl ether to cyclic thioacetal carbonate.
Next, the control of the molecular weights of the degraded products was examined by varying the concentration of cyclic thioacetal carbonate without changing the concentrations of the vinyl ether and initiator. The feed ratio of EVE to 22-CTAC was varied from 10 to 20 and 40 ([EVE]0/[22-CTAC]0 = 10, 20, and 40), whereas that of EVE to 1 was kept constant at 200 ([EVE]0/[1]0 = 200). The consumption of 22-CTAC was much faster than that of EVE in all cases (Fig. S15†), resulting in copolymers (Fig. S16†). The SEC analysis of the products revealed that the molecular weights of the obtained copolymers were nearly constant (Mn ∼ 20000) (Fig. 6A).
 | ||
| Fig. 6 Effect of varying [EVE]0/[22-CTAC]0 with constant [EVE]0/[1]0 on Mn values and SEC curves (B and C) of the polymers obtained by cationic DT copolymerizations of EVE and 22-CTAC before and after degradation: [EVE]0/[22-CTAC]0/[1]0/[ZnCl2]0 = 4000/100, 200, 400/20/4.0 mM in CH2Cl2/n-hexane/Et2O (20/10/10) at −40 °C. [Thioacetal unit]0/[AgNO3]0 = 5.0/50 mM in THF/H2O at 20 °C. [Carbonate]0/[NaOCH3]0 = 5.0/50 mM in CH3OH at 60 °C. | ||
AgNO3 and NaOCH3 were then similarly used for selective degradation. All SEC curves of the degraded products shifted to low-molecular-weight regions with decreasing dispersity (Fig. 6B and C). Here, the shifts became more remarkable as the 22-CTAC feed increased, and they were nearly independent of the catalysts (AgNO3 and NaOCH3) used for degradation. Furthermore, the Mn values of the degraded products were close to the theoretical values in which one thioacetal bond generated one poly(EVE) segment and linearly increased with the feed ratio of EVE to 22-CTAC. Thus, another series of dual-stimuli degradable polymers, with the same molecular weights determined by the fixed feed ratio of vinyl ether to initiator, could be degraded into different molecular weights that were tunable by the varied feed ratios of vinyl ether to cyclic thioacetal carbonate.
These results indicated that dual-stimuli degradable polymers with tunable molecular weights before and after degradation could be synthesized by cationic DT copolymerization of vinyl ether and macrocyclic thioacetal carbonate, which has two thioacetal bonds and one carbonate bond.
Effect of cyclic thioacetal carbonate: 26-CTACvs.22-CTAC
The other macrocyclic thioacetal carbonate (26-CTAC) with two carbonate and two thioacetal bonds in a larger ring was similarly copolymerized with EVE to synthesize another series of dual-stimuli degradable poly(EVE). In contrast to 22-CTAC, 26-CTAC possesses another carbonate bond originating from the dithiol unit (DT-2) as the starting material in addition to the other carbonate bond originating from the divinyl ether unit (DVE-1). Consequently, the base-induced degradation of the polymers produced from 26-CTAC could reduce the molecular weight of the degraded products to half of that of those from 22-CTAC.The cationic DT copolymerization of 26-CTAC and EVE was similarly examined using the 1/ZnCl2 system. The consumption of 26-CTAC occurred more rapidly than that of EVE (Fig. 7A), as in the case of 22-CTAC. The SEC curves shifted to high-molecular-weight regions as the polymerization proceeded (Fig. 7B). The Mn of the main peak increased in direct proportion to monomer conversion and was similar to the calculated value (Fig. S17†). The 1H NMR spectrum of the main-peak products purified by preparative SEC revealed two kinds of carbonate bonds and one kind of thioacetal bond originating from 26-CTAC (Fig. S18A†). The average numbers of the incorporated thioacetal bonds (2m) and carbonate bonds (2m) calculated from the respective peak area ratios were both 16. The number-average degree (n) of poly(EVE) segments per thioacetal or carbonate bond was 10 (Fig. S18A†). These values were similar to the theoretical values determined by the feed ratios. Thus, 26-CTAC similarly underwent cationic ring-opening copolymerization with EVE to produce polymers with twice the number of carbonate bonds in the polymer chains as those obtained with 22-CTAC.
 | ||
| Fig. 7 Time-conversion curve (A) and SEC curves of the polymers obtained in cationic DT copolymerization of EVE with 26-CTAC before (B) and after (C) degradation with AgNO3 and NaOCH3: [EVE]0/[26-CTAC]0/[1]0/[ZnCl2]0 = 4000/200/20/8.0 mM in CH2Cl2/n-hexane/Et2O (20/10/10) at −40 °C. [Thioacetal unit]0/[AgNO3]0 = 5.0/50 mM in THF/H2O at 20 °C. [Carbonate]0/[NaOCH3]0 = 5.0/50 mM in CH3OH at 60 °C. | ||
The selective degradation of the thioacetal and carbonate bonds was then investigated using AgNO3 and NaOCH3 solutions, respectively. In both cases, the SEC curves of the products shifted to the low-molecular-weight region and became narrower (Fig. 7C). In particular, the Mn value after degradation with NaOCH3 was reduced to approximately half of that obtained with AgNO3. This result was consistent with the designed polymer structure and indicated that the dual-stimuli degradable poly(vinyl ether)s were rationally designed on the basis of the structure of the macrocyclic thioacetal carbonates.
Various vinyl ethers for cationic copolymerization with 22-CTAC
To adapt the dual-stimuli degradable poly(vinyl ether)s, other vinyl ethers, such as isobutyl (IBVE) and 2-methoxyethyl vinyl ether (MOVE), which are typical hydrophobic and hydrophilic monomers widely used in cationic polymerizations, were copolymerized with 22-CTAC. As in the case of EVE, IBVE and MOVE were nearly quantitatively consumed after rapid consumption of 22-CTAC (Fig. 8). In both cases, the Mn values of the obtained copolymers increased in direct proportion to monomer conversion and were similar to the theoretical values (Fig. S20 and S21†). The Mn of the final products reached 20000 in this case.
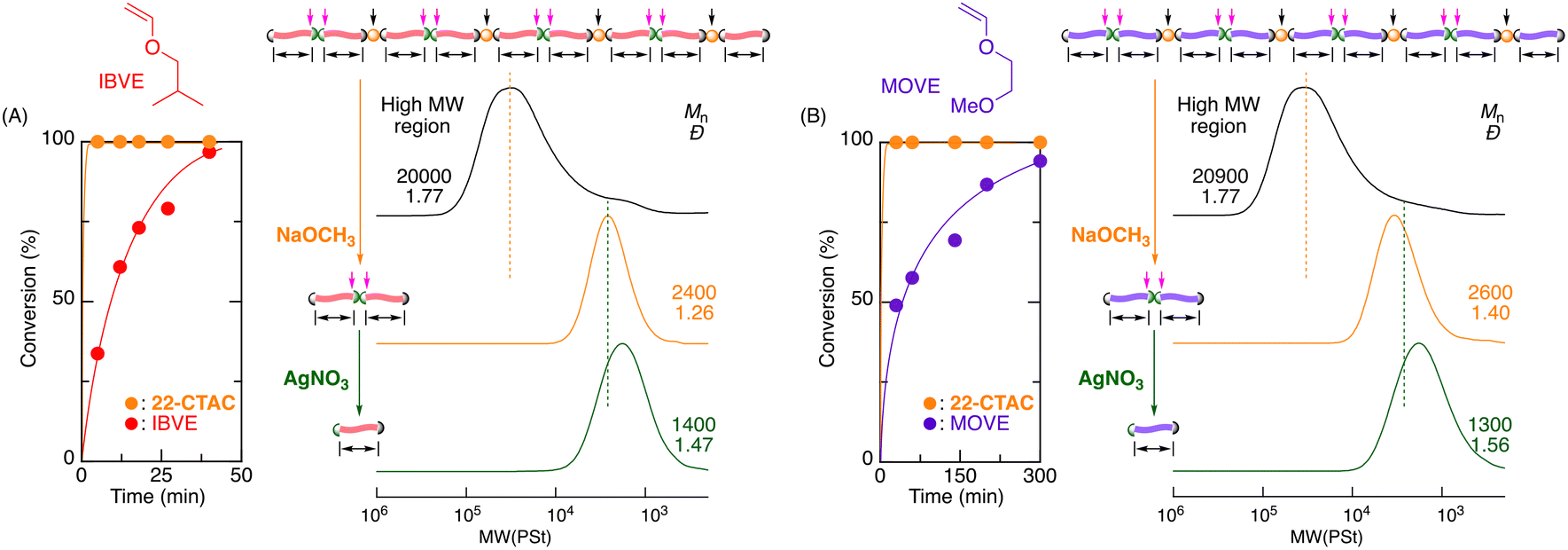 | ||
| Fig. 8 Cationic DT copolymerization of IBVE (A) and MOVE (B) with 22-CTAC and SEC curves before and after stepwise degradation: [VE]0/[22-CTAC]0/[1]0/[ZnCl2]0 = 4000/200/20/4.0 mM in CH2Cl2/n-hexane/Et2O (20/10/10) at −40 °C. [Thioacetal unit]0/[AgNO3]0 = 5.0/50 mM in THF/H2O at 20 °C. [Carbonate]0/[NaOCH3]0 = 5.0/50 mM in CH3OH at 60 °C. | ||
The obtained copolymers were also analyzed by 1H NMR. The broad large peaks were attributed to each vinyl ether unit and relatively sharp peaks derived from the ring-opened 22-CTAC units (Fig. S22 and S23†). The average numbers of thioacetal (2m) and carbonate bonds (m) in each copolymer were 18 and 9 for IBVE and 20 and 10 for MOVE, respectively. In addition, the number-average degrees (n) of the poly(IBVE) and poly(MOVE) segments were 10 and 14, respectively, and were similar to the theoretical values based on the feed ratio of the monomers to the initiator. Thus, these vinyl ethers were also successfully copolymerized with 22-CTAC.
The obtained copolymers were then subjected to stepwise degradation using NaOCH3 and AgNO3 sequentially. In both cases, the first reaction with NaOCH3 significantly shifted the SEC curves to low-molecular-weight regions, where the Mn values of the products decreased to approximately 2000 and became narrower (Fig. 8). The second reaction with AgNO3 resulted in Mn values approximately half those of the first products. Thus, cyclic thioacetal carbonates could be widely used for the synthesis of various dual-stimuli degradable poly(vinyl ether)s with different side chains.
Multiblock copolymers with dual-stimuli degradability
Finally, the one-pot synthesis of dual-stimuli degradable multiblock poly(vinyl ether)s was investigated via the only one-time addition of a second monomer (B) to the copolymerization of the first vinyl ether (A) and 22-CTAC. Herein, the main-chain thioacetal bond formed in the first copolymerization works as the in-chain dormant bond, into which the second monomer was schematically inserted to form the AB diblock copolymer segments connected by both thioacetal and carbonate bonds. Moreover, as shown in the scheme in Fig. 9, the carbonate bond should be located in the middle of the first segment (poly(A)), whereas the two thioacetal bonds should be located in the middle of the second segment (poly(B)). The selective degradation of the copolymers using AgNO3 and NaOCH3 should result in different triblock copolymers, i.e., ABA and BAB copolymers, respectively. | ||
| Fig. 9 One-pot synthesis of dual-degradable multiblock copolymers linked by thioacetal bonds and degradation with AgNO3 (A) and NaOCH3 (B) into the triblock copolymers: [IBVE]0/[EVE]add/[22-CTA]0/[1]0/[ZnCl2]0 = 2000/2000/200/20/4.0 mM in CH2Cl2/n-hexane/Et2O (20/10/10) at −40 °C. [Thioacetal unit]0/[AgNO3]0 = 5.0/50 mM in THF/H2O at 20 °C. [Carbonate]0/[NaOCH3]0 = 5.0/50 mM in CH3OH at 60 °C. | ||
To demonstrate this feasibility, IBVE was chosen as the first monomer (A) and copolymerized with 22-CTAC using the 1/ZnCl2 initiating system. After quantitative consumption of IBVE, EVE was added to the reaction solution as the second monomer (B) and was also smoothly consumed (Fig. S26†). After the addition of EVE, the SEC curves (black solid lines in Fig. 9) shifted to the high-molecular-weight regions; these results indicated that EVE was copolymerized into the main-chain thioacetal of the poly(IBVE) segments to form the block copolymer segments. The 1H NMR analysis further supported the formation of block copolymers since the peaks attributed to both EVE and IBVE units were observed in addition to the peaks derived from the ring-opened 22-CTAC unit (Fig. S27†). The number-average degrees of poly(IBVE) and poly(EVE) segments were calculated from the integral ratios of each peak and were both 8, and the average number of thioacetal groups (2m) in the main chain was 11. These results showed the formation of the multiblock copolymer, in which 11 block copolymer segments with 8 IBVE and EVE units on average were connected with thioacetal and carbonate bonds.
The selective degradations of the first polymer and the multiblock copolymer were also investigated using AgNO3 and NaOCH3 separately. In the case of AgNO3, both SEC curves of the products shifted to low-molecular-weight regions (green dashed lines) and became narrower (Fig. 9A). In particular, the SEC curve obtained from the multiblock copolymer was located in higher-molecular-weight regions than that obtained from the first polymer. Similar results were obtained for NaOCH3 (orange dashed lines). Furthermore, the Mn values of the degraded products were independent of the catalyst. The 1H NMR analysis also supported the selective degradation reactions. The peak derived from the thioacetal groups disappeared with AgNO3 along with the appearance of aldehyde terminals, whereas the peak of the methylene protons adjacent to the carbonate group disappeared with NaOCH3 (Fig. S28†). In contrast, the carbonate and thioacetal groups remained unchanged with AgNO3 and NaOCH3, respectively. Thus, the products degraded with AgNO3 were ABA triblock polymers with carbonate bonds in the middle of the B-segment, whereas those obtained with NaOCH3 were BAB triblock polymers with thioacetal bonds in the middle of the A-segment. These results indicated that the block copolymerization of different vinyl ethers using macrocyclic thioacetal carbonate enabled the one-pot synthesis of dual-degradable multiblock copolymers; ultimately, this resulted in the ABA and BAB triblock polymers by the selective orthogonal degradation.
Conclusions
The synthesis of dual-stimuli degradable poly(vinyl ether)s with thioacetal and carbonate bonds evenly distributed in the main chain was achieved by cationic DT copolymerization of vinyl ethers and the design of macrocyclic thioacetal carbonates; these were prepared by a cationic thiol–ene reaction between divinyl ether and dithiol containing carbonate bonds. The macrocyclic thioacetal carbonates underwent efficient ring-opening cationic polymerization to generate the main-chain thioacetal and carbonate bonds; here, the macrocyclic thioacetal carbonate served as dormant species for the cationic DT polymerization of the vinyl ethers to control the total and segmental molecular weights. The main-chain thioacetal and carbonate bonds were selectively degraded to low-molecular-weight products with narrow dispersities using acid and base catalysts, respectively. Furthermore, multiblock copolymers with dual degradability were synthesized by the one-time addition of the second monomer and were degraded into ABA and BAB triblock copolymers with different sequences by the orthogonal degradation. Furthermore, the carbonate bonds in macrocyclic thioacetal carbonate could be replaced with other cleavable bonds; this aspect will advance multi-degradable polymers and further contribute to the macromolecular design of sustainable polymer materials.Author contributions
M. U., K. M., and M. K. designed the experiments. M. U. and K. M. conducted the experiments. M. U., K. M., and M. K. anaylzed the results. M. U. and M. K. prepared the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a JSPS KAKENHI Grant-in-Aid for Scientific Research (C) (no. JP22K05209) for M. U. and for Scientific Research (A) (no. JP22H00333 and JP25H00894) for M. K., a JST CREST (no. JPMJCR22L2) for M. U., and a project (JPNP18016) commissioned by the New Energy and Industrial Technology Development Organization (NEDO). The authors would like to thank Hironobu Watanabe and Chihiro Homma for fruitful discussions and valuable comments on this research.References
- J. R. Jambeck, R. Geyer, C. Wilcox, T. R. Siegler, M. Perryman, A. Andrady, R. Narayan and K. L. Law, Science, 2015, 347, 768–771 CrossRef CAS PubMed.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- A. Chamas, H. Moon, J. Zheng, Y. Qiu, T. Tabassum, J. H. Jang, M. Abu-Omar, S. L. Scott and S. Suh, ACS Sustainable Chem. Eng., 2020, 8, 3494–3511 CrossRef CAS.
- G.-X. Wang, D. Huang, J.-H. Ji, C. Völker and F. R. Wurm, Adv. Sci., 2020, 8, 2001121 CrossRef PubMed.
- D. E. Fagnani, J. L. Tami, G. Copley, M. N. Clemons, Y. D. Y. L. Getzler and A. J. McNeil, ACS Macro Lett., 2021, 10, 41–53 CrossRef CAS PubMed.
- S. M. Clouthier, J. Li, J. Tanaka and W. You, Polym. Chem., 2024, 15, 17–21 RSC.
- C.-C. Chang and T. Emrick, Macromolecules, 2014, 47, 1344–1350 CrossRef CAS.
- K. Saito, F. Eisenreich and Ž. Tomović, Macromolecules, 2024, 57, 8690–8697 CAS.
- V. Delplace and J. Nicolas, Nat. Chem., 2015, 7, 771–784 CrossRef CAS PubMed.
- J. Pan, X. Ai, C. Ma and G. Zhang, Acc. Chem. Res., 2022, 55, 1586–1598 CrossRef CAS PubMed.
- J. Zheng, Z. M. Png, X. C. N. Quek, X. J. Loh and Z. Li, Green Chem., 2023, 25, 8903–8934 RSC.
- C. Lefay and Y. Guillaneuf, Prog. Polym. Sci., 2023, 147, 101764 CrossRef CAS.
- L. Wu, B. Rondon, S. Dym, W. Wang, K. Chen and J. Niu, Prog. Polym. Sci., 2023, 145, 101736 CrossRef CAS.
- M. Kamigaito, Bull. Chem. Soc. Jpn., 2024, 97, uoae069 CrossRef CAS.
- S. Tang, F. W. Seidel and K. Nozaki, Angew. Chem., Int. Ed., 2021, 60, 26506–26510 CrossRef CAS PubMed.
- M. Mizutani, K. Satoh and M. Kamigaito, J. Am. Chem. Soc., 2010, 132, 7498–7507 CrossRef CAS PubMed.
- T. Kimura, K. Kuroda, H. Kubota and M. Ouchi, ACS Macro Lett., 2021, 10, 1535–1539 CrossRef CAS PubMed.
- J. B. Garrison, R. W. Hughes and B. S. Sumerlin, ACS Macro Lett., 2022, 11, 441–446 CrossRef CAS PubMed.
- S. Yamamoto, T. Kubo and K. Satoh, J. Polym. Sci., 2022, 60, 3435–3446 CrossRef CAS.
- H. S. Wang, N. P. Truong, Z. Pei, M. L. Coote and A. Anastasaki, J. Am. Chem. Soc., 2022, 144, 4678–4684 CrossRef CAS PubMed.
- S. Oh and E. E. Stache, J. Am. Chem. Soc., 2022, 144, 5745–5749 CrossRef CAS PubMed.
- L. H. Kugelmass, C. Tagnon and E. E. Stache, J. Am. Chem. Soc., 2023, 145, 16090–16097 CrossRef CAS PubMed.
- W. J. Bailey, Polym. J., 1985, 17, 85–95 CrossRef CAS.
- F. Sanda and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 265–276 CrossRef CAS.
- A. Tardy, J. Nicolas, D. Gigmes, C. Lefay and Y. Guillaneuf, Chem. Rev., 2017, 117, 1319–1406 CrossRef CAS PubMed.
- T. Pesenti and J. Nicolas, ACS Macro Lett., 2020, 9, 1812–1835 CrossRef CAS PubMed.
- A. W. Jackson, Polym. Chem., 2020, 11, 3525–3545 RSC.
- Y. Deng, F. Mehner and J. Gaitzsch, Macromol. Rapid Commun., 2023, 44, e2200941 CrossRef PubMed.
- W. J. Bailey, Z. Ni and S.-R. Wu, Macromolecules, 1982, 15, 711–714 CrossRef CAS.
- T. Zeng, W. You, G. Chen, X. Nie, Z. Zhang, L. Xia, C. Hong, C. Chen and Y. You, iScience, 2020, 23, 100904 CrossRef CAS PubMed.
- A. Tardy, N. Gil, C. M. Plummer, C. Zhu, S. Harrisson, D. Siri, J. Nicolas, D. Gigmes, Y. Guillaneuf and C. Lefay, Polym. Chem., 2020, 11, 7159–7169 RSC.
- R. A. Evans, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1994, 27, 7935–7937 CrossRef CAS.
- R. A. Evans and E. Rizzardo, Macromolecules, 1996, 29, 6983–6989 CrossRef CAS.
- J. M. Paulusse, R. J. Amir, R. A. Evans and C. J. Hawker, J. Am. Chem. Soc., 2009, 131, 9805–9812 CrossRef CAS PubMed.
- H. Huang, B. Sun, Y. Huang and J. Niu, J. Am. Chem. Soc., 2018, 140, 10402–10406 CrossRef CAS PubMed.
- N. M. Bingham and P. J. Roth, Chem. Commun., 2019, 55, 55–58 RSC.
- R. A. Smith, G. Fu, O. McAteer, M. Xu and W. R. Gutekunst, J. Am. Chem. Soc., 2019, 141, 1446–1451 CrossRef CAS PubMed.
- G. R. Kiel, D. J. Lundberg, E. Prince, K. E. L. Husted, A. M. Johnson, V. Lensch, S. Li, P. Shieh and J. A. Johnson, J. Am. Chem. Soc., 2022, 144, 12979–12988 CrossRef CAS PubMed.
- O. Ivanchenko, S. Mazières, S. Harrisson and M. Destarac, Polym. Chem., 2022, 13, 5525–5529 RSC.
- R. Kamiki, T. Kubo and K. Satoh, Macromol. Rapid Commun., 2023, 44, e2200537 CrossRef PubMed.
- M. Pięta, V. B. Purohit, P. Paneth, J. Pietrasik, L. Li and C. M. Plummer, Polym. Chem., 2023, 14, 3872–3880 RSC.
- A. Kazama and Y. Kohsaka, Polym. Chem., 2019, 10, 2764–2768 RSC.
- X. Y. Oh, Y. Ge and A. Goto, Chem. Sci., 2021, 12, 13546–13556 RSC.
- A. Kanazawa, S. Kanaoka and S. Aoshima, J. Am. Chem. Soc., 2013, 135, 9330–9333 CrossRef CAS PubMed.
- T. Shirouchi, A. Kanazawa, S. Kanaoka and S. Aoshima, Macromolecules, 2016, 49, 7184–7195 CrossRef CAS.
- A. Kanazawa and S. Aoshima, Macromolecules, 2020, 53, 5255–5265 CrossRef CAS.
- K. Maruyama, A. Kanazawa and S. Aoshima, Macromolecules, 2022, 55, 4034–4045 CrossRef CAS.
- A. Katto, S. Aoshima and A. Kanazawa, Macromolecules, 2024, 57, 6255–6266 CrossRef CAS.
- T. Shen, K. Chen, Y. Chen and J. Ling, Macromol. Rapid Commun., 2023, 44, e2300099 CrossRef PubMed.
- Y. Takahashi, A. Kanazawa and S. Aoshima, Macromolecules, 2023, 56, 4198–4207 CrossRef CAS.
- A. E. Neitzel, L. Barreda, J. T. Trotta, G. W. Fahnhorst, T. J. Haversang, T. R. Hoye, B. P. Fors and M. A. Hillmyer, Polym. Chem., 2019, 10, 4573–4583 RSC.
- H. Watanabe and M. Kamigaito, J. Am. Chem. Soc., 2023, 145, 10948–10953 CrossRef CAS PubMed.
- O. Ivanchenko and M. Destarac, ACS Macro Lett., 2024, 13, 47–51 CrossRef CAS PubMed.
- Y. Ishido, R. Aburaki, S. Kanaoka and S. Aoshima, Macromolecules, 2010, 43, 3141–3144 CrossRef CAS.
- Y. Ishido, A. Kanazawa, S. Kanaoka and S. Aoshima, Macromolecules, 2012, 45, 4060–4068 CrossRef CAS.
- M. Kawamura, A. Kanazawa, S. Kanaoka and S. Aoshima, Polym. Chem., 2015, 6, 4102–4108 RSC.
- T. Nara, A. Kanazawa and S. Aoshima, Macromolecules, 2022, 55, 6852–6859 CrossRef CAS.
- K. Kuroda and M. Ouchi, Angew. Chem., Int. Ed., 2024, 63, e202316875 CrossRef CAS PubMed.
- M. Uchiyama, M. Imai and M. Kamigaito, Polym. J., 2024, 56, 359–368 CrossRef CAS.
- Y. Chen, C. Yue, B. An, S. Liu, L. Zhou, C.-L. Ji and Y. Li, Macromolecules, 2024, 57, 4918–4925 CrossRef CAS.
- F. Kawai, Appl. Microbiol. Biotechnol., 1993, 39, 382–385 CrossRef CAS.
- T. Hayashi, M. Mukouyama, K. Sakano and Y. Tani, Appl. Environ. Microbiol., 1993, 59, 1555–1559 CrossRef CAS PubMed.
- S. M. Barbon, M. C. D. Carter, L. Yin, C. M. Whaley, V. C. Albright and R. E. Tecklenburg, Macromol. Rapid Commun., 2022, 43, e2100773 CrossRef PubMed.
- M. Uchiyama, K. Satoh and M. Kamigaito, Angew. Chem., Int. Ed., 2015, 54, 1924–1928 CrossRef CAS PubMed.
- M. Uchiyama, K. Satoh and M. Kamigaito, Macromolecules, 2015, 48, 5533–5542 CrossRef CAS.
- M. Uchiyama, K. Satoh and M. Kamigaito, Polym. Chem., 2016, 7, 1387–1396 RSC.
- M. Kamigaito, K. Satoh and M. Uchiyama, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 243–254 CrossRef CAS.
- M. Kamigaito and M. Sawamoto, Macromolecules, 2020, 53, 6749–6753 CrossRef CAS.
- M. Uchiyama, K. Satoh and M. Kamigaito, in RAFT Polymerization: Methods, Synthesis, and Applications, ed. G. Moad and E. Rizzardo, Wiley-VCH GmbH, Weinheim, 2021, pp. 1171–1194 Search PubMed.
- M. Uchiyama, K. Satoh and M. Kamigaito, Prog. Polym. Sci., 2022, 124, 101485 CrossRef CAS.
- M. Uchiyama, M. Sakaguchi, K. Satoh and M. Kamigaito, Chin. J. Polym. Sci., 2019, 37, 851–857 CrossRef CAS.
- M. Uchiyama, M. Osumi, K. Satoh and M. Kamigaito, Macromol. Rapid Commun., 2021, 42, 2100192 CrossRef CAS PubMed.
- M. Matsuda, M. Uchiyama, Y. Itabashi, K. Ohkubo and M. Kamigaito, Polym. Chem., 2022, 13, 1031–1039 RSC.
- M. Uchiyama, K. Satoh and M. Kamigaito, ACS Macro Lett., 2016, 5, 1157–1161 CrossRef CAS PubMed.
- M. Uchiyama, K. Satoh and M. Kamigaito, Giant, 2021, 5, 100047 CrossRef CAS.
- M. Uchiyama, D. Watanabe, Y. Tanaka, K. Satoh and M. Kamigaito, J. Am. Chem. Soc., 2022, 144, 10429–10437 CrossRef CAS PubMed.
- R. J. Sifri, A. J. Kennedy and B. P. Fors, Polym. Chem., 2020, 11, 6499–6504 RSC.
- P. C. Knutson, A. J. Teator, T. P. Varner, C. T. Kozuszek, P. E. Jacky and F. A. Leibfarth, J. Am. Chem. Soc., 2021, 143, 16388–16393 CrossRef CAS PubMed.
- Z. Yang, W. Xiao, X. Zhang and S. Liao, Polym. Chem., 2022, 13, 2776–2781 RSC.
- M. Uchiyama, Y. Murakami, K. Satoh and M. Kamigaito, Angew. Chem., Int. Ed., 2023, 62, e202215021 CrossRef CAS PubMed.
- M. Uchiyama, M. Osumi, K. Satoh and M. Kamigaito, Angew. Chem., Int. Ed., 2020, 59, 6832–6838 CrossRef CAS PubMed.
- T. H. Fife and L. K. Jao, J. Am. Chem. Soc., 1969, 91, 4217–4220 CrossRef CAS.
- J. L. Jensen and W. P. Jencks, J. Am. Chem. Soc., 1979, 101, 1476–1488 CrossRef CAS.
- D. P. N. Satchell, Chem. Soc. Rev., 1977, 6, 345–371 RSC.
- D. P. N. Satchell and R. S. Satchell, Chem. Soc. Rev., 1990, 19, 55–81 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed information on materials, measurements, polymerization and degradation procedures, and additional polymer characterization data (SEC and NMR). See DOI: https://doi.org/10.1039/d5py00054h |
| This journal is © The Royal Society of Chemistry 2025 |