
Chemically crosslinked yet reprocessable epoxidized natural rubber via thermo-activated disulfide rearrangements†
L.
Imbernon
,
E. K.
Oikonomou
,
S.
Norvez
* and
L.
Leibler
Matière Molle et Chimie, ESPCI ParisTech – CNRS, UMR-7167, PSL Research University, 10 rue Vauquelin, 75005 Paris, France. E-mail: sophie.norvez@espci.fr
First published on 30th April 2015
Abstract
The thermo-activated reprocessing ability of a chemically crosslinked epoxidized natural rubber (ENR) is investigated. Disulfide groups are introduced into the rubber using dithiodibutyric acid (DTDB) as a crosslinker. DTDB-cured ENR presents strong elastomeric properties due to the high molecular weight of the rubber and efficient crosslinking. The material behaves like a standard natural rubber until 100 °C and becomes reprocessable above 150 °C, owing to disulfide rearrangements. After grinding to a fine powder, the crosslinked rubber can be reprocessed and recovers most of its initial mechanical properties. The heat-triggered disulfide rearrangements are characterized in terms of stress relaxation, creep/recovery, adhesion tests, and swelling experiments. Comparison is made with ENR samples crosslinked by permanent links.
Introduction
The rubber industry faces a major recycling problem due to the irreversibility of the crosslinked networks. Indeed, to exhibit good mechanical properties such as elasticity and high toughness as well as solvent resistance, rubbers must be chemically crosslinked. Afterwards however, the chemical network prevents the reshaping or recycling of rubbery objects. This irreversibility problem common to all thermoset polymers draws a lot of attention from the chemistry community. Covalently adaptable networks exhibiting reversible covalent chemistry appear to be a possible way to overcome this issue.1–5 However, these networks can potentially be depolymerized under certain conditions (for example, heat). Vitrimers, recently introduced by Leibler and co-workers, are a new class of polymers.6 The novelty comes from the fact that vitrimers are fully processable but still insoluble materials. This behaviour is essentially different from those of two hitherto identified classes of polymers: thermoplastics, which are malleable and soluble, and thermosets (like rubbers), which are insoluble and cannot be reshaped once synthesized. Vitrimers consist of a network containing exchangeable bonds. Exchange reactions like transesterification,6–8 olefin metathesis,9 or vinylogous transamination10 are used instead of reversible bonds. The number of crosslinking points and the average functionality of the network remain constant, making vitrimers fully malleable and mendable but still insoluble materials.6–8 Shape is fixed by cooling, which slows down exchange reactions and quenches the topology of the network. The vitrimer transition is analogous to a glass transition but the topology of the network rather than the density of the material is quenched at the transition.Around 1840, Goodyear and Hancock independently reinvented rubber by introducing the vulcanization process, involving combination with sulphur and heating. Vulcanization is thus an old industrial process, but much of its chemistry still remains unclear. Nevertheless, there is general agreement that the outstanding mechanical properties, in particular fatigue resistance, of sulphur-vulcanized rubbers are associated with polysulfide rearrangements. Very few papers, mostly directed towards industrial applications, have reported the recycling of sulphur-vulcanized NR by compression moulding of pulverized rubber waste11–13 but no mechanistic studies were described. Model systems containing disulfide bonds have attracted considerable interest recently to produce self-healing materials under various stimuli like heat, light or redox conditions.14–24 Only a limited number of studies however have focused on chemically crosslinked solvent-free polymer networks. Amamoto et al. showed the complete self-healing of a crosslinked polyurethane by exposure to visible light at room temperature.20 The network contained thiuram disulfide moieties in the main chain which permitted the reshuffling of the bonds. Recently, the Odriozola group reported a synthetic polyurethane that self-healed without external stimuli via rearrangement of aromatic disulfide bonds.22,23 Self-healing occurred at room temperature, partly due (50%) to the presence of hydrogen bonds. In the meantime, Lei et al. showed that tributylphosphine catalyzed the self-healing of a polysulfide network at room temperature.24 Heat was also used as a stimulus as shown by Klumperman, Goossens and co-workers.15 Their disulfide-based epoxy resin produced a healable thermoset system that regained 100% of the original mechanical properties after healing for 1 hour at 60 °C. The self-healing was mostly attributed to thiol–disulfide reversible reshuffling.16 Yet, all these systems present relatively weak elastomeric properties. At the rupture point, the strain is usually limited to 200% and the stress inferior to 1 MPa, where a standard industrial rubber may be expected to reach at least 500% elongation and 10 MPa stress before reinforcement by fillers.25 Using the disulfide reversible chemistry in a natural rubber presenting intrinsic strong elastomeric properties could combine reprocessing ability and good mechanical behaviour.
Widely used in the rubber industry, Epoxidized Natural Rubber (ENR) is a unique elastomer obtained by epoxidation of natural rubber harvested from Hevea trees. ENR mechanical properties are very similar to those of NR up to 50 mol% epoxidation.25 Owing to the polar nature of the oxirane groups, ENR demonstrates interesting additional properties, such as low gas permeability26 or oil resistance,27 as well as enhanced compatibility with polymers bearing polar groups.28,29 Self-healing properties after ballistic damage were reported in a low molecular weight ENR slightly crosslinked by dicumylperoxide.30 The self-healing was attributed to the small chain inter-diffusion process. Self-healing properties decreased and rapidly disappeared as the crosslinking density increased, since the crosslinking hindered the diffusion process. Here we investigate reprocessing properties in a highly crosslinked high molecular weight ENR exhibiting strong elastomeric properties.
Our group recently showed that ENR can be efficiently crosslinked by dicarboxylic acids in the presence of 1,2-dimethylimidazole acting as an accelerator.31–33 The crosslinking reaction of this bio-based polymer relies on the opening of the epoxy rings by the acid functions, resulting in the formation of β-hydroxy esters along the main chains.34 This new type of crosslinking competes efficiently with conventional vulcanizations using sulphur or peroxides.35 It also offers the opportunity to introduce different chemical functions in the rubber by way of the carboxylic diacid crosslinker. In this work, a disulfide function was incorporated into ENR using dithiodibutyric acid (DTDB) as the crosslinker (Fig. 1). Thermo-activated disulfide rearrangements in the crosslinked rubber are studied by stress-relaxation and creep/recovery experiments. Comparison with ENR crosslinked by an aliphatic diacid shows the importance of disulfide bonds in the relaxation process, which allows partial recycling of the rubber.
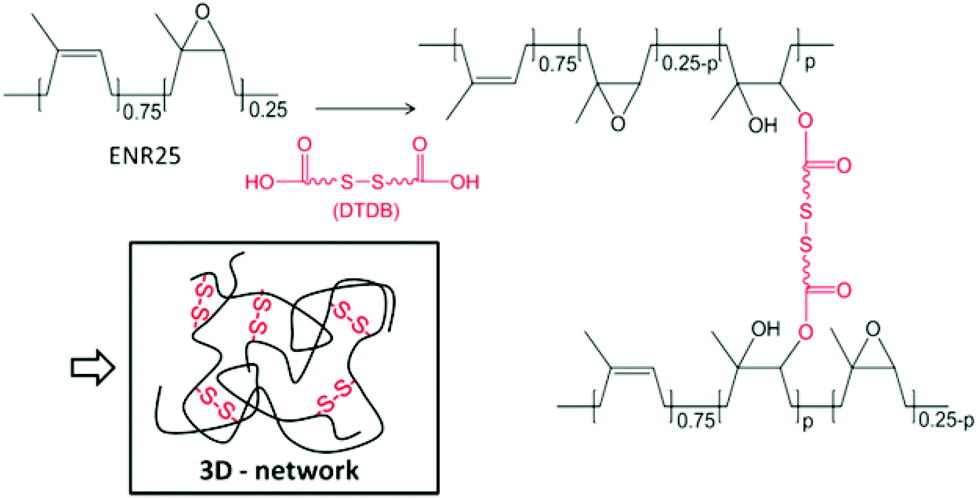 | ||
| Fig. 1 Crosslinking of ENR25 (25 mol% epoxidized): schematic representation of the disulfide containing network. The ratio of the epoxy function per diacid molecule is fixed at 25 to provide the network with optimal mechanical properties.31 | ||
Experimental section
Materials
ENR25, containing 25 mol% epoxy groups, and kaolin were kindly provided by Hutchinson SA Research Center, France. Dithiodibutyric acid (DTDB, 95%) and 1,2-dimethylimidazole (DMI, 98%) were obtained from Sigma-Aldrich and dodecanedioic acid (DA, 99%) was purchased from Acros.Blend preparation
Mixtures of ENR25, diacids, and DMI were prepared in a Haake Polydrive mixer at 40 °C for about 12 min, following a protocol already described.31 The amount of diacid was fixed to one molecule of diacid per 100 monomers, that is to say, one per 25 epoxy groups for ENR25. DMI, a liquid under the mixing conditions (mp ≈ 38 °C), was premixed with kaolin (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, w/w), an inert filler, to facilitate its incorporation into the rubber. After blending, samples were cured in a CARVER press under 8 tons pressure at 180 °C for an appropriate time (time determined by rheology experiment, about 40 min in both cases).
:2, w/w), an inert filler, to facilitate its incorporation into the rubber. After blending, samples were cured in a CARVER press under 8 tons pressure at 180 °C for an appropriate time (time determined by rheology experiment, about 40 min in both cases).
Crosslinking process
The cross-linking process was followed using an Anton Paar MCR 501 rheometer, in plate–plate geometry (25 mm diameter, 1 Hz frequency, 0.1% strain). Samples of 26 mm diameter were cut into 1.5 mm thick uncured sheets pressed at 80 °C. To ensure good contact between the rubber disc and the plates, an axial force of 10 N was first applied to the sample for 5 min before the experiment. The axial force was then maintained at 2 N by adjusting the gap during the experiment. Under these conditions, results of the measurements were comparable with those obtained in a Moving Die Rheometer.31 The temperature was quickly raised from 30 to 180 °C, and then the sample was left under oscillatory shear at 180 °C for curing.Stress relaxation
After in situ curing, the samples were analyzed by stress relaxation experiments in an Anton Paar rheometer. The temperature was fixed at 180 °C and a 1% strain step was applied. The resulting relaxation modulus was monitored over time.Reprocessing procedure
A piece of crosslinked rubber was reduced to a fine powder using a Fritsch Pulverisette 14. Liquid nitrogen was used to process below the Tg of the sample and facilitate grinding. The obtained powder was then heated at 180 °C for 40 min in a Carver press under 8 tons pressure. Dumbbells for tensile tests were punched out from the cured material. The same experiment was conducted on DTDB- and DA-cured ENR (Fig. S1a in ESI†).Strain recovery profile
Strain-recovery profiles were recorded using a DMA Q800 series TA Instrument in tension-film mode. Rectangular samples (5 × 20 × 1.6 mm) were cut in a film of cured ENR. At a fixed temperature, the force applied to the sample was alternatively fixed to 0.5 and 0 N. In a typical simple test a 0.5 N force was applied for 300 min before a 300 min recovery period (0 N). For cyclic creep/recovery, periods of 15 min were chosen. In each case, the corresponding strain profile was recorded over time.Adhesion measurements
Lap-shear geometry was adopted for adhesion measurements. Rectangular samples (5 × 100 × 1.5 mm) were cut in a cured film and superimposed on 20 mm length. This lap joint was then put under pressure with a controlled 30% deformation in a home-made setup and left in a vacuum oven at 180 °C for different adhesion times. The resulting lap shear specimen was tested on an Instron machine with a crosshead speed of 100 mm min−1, at room temperature, with no humidity control, until complete peeling or break. Rectangular samples of the same geometry but without an overlapping region were tested under the same conditions to determine the Young's modulus E of the material. E was taken as the slope of the stress/strain curve at low strains and was used in the calculation of adhesion energies.Tensile tests
The stress–strain behaviour was studied using an Instron machine with a crosshead speed of 500 mm min−1, at room temperature, with no humidity control, until break. Dumbbells of 10 mm effective length and 2 mm width were cut into a 1.5 mm-thick cured rubber sheet (about 2 mm for reprocessed samples) (Fig. S1b and c†). The strain was followed using a video extensometer. At least five samples (see Fig. S2†) were tested for each material. To calculate the ultimate tensile properties, the average of the stress and strain at break on all tested specimens was taken.Swelling experiments
Toluene was used as a good solvent of ENR for the simple swelling experiments.The swelling ratio Q is defined as

For the reductive swelling experiments, tetrahydrofuran (THF) was preferred to toluene because of its miscibility with water. The samples (1.5 mm thickness discs of 5 mm diameter) were immersed in THF and their weight evolution was followed over time. When the equilibrium swelling was reached, a small excess of tri-n-butylphosphine (TBP) and a few drops of water were added under magnetic stirring.
Gel permeation chromatography
Molecular weights and dispersities were determined using SEC, with a Malvern GPC1000 pump and a Viscotek TDA 305 triple detection array with three thermostatically controlled columns (LT5000L). Tetrahydrofuran (THF) was used as the eluent at 35 °C and at a flow rate of 1 mL min−1. The apparatus was used in a single detection mode and the apparent molecular weights (Mn and Mw) and dispersities (Mw/Mn) were obtained with polystyrene conventional calibration.Nuclear magnetic resonance spectroscopy
1H NMR spectra were recorded in CDCl3 using a Bruker Avance 400 spectrometer (400 MHz).Results and discussion
DTDB is an efficient crosslinker of ENR, comparable with DA
Crosslinking of ENR by DTDB is followed in a rheometer (Fig. 2). Comparison is made with dodecanedioic acid (DA), an aliphatic crosslinker which contains no disulfide bond.31,32 After a fast heating ramp from 25 to 180 °C, the temperature is maintained at 180 °C for crosslinking. During the temperature ramp, the elastic modulus G′ first decreases due to softening of the rubber. At 180 °C, the sharp increase of G′ shows that the crosslinking reaction takes place. Kinetics of crosslinking are very similar for both materials, as indicated by the identical G′ initial slopes. A swelling experiment in toluene indicated that after curing for 40 min at 180 °C, both networks have similar crosslinking densities (the swelling ratio Q is 3.0 ± 0.1 for DA-cured and 3.3 ± 0.2 for DTDB-cured samples). The crosslinking process in the case of DTDB is thus believed to be similar to that observed with aliphatic diacid containing no disulfide bonds: diacids open the epoxy groups to create β-hydroxyesters along the main chains.34 If this was strictly the case, the network should be the one drawn in Fig. 1 and it should be possible to fully solubilize the rubber by selectively breaking disulfide bonds. | ||
| Fig. 2 Kinetic study of the curing at 180 °C in the rheometer. ENR25 + DTDB is compared with the reference material (ENR25 + DA). | ||
Two important features arise from this rheological experiment (Fig. 2): the crosslinking reaction is complete in about half an hour at 180 °C, and the resulting material exhibits a shear modulus in the range of 1 MPa, which ranks our system among standard unfilled rubbers used in industry. To test the presence of disulfide bonds after the curing process at 180 °C, reductive swelling experiments were conducted in THF. A piece of rubber was swollen in the good solvent until equilibrium and then a reductive agent was added. Trialkylphosphines, R3P, are known to quantitatively reduce organic disulfides into thiols in the presence of water (eqn (1)).
| R3P + R′SSR′ + H2O → R3P=O + 2R′SH | (1) |
After equilibrium swelling in pure THF, an excess of tri-n-butylphosphine (TBP) and water (to the theoretical number of disulfide bonds) was added and the additional swelling (if any) measured (Fig. 3). After the addition of the reductive agent, rubber behaves very differently as it contains disulfides or not. The swelling ratio of a DA-cured network remains stable whereas the DTDB-cured network swells a lot, due to the reduction of disulfide crosslinks into separate thiols. We checked that when no TBP was added, the DTDB-cured network behaved exactly like the reference material. This confirms that disulfide bonds are still present after crosslinking at 180 °C. The very large swelling of the disulfide-containing network weakens the material a lot. Under magnetic stirring, the piece of rubber finally dissolves. Gel permeation chromatography and 1H-NMR indicated that the material obtained after swelling was degraded (chains much shorter than in the initial ENR, see Fig. S4†), preventing quantitative weight measurements and estimation of the permanent/reversible bonds ratio in the system. However, it is very likely that a few permanent bonds are created during the curing process. When increasing the temperature, disulfide bonds can break either into radicals or into thiols that can react with another disulfide bond (reversible covalent chemistry). But thiyl radicals can also react with the double bonds of ENR (as for classical sulphur vulcanization of NR). This kind of side reaction creates permanent crosslinks (monosulfide bonds). The high temperature (180 °C) necessary for crosslinking ENR by dicarboxylic acids favours these side reactions. Thus, after curing, we likely obtain a dual network mainly composed of disulfide crosslinks, but which most probably also contains a few permanent crosslinks.
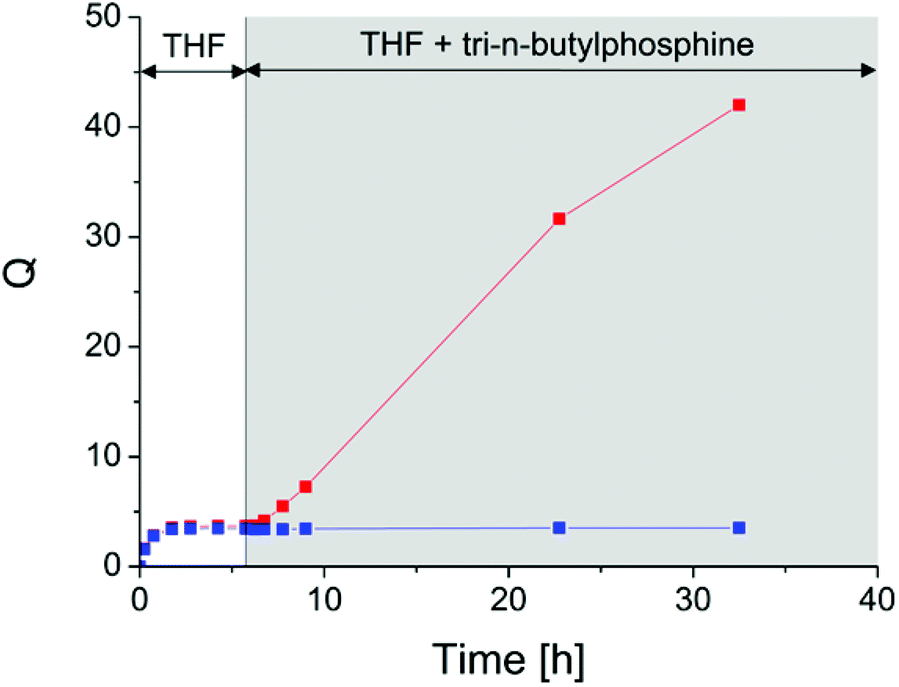 | ||
| Fig. 3 Reductive swelling experiment (red: DTDB-cured sample, blue: DA-cured sample). The reductive agent was introduced after 6 hour equilibration in pure THF. | ||
The presence of disulfide bonds enables network rearrangements
Stress relaxation experiments were conducted at high temperature after in situ curing inside the rheometer. Relaxation modulus versus time is plotted in Fig. 4. The relaxation of ENR crosslinked with dodecanedioic acid (DA) is presented for comparison. In the latter system, the network has no possibility of rearrangement as it does not contain disulfide bonds. In Fig. 4a, the sample containing disulfide bonds exhibits a substantial relaxation of stress whereas the control material shows a mostly flat curve. The in situ curing of the sample prevents any slipping effect during stress relaxation experiments. The slight relaxation observed in the control material is likely due to the diffusion of dangling chains.36 Another possibility could be ester linkages undergoing transesterification. However, transesterification is not favoured in our system because of the scarcity and little availability of free hydroxyl groups,7 as well as the absence of a transesterification catalyst.8 The difference in behaviour between the DTDB- and DA-cured networks indicates a major contribution of disulfide bonds to the relaxation process. Nevertheless, the relaxation of the DTDB-cured sample is not complete within the two hour experiment, likely because side reactions at high temperature generate non-reversible bonds. In Fig. 4b, we compare two successive stress relaxations on the same DTDB-cured sample. The second relaxation is very similar to the first one. The initial modulus is the same and then a substantial relaxation is observed, indicating that the material still contained a lot of disulfide bonds after the first relaxation experiment. However, the second relaxation seems slower, the corresponding relaxation modulus remaining higher than during the first relaxation experiment. The slowdown of the relaxation dynamics may be associated with the consumption of some reversible bonds by side reactions happening at high temperatures. The shear modulus G′ is still very similar before and after the relaxation experiment, indicating that the number of crosslinks did not increase much. This tends to prove that the effect of side reactions stays limited at this time scale. | ||
| Fig. 4 Stress relaxation experiment at 180 °C after curing in the rheometer. (a) Comparison between DA- and DTDB-cured materials, and (b) the first stress relaxation (filled symbols) and the second stress relaxation (void symbols) of DTDB-cured ENR. | ||
To complete stress relaxation data, creep experiments were performed in the DMA. Chemically crosslinked rubbers usually recover their original shape after stretching. Force-controlled creep/recovery tests at different temperatures were performed on the DTDB-cured rubber (Fig. 5a). The results of the same tests for DA-cured samples are plotted for comparison (Fig. 5b). Both rubbers can withstand temperatures as high as 100 °C without residual deformation, but their behaviour at higher temperature greatly differs. The slight increase of the strain observed for the control material at 150 °C and 180 °C can probably be related to some degradation of the material. On the other hand the disulfide-containing material deforms much more and faster. This pronounced elongation may be associated with rearrangements of disulfide bonds activated at high temperature. Yet, the slope of deformation decreases with time when the experiment is conducted at 180 °C. This phenomenon is the mark of the side reactions that impede disulfide rearrangements. It is much more present and rapid at 180 °C, indicating the effect of temperature on the probability and rate of side reactions.
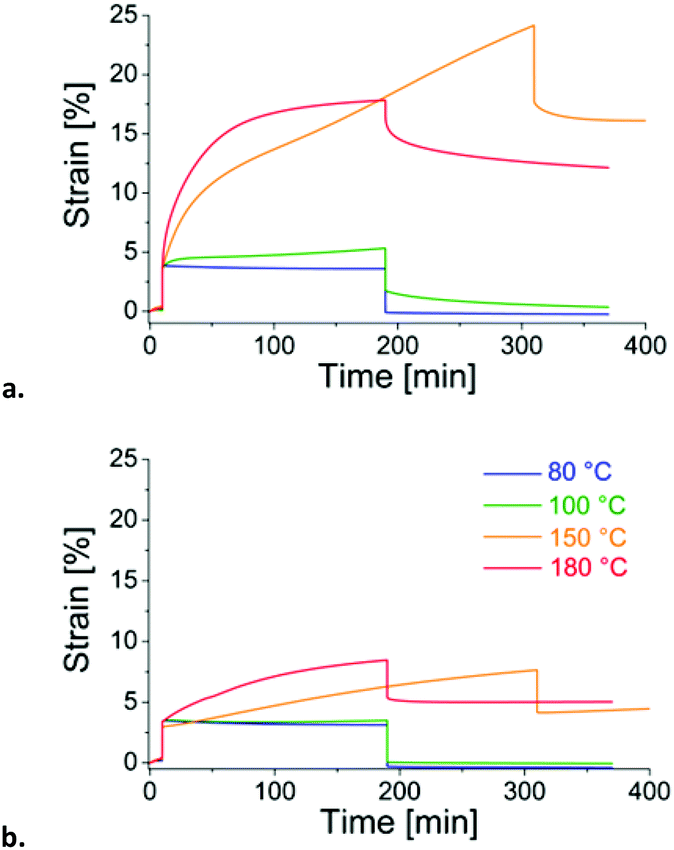 | ||
| Fig. 5 Creep/recovery tests at different temperatures. (a) DTDB-cured ENR and (b) DA-cured ENR. A force of 0.5 N is applied for 180 min and then released. | ||
Cyclic strain/recovery experiments were performed before, during, and after heating to test the reversibility of the dynamic behaviour with temperature (Fig. 6). As expected from the previous experiment, at 30 °C our material behaves like a standard rubber and recovers its original shape after removing the force. The material response is reproducible whatever the number of solicitations (three cycles presented here). After equilibrating the temperature at 150 °C for 5 min, the response of the material to the same experiment is completely different. First, an increase in strain during heating and equilibration should be noticed, likely due to thermal expansion of the material. Upon the application of a 0.5 N force, the rubber deforms about 10% and does not recover its initial length after removing the applied force. After three cycles, the strain of the unstretched material has increased from 3 to 8% thanks to the rearrangements of the network. However, the deformation seems to decrease at each cycle. This is probably caused by side reactions that impede the material from reshaping. Repeating the experiment after decreasing the temperature back to 30 °C, the response of the material is the same as during the first three cycles (with the remaining strain of 5% due to the rearrangements that occurred at 150 °C). The network has stopped rearranging, the material behaves like a classical chemically crosslinked rubber again. Therefore, it is possible to activate the rearrangement of the network by heating and stop it completely by cooling.
 | ||
| Fig. 6 Cyclic strain/recovery profiles on ENR25 crosslinked with DTDB at different temperatures (black: applied force; red: strain response of the material). | ||
The rearrangements of the network can promote good adhesion behaviour
We showed that the temperature activates chemical rearrangements in a disulfide-containing ENR network. The rearrangements are a combination of disulfide rearrangements and side reactions that both result in the creation of new crosslinks. Adhesion experiments were conducted to quantify a possible mendability of the crosslinked rubber. Lap shear geometry was selected as it is well documented37 and easy to implement. Lap shear measurements are independent of the length of the joint as long as it is more than about four times the joint thickness,37 so a length of 20 mm was selected for our 3 mm thickness samples. After an adhesion time of 30 min at 180 °C (Fig. 7a), DA-cured samples rapidly peel and the maximum force is less than 5 N, whereas for DTDB-cured samples, the force goes up to 20 N, where the sample breaks instead of peeling. The adhesion between DTDB-cured samples is thus much higher compared to the reference material. The adhered joints were swollen into a good solvent to characterize the physical or chemical nature of the adhesion. The DA-cured joint rapidly lifts off upon swelling whereas the DTDB-cured joint swells a lot without breaking, showing the presence of chemical bonds between the two pieces of rubber. Chemical rearrangements create new bonds between the samples and are responsible for the good adhesion properties.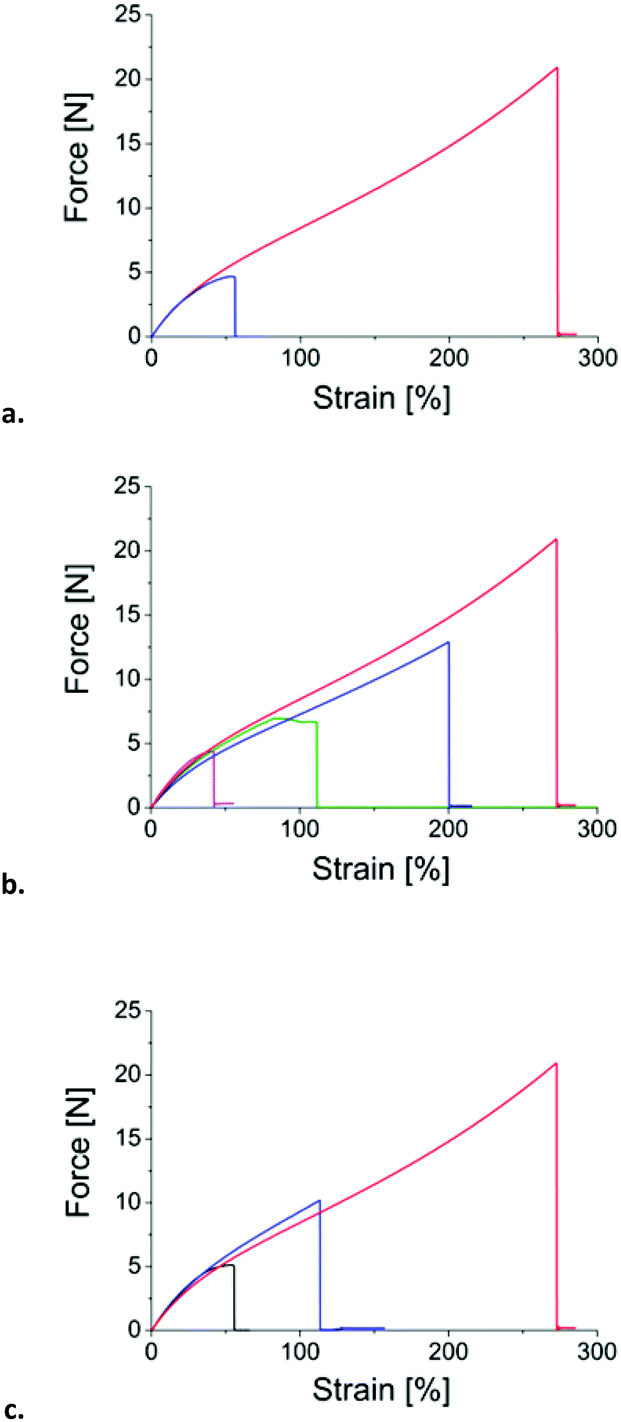 | ||
| Fig. 7 Lap shear tests on cured rubbers: (a) comparison between DA- (blue) and DTDB- (red) cured rubbers, adhesion for 30 min at 180 °C, (b) DTDB-cured rubber adhered at 180 °C for varying times (pink: 5 min, green: 10 min, blue: 20 min and red: 30 min), (c) DTDB-cured rubber adhered for 30 min at different temperatures (black: 100 °C, blue: 150 °C and red: 180 °C). | ||
The effect of contact time and temperature was investigated on DTDB-cured rubber. Joints adhered for different times at 180 °C were tested; the corresponding force/strain profiles are plotted in Fig. 7b. The temperature was also varied keeping the contact time constant at 30 min (Fig. 7c). At short adhesion times or low temperatures, as for DA-cured rubber, the samples completely peel, enabling to calculate an adhesion energy using Kendall's formula (eqn (2)):37
 | (2) |
| Sample | DTDB-cured | DA-cured | |||||
|---|---|---|---|---|---|---|---|
| Temperature | 180 °C | 150 °C | 100 °C | 180 °C | |||
| Adhesion time (min) | 5 | 10 | 20 | 30 | 30 | 30 | 30 |
| Adhesion energy (J m−2) | 70 | 230 | Sample breaks | Sample breaks | Sample breaks | 80 | 90 |
Thermo-activated adhesion enables recycling of the rubber
For recycling experiments, the DTDB-cured sample was reduced to a fine powder using a variable speed rotor mill and then heated at 180 °C for 40 min in a heating press (Fig. 8). The same reprocessing experiment was conducted on a DA-cured sample for comparison. After the heat treatment, the integrity of the DA-crosslinked sample is not recovered (see Fig. S1†). Specimens could still be cut and tested showing that the “reformed” material has completely lost its properties after grinding and heating. The behaviour of the disulfide-containing network is completely different. The integrity of the sample is fully restored after heating as shown in Fig. 8a. Most of the mechanical properties are recovered: the reprocessed sample shows about 50% of the initial stress and 80% of the initial strain at break (Fig. 8b). After fine grinding of the rubber, when grains of powder are pressed together at high temperature, rearrangements occur. Under these conditions, chains start to interpenetrate and chemical reactions will then “mend” the grains together. Side reactions generating permanent bonds limit the rearrangements inside each grain of powder, but they take part in the mending process creating bonds between and inside particles, even if the created bonds are not dynamic anymore. All this process can be seen as relaxation allowing intimate adhesion of the powder grains. The properties of both samples are listed in Table 2. It is worth noting that for DTDB-cured material, the Young's modulus remains identical after reforming the sample (Fig. S5†). Swelling experiments in toluene (Table 2 and Fig. S3†) confirmed that the crosslinking densities before and after reforming are comparable. | ||
| Fig. 8 Recycling experiment. (a) The cured rubber is finely ground and then reshaped by compression moulding. (b) Tensile test before and after the recycling experiment (black: DTDB-cured; grey: DA-cured; dashed: before; line: after). | ||
| Stress at break [MPa] | Strain at break | Young's modulus [MPa] | Equilibrium swelling ratio Q | |
|---|---|---|---|---|
| DTDB_virgin | 12 ± 2 | 5.3 ± 0.2 | 1.67 ± 0.02 | 3.5 |
| DTDB_reprocessed | 5 ± 1 | 4.2 ± 0.2 | 1.66 ± 0.02 | 3.3 |
| DA_virgin | 10 ± 1 | 4.6 ± 0.2 | ||
| DA_reprocessed | 1.0 ± 0.1 | 0.6 ± 0.1 |
Conclusions
A dynamic covalent network was obtained by crosslinking ENR with a disulfide-containing carboxylic diacid. The resulting rubber was shown to be a dual network containing mostly disulfide crosslinks (considered as dynamic) and a few permanent crosslinks resulting from unavoidable side reactions. The dynamic nature of the disulfide bonds was highlighted through stress relaxation, creep and adhesion experiments. The network presents strong elastomeric properties at temperatures as high as 100 °C and is able to rearrange at higher temperatures (above 150 °C). The processing of the rubber at high temperature is limited because of the dual nature of the network, but even after several cycles at high temperature rearrangements still occur. The material can be recycled and recovers most of its mechanical properties after grinding to a fine powder and remoulding. A compromise is obtained: despite the formation of some permanent bonds, the object, even used at high temperatures, may be reshaped at the end of its life cycle. This could be optimized by the use of a catalyst for example. Furthermore, the good adhesion properties of this rubber should permit the repairing of cracks by heating and thus lead to fatigue improvement. Our comparison of DA- and DTDB-cured systems shows the importance of the disulfide rearrangements in the stress relaxation process, the adhesion properties and recycling capability of DTDB-cured ENR rubber.Other kinds of chemical functions could be introduced into ENR via the internal chain of a carboxylic diacid. We thus can envision developing rubbers sensitive to other stimuli than temperature or even materials having a fully reversible behaviour if side reactions can be avoided.
Acknowledgements
This research was supported by the European Seventh Framework NMP programme, the SHINE project – grant agreement 309450-2. Benoît le Rossignol from Hutchinson SA, Montargis Research Centre, France, is warmly acknowledged for the gift of ENR25. The authors also thank Michel Cloître and Renaud Nicolaÿ for helpful discussions on rheological measurements and disulfide chemistry, respectively, as well as Erwan Chabert for his help in adhesion sample preparation.Notes and references
- C. N. Bowman and C. J. Kloxin, Angew. Chem., Int. Ed., 2012, 51, 4272–4274 CrossRef CAS PubMed.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 CrossRef CAS PubMed.
- G. Deng, C. Tang, F. Li, H. Jiang and Y. Chen, Macromolecules, 2010, 43, 1191–1194 CrossRef CAS.
- C. J. Kloxin and C. N. Bowman, Chem. Soc. Rev., 2013, 42, 7161–7173 RSC.
- R. J. Wojtecki, M. A. Meador and S. J. Rowan, Nat. Mater., 2011, 10, 14–27 CrossRef CAS PubMed.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed.
- M. Capelot, D. Montarnal, F. Tournilhac and L. Leibler, J. Am. Chem. Soc., 2012, 134, 7664–7667 CrossRef CAS PubMed.
- M. Capelot, M. M. Unterlass, F. Tournilhac and L. Leibler, ACS Macro Lett., 2012, 1, 789–792 CrossRef CAS.
- Y.-X. Lu, F. Tournilhac, L. Leibler and Z. Guan, J. Am. Chem. Soc., 2012, 134, 8424–8427 CrossRef CAS PubMed.
- W. Denissen, G. Rivero, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. Du Prez, Adv. Funct. Mater., 2015, 25, 2451–2457 CrossRef CAS PubMed.
- B. Adhikari, D. De and S. Maiti, Prog. Polym. Sci., 2000, 25, 909–948 CrossRef CAS.
- E. Bilgili, A. Dybek, H. Arastoopour and B. Bernstein, J. Elastomers Plast., 2003, 35, 235–256 CrossRef CAS PubMed.
- A. Gugliemotti, C. Lucignano and F. Quadrini, Plast., Rubber Compos., 2012, 41, 40–46 CrossRef CAS PubMed.
- M. Le Neindre and R. Nicolay, Polym. Chem., 2014, 5, 4601–4611 RSC.
- J. Canadell, H. Goossens and B. Klumperman, Macromolecules, 2011, 44, 2536–2541 CrossRef CAS.
- M. Pepels, I. Filot, B. Klumperman and H. Goossens, Polym. Chem., 2013, 4, 4955–4965 RSC.
- U. Lafont, H. van Zeijl and S. van der Zwaag, ACS Appl. Mater. Interfaces, 2012, 4, 6280–6288 CAS.
- H. Otsuka, S. Nagano, Y. Kobashi, T. Maeda and A. Takahara, Chem. Commun., 2010, 46, 1150–1152 RSC.
- J. A. Yoon, J. Kamada, K. Koynov, J. Mohin, R. Nicolaÿ, Y. Zhang, A. C. Balazs, T. Kowalewski and K. Matyjaszewski, Macromolecules, 2012, 45, 142–149 CrossRef CAS.
- Y. Amamoto, H. Otsuka, A. Takahara and K. Matyjaszewski, Adv. Mater., 2012, 24, 3975–3980 CrossRef CAS PubMed.
- R. Nicolaÿ and Y. Kwak, Isr. J. Chem., 2012, 52, 288–305 CrossRef PubMed.
- A. Rekondo, R. Martin, A. Ruiz de Luzuriaga, G. Cabanero, H. J. Grande and I. Odriozola, Mater. Horizons, 2014, 1, 237–240 RSC.
- R. Martin, A. Rekondo, A. Ruiz de Luzuriaga, G. Cabanero, H. J. Grande and I. Odriozola, J. Mater. Chem. A, 2014, 2, 5710–5715 CAS.
- Z. Q. Lei, H. P. Xiang, Y. J. Yuan, M. Z. Rong and M. Q. Zhang, Chem. Mater., 2014, 26, 2038–2046 CrossRef CAS.
- C. K. L. Davies and S. V. Wolfe, Polymer, 1983, 24, 107–113 CrossRef CAS.
- T. Johnson and S. Thomas, Polymer, 1999, 40, 3223–3228 CrossRef CAS.
- A. S. Hashim and S. Kohjiya, KGK, Kautsch, Gummi Kunstst., 1993, 46, 208–213 CAS.
- A. N. Bibi, D. A. Boscott, T. Butt and R. S. Lehrle, Eur. Polym. J., 1988, 24, 1127–1131 CrossRef CAS.
- S. N. Koklas, D. D. Sotiropoulou, J. K. Kallitsis and N. K. Kalfoglou, Polymer, 1991, 32, 66–72 CrossRef CAS.
- M. A. Rahman, L. Sartore, F. Bignotti and L. Di Landro, ACS Appl. Mater. Interfaces, 2013, 5, 1494–1502 CAS.
- M. Pire, S. Norvez, I. Iliopoulos, B. Le Rossignol and L. Leibler, Polymer, 2010, 51, 5903–5909 CrossRef CAS PubMed.
- M. Pire, S. Norvez, I. Iliopoulos, B. Le Rossignol and L. Leibler, Polymer, 2011, 52, 5243–5249 CrossRef CAS PubMed.
- L. Imbernon, M. Pire, E. K. Oikonomou and S. Norvez, Macromol. Chem. Phys., 2013, 214, 806–811 CrossRef CAS PubMed.
- M. Pire, C. Lorthioir, E. K. Oikonomou, S. Norvez, I. Iliopoulos, B. Le Rossignol and L. Leibler, Polym. Chem., 2012, 3, 946 RSC.
- M. Pire, S. Norvez, I. Iliopoulos, B. Le Rossignol and L. Leibler, Compos. Interfaces, 2014, 21, 45–50 CrossRef CAS PubMed.
- R. Chasset and P. Thirion, Proceedings of the Conference on Physics of Non-Crystalline Solids, North Holland Publishing Co., Amsterdam, 1965.
- K. Kendall, J. Phys. D: Appl. Phys., 1975, 8, 512–522 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Some test procedures and characterization. See DOI: 10.1039/c5py00459d |
| This journal is © The Royal Society of Chemistry 2015 |