
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotocrosslinkable starch cinnamyl ethers as bioinspired bio-based polymers†
Simona
Petroni‡
 a,
Sara Fernanda
Orsini‡
b,
Daniele
Bugnotti
c,
Emanuela
Callone
c,
Sandra
Dirè
cd,
Luca
Zoia
e,
Roberta
Bongiovanni
fg,
Sara
Dalle Vacche
fg,
Alessandra
Vitale
fg,
Luisa
Raimondo
b,
Adele
Sassella
b,
Pietro
Mariani
b,
Massimiliano
D’Arienzo
*b and
Laura
Cipolla
*a
a,
Sara Fernanda
Orsini‡
b,
Daniele
Bugnotti
c,
Emanuela
Callone
c,
Sandra
Dirè
cd,
Luca
Zoia
e,
Roberta
Bongiovanni
fg,
Sara
Dalle Vacche
fg,
Alessandra
Vitale
fg,
Luisa
Raimondo
b,
Adele
Sassella
b,
Pietro
Mariani
b,
Massimiliano
D’Arienzo
*b and
Laura
Cipolla
*a
aDepartment of Biotechnology and Biosciences, University of Milano – Bicocca, P.za della Scienza 2, 20126 Milano, Italy. E-mail: laura.cipolla@unimib.it
bDepartment of Materials Science, University of Milano – Bicocca, Via R. Cozzi 55, 20125 Milano, Italy
c“Klaus Müller” Magnetic Resonance Lab., Department of Industrial Engineering, University of Trento, Via Sommarive 9, 38123 Trento, Italy
dDepartment Industrial Engineering, University of Trento, Via Sommarive 9, 38123 Trento, Italy
eDepartment of Earth and Environmental Sciences, University of Milano-Bicocca, Building U01, Piazza della Scienza 1, 20126 Milan, Italy
fDepartment of Applied Science and Technology, DISAT, Politecnico di Torino, Corso Duca degli Abruzzi 24, 10129 Torino, Italy
gConsorzio Interuniversitario Nazionale per la Scienza e Tecnologia dei Materiali, (INSTM) Via G. Giusti, 9, 50121 Firenze, Italy
First published on 27th November 2024
Abstract
A novel starch-based ether bearing cinnamyl functionalities, conferring photo-crosslinking properties, is synthesised by reaction with cinnamyl chloride in the presence of sodium hydroxide. Natural yuca was selected as a sustainable source of starch. Three different molar equivalents of reagents are used, affording starch-cinnamyl ethers with different degrees of substitution, ranging from 0.09 to 1.24, as determined by liquid phase nuclear magnetic resonance (NMR). The double bonds in the cinnamyl moieties show reactivity towards photodimerization upon irradiation at 254 nm, affording a novel cross-linked bio-inspired polymer. The formation of the covalent ether linkage and the [2+2] cycloaddition of the cinnamyl units are confirmed by a combination of spectroscopic techniques, including solid state NMR. The materials are further characterized by gel permeation chromatography (GPC), thermogravimetric analysis (TGA), and X-ray diffraction analysis (XRD). Starch-cinnamyl ethers with a DS of 0.09 are water soluble, and suitable for the preparation of transparent films potentially exploitable for biodegradable packaging materials.
1. Introduction
In the last few decades, significant academic and industrial research has been directed towards the development of innovative bio-based polymers from renewable and biodegradable biomass resources.1–3 Owing to its high availability, low cost, high biocompatibility and biodegradability, renewability, good film-forming ability and processability, about 50% of bioplastics currently in use are derived from starch.4–7 Furthermore, research is fostering the development of starch-based innovative products, to be applied in the expanding fields of biomaterials, regenerative medicine and therapeutic area (e.g., hydrogels, aerogels and biofoams).8 The richness of hydroxyl groups makes starch easy to modify in terms of structure and functional properties exploiting chemical or enzymatic methods.9,10Despite the widespread use of starch-derived products in the market, moisture sensitivity and brittleness remain challenges to be overcome. To this aim, acetylation,11,12 blending with synthetic polymers (e.g. polyethylene, polyvinyl acetate or styrene butadiene rubber), incorporation of nanometric bio-fillers,13 and modification with cross-linking units which induce network formation14–16 have been employed to improve the tensile strength and the hydrophobicity of starch. However, poor compatibility among starch, filler and synthetic polymers, non-biodegradability or even toxicity of some incorporated compounds raise concerns about the application of these materials in specific fields, such as food packaging.1,17 In this scenario, the use of naturally occurring crosslinking agents has recently attracted increased attention as a promising route to tackle environmental and health concerns, along with economic issues.14 Among them, photo-catalyzed reactions are particularly useful, since they proceed with high selectivity and efficiency without producing toxic side products. Due to natural abundance, cinnamyl moieties (3-phenylprop-2-en-1-yl moieties) can be fished out as a promising option. Many cinnamyl derivatives are well-known antioxidants and are supposed to have several benefits due to their strong free radical scavenging behavior, antibacterial, antiviral and antifungal properties.18 Cinnamyl derivatives can act as cross-linking agents, due to their well-known ability to undergo a reversible [2+2] cycloaddition under UV light at a specific wavelength, without the need for additional chemicals as photoinitiators.19,20 This has been extensively exploited for the production of several photo-cross-linkable polymers, such as poly(vinyl cinnamate) applied in liquid crystal displays, in tissue engineering,21 and in cellulose-based organic transistors.22–24 Recently, cinnamyl units have also been employed in the so-called “smart materials”, whose molecular structure and subsequent properties can be changed in response to external stimuli such as light, heat, and pH, imparting them interesting and programmable properties for advanced applications.25,26 In these systems, cinnamyl derivatives allow replacing toxic and non-sustainable adducts such as anthracene, thiols, or azo compounds;27–29 furthermore, the photoactivated cross-linking polymerization represents an eco-friendly procedure as it does not require either heating or solvents.30–33 Very few studies in the literature have reported the synthesis of cinnamyl moieties grafted to starch,34,35 while, as far as we are aware, there is only one example of the photocross-linking reaction performed on such derivatives.36 The present study proposes a synthetic protocol towards new starch-based polymers bearing cinnamyl functionalities, where for the first time the grafting strategy involves a cinnamyl etherification reaction, in place of the more widespread cinnamoyl esterification (Scheme 1). The ether bond is known to be chemically more stable, and less polar than its ester counterpart; in addition, it may modify the structural and mechanical properties of the overall structure due to changes in carbon hybridization and conjugation system.37
 | ||
| Scheme 1 Acylation and etherification strategies towards cinnamyl starch derivatives (in red the extended conjugated p-system). | ||
The etherification reaction is studied under different conditions, and the ability of starch-cinnamyl derivatives to undergo photo-induced cross-linking is investigated by UV-Vis spectroscopy and, for the first time on such derivatives, by nuclear magnetic resonance spectroscopy (NMR), allowing us to gain interesting insights into the reactivity of the system. Finally, the starch-cinnamyl ether was exploited for the preparation of transparent films by the solvent casting method in view of potential application as transparent biodegradable packaging materials.
2. Materials and methods
2.1 Materials
Before use, yuca (or cassava) starch powder from Colombia (almidon de yuca dulce produced by Cimpa s.a.s.) was dried in a static oven for 48 h at 65 °C; the average weight loss was between 9% and 11%. Cinnamyl chloride (3-chloropropenyl)benzene (CINN-Cl, CAS 2687-12-9, purity 95%), [AMIM]Cl (1-allyl-3-methylimidazolium chloride, CAS 65039-10-3, purity ≥ 97%), pyridine (CAS 110-86-1, purity 99.8%) and benzoyl chloride (PhCOCl, CAS 98-88-4, purity 99%), anhydrous NaOH (pellets, purity grade ≥ 98%), and tetrahydrofuran (THF, inhibitor-free, HPLC grade ≥ 99.9%) were purchased from Sigma-Aldrich and used without any further purification. Acetone (CAS 67-64-1, 96.6% purity grade), and analytical grade ethanol (EtOH, CAS 64-17-5) were purchased from Thermo Fisher Scientific. Anhydrous dimethyl sulfoxide (DMSO, CAS 67-68-5, analytical grade) was purchased from VWR International. Ultrapure water was obtained using the Milli-Q® system with residual conductivity of 13 μS cm−1. DMSO-d6 (CAS 2206-27-1, 99.5 atom% D) and trifluoroacetic acid-d (TFA-d, CAS 599-00-8, 99.5 atom% D) for NMR spectroscopy were purchased from Acros Organics. Thin layer chromatography (TLC) was performed on silica gel 60 F254 coated glass plates (Merck), using a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.5 CHCl3:EtOH mixture as the eluent and visualized with an UV lamp at a 254 nm wavelength.
:0.5 CHCl3:EtOH mixture as the eluent and visualized with an UV lamp at a 254 nm wavelength.
2.2 General procedure for the synthesis of starch cinnamyl ether derivatives
Three different starch/NaOH/cinnamyl chloride ratios were used in order to obtain different substitution degrees (DS). The ratios are calculated on the mmol of anhydroglucose units (AGU, MW = 162.14 g mol−1).Typically, 1 g (6.16 mmol AGU) of dried yuca starch is suspended in 16 mL of dry DMSO (c = 62.5 g L−1) in a round-bottom flask and heated to 90 °C with an oil bath under magnetic stirring until dissolution (3 h). The solution is then cooled to room temperature and powdered NaOH, previously suspended in 2 mL of the reaction mixture, is added (6.16 mmol or 18.48, Table 2). The mixture is homogenized for 1 hour. Cinnamyl chloride (0.164 g, 0.940 g, or 4.236 g) is added dropwise, and the reaction is kept under stirring for 24 hours at room temperature. The solution is transferred into a plastic centrifuge tube, cold acetone (30 mL) is added and gently mixed. After 10 min the crude product precipitates, the suspension is centrifuged (5000 rpm, 20 °C, 10 min), the solvents are carefully removed and the solids (white powder) are washed under magnetic stirring for 10 min with a mixture of 30 mL of acetone and 5 mL of deionized water in order to remove residual organics (cinnamyl chloride or by-products derived thereof) and inorganics (NaOH and NaCl); the suspension is centrifuged (5000 rpm, 20 °C, 10 min), the solvents are carefully removed and the washing is repeated once more with a fresh acetone:water mixture, followed by a final washing with pure acetone (30 mL). The collected solids are dried under vacuum in a desiccator. TLC of mother liquors was used to monitor washing effectiveness in removing organic by-products.
2.3 Starch cinnamyl ether film preparation
450 mg of cinnamyl-starch ether (DS 0.09, CS1) are dissolved in 10 mL of deionized water at 90 °C under magnetic stirring for 1 hour. After complete dissolution, 2 mL of the solution are cast at room temperature on a Petri dish of low-density polyethylene. The film is peeled-off, and dried at room temperature in air for 3 days.2.4 Materials characterization
:3 EtOH:H2O mixture. The solids are recovered by centrifugation (3000 rpm, 15 min), after removal of the supernatant. In order to fully remove the ionic liquid, pyridine salts and excess reagents, the precipitate is vortexed in a 7:3 fresh mixture of EtOH:H2O (10 mL, 10 min) followed by centrifugation (3000 rpm, 15 min). These steps are repeated twice. The white solid is dried under reduced pressure. 1 mg of perbenzoylated starch is dissolved in THF and filtered with a GHP 0.45 μm Acrodis syringe filter for the GPC analysis.37 The analyses are performed with a HP1100 series liquid chromatography connected to a HP 1040 UV photodetector at a wavelength of 240 nm. The injector has a Rheodyne loop valve with a loop capacity of 20 μL. The GP-column system was composed as follows (according to the solvent flow direction): Agilent PLgel 5 μm (500 Å), Agilent PLgel 5 μm (1000 Å) and Agilent PLgel 5 μm (10000 Å). THF at a flow rate of 1 mL min−1 was flushed. PL Polymer Standards of Polystyrene from Polymer Laboratories are used for calibration. The evaluation of the number-average molecular weight (Mn) and the weight-average molecular weight (Mw) of the samples is performed. Moreover, the ratio I = Mw/Mn, defined as the dispersity index is also calculated. The reported Mn and Mw values are the average of three analyses (standard error Mw: 500 g mol−1; Mn: 100 g mol−1).
The DS is determined by the ratio between the normalized integration area of the aromatic protons (APh/5) and the integration areas of the anomeric protons Aα(1,4) + Aα(1,6), following eqn (1).
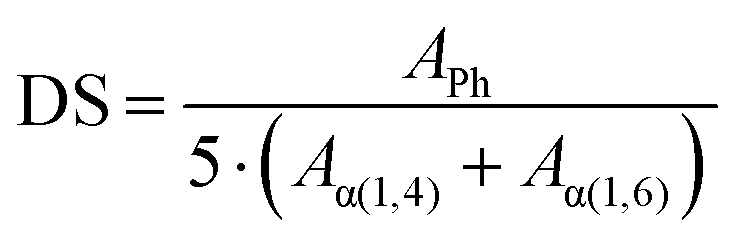 | (1) |
 | (2) |
3. Results and discussion
Starch is a natural polymer characterized by two components, the linear α(1,4)-polysaccharide amylose, and the branched α(1,6)-polysaccharide amylopectin. For the present work, yuca (or cassava) was selected as the starch source, since its cultivation is sustainable, high yielding and it does not require demanding agricultural practices.40 The starch source defines starch composition in terms of the two components, which will have impact on starch functionalization. For yuca starch, the literature reports41 a 17% amylose content and 83% amylopectin. Besides a comprehensive characterization of the native polysaccharide, the structure and the functionalized starch cinnamyl ether were carefully studied by X-ray diffraction (XRD), attenuated total reflection Fourier transform infrared (ATR-FTIR) spectroscopy, liquid and solid-state nuclear magnetic resonance (NMR), and thermogravimetric analysis (TGA). To prove the efficacy of the proposed synthetic strategy in supplying a photo-cross-linkable material, the photochemical properties of the starch cinnamyl ether have been carefully checked by UV-vis spectroscopy, and by NMR. Finally, considering the high demand for transparent biodegradable materials in the frame of packaging products,42 a proof of concept of the potential applicability of the novel bio-based polyether in the preparation of transparent and homogeneous films is reported.3.1 Characterization of yuca starch
Since the extent of substitution with the cinnamyl moieties will be influenced by starch composition, a preliminary characterization of yuca starch was performed. Pristine yuca starch was characterized in terms of its number-average molecular weight, weight-average molecular weight, and dispersity index by GPC analysis, together with its degree of branching by solution 1H-NMR.The determination of the average molecular weight of the two components is usually performed through GPC chromatography after derivatization.43 Thus, a preliminary benzoylation of the analyte both for the complete solubilization in the chromatographic solvent and for the UV detection was performed (Scheme 2), using the ionic liquid [AMIM]Cl as the solvent, under dark conditions.
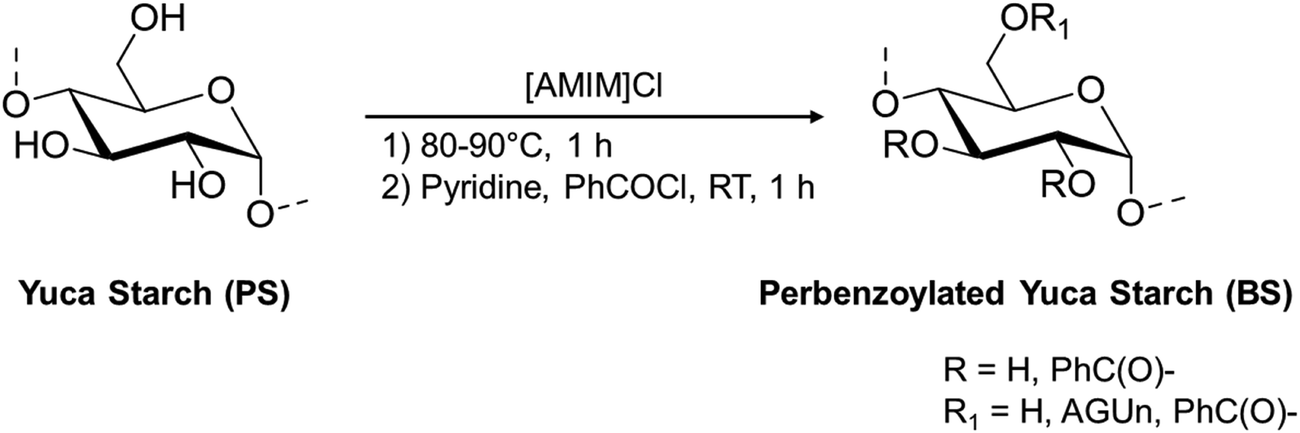 | ||
| Scheme 2 Benzoylation procedure of pristine starch PS. | ||
GPC analysis (Fig. 1 and Table 1) allowed the determination of the number-average molecular weight (Mn), the weight-average molecular weight (Mw) and the peak molecular weight (Mp), which is defined as the molecular weight at the maximum absorbance, along with the polydispersity index (I).
 | ||
| Fig. 1 GPC chromatogram of yuca starch and the corresponding peak deconvolution. | ||
| Yuca starch profile | 1 | 2 | 3 | |
|---|---|---|---|---|
| Area (%) | — | 47 | 33 | 20 |
| M p | 1.4 × 106 | 6.8 × 105 | 1.5 × 106 | 8.5 × 106 |
| M n | 4.8 × 106 | 8.7 × 105 | 1.6 × 106 | 1.45 × 107 |
| M w | 6.1 × 107 | 1.1 × 106 | 1.8 × 106 | 6.3 × 107 |
| I | 12.8 | 1.3 | 1.2 | 4.3 |
The GPC profile (Fig. 1) displayed a polymodal distribution, thus the curve was deconvoluted by a Gaussian distribution, affording three different contributions (profiles 1, 2, 3, Fig. 1); the area (%) for each contribution is reported in Table 1.
The deconvoluted peaks 1, 2, and 3 correspond to molecular weight (Mw) values of 1.1 × 106, 1.8 × 106 and 6.3 × 107 g mol−1, respectively. The higher Mw peak (profile 3) was related to the amylopectin component, in agreement with the literature data, reporting values between 107 and 109 g mol−1.44–46 Amylose has a relatively lower Mw compared with amylopectin, ranging from 105 to 106 g mol−1. Accordingly, profile 2 was mainly (but not uniquely) attributed to higher Mw amylose, whereas profile 1 was considered to comprise mostly (but not entirely) lower Mw amylose.47
The yuca starch degree of branching calculated with respect to the anhydroglucose unit was determined by solution 1H-NMR as described in the Materials and methods section and resulted in 3.7% (see Fig. 4); as a consequence, a maximum degree of substitution of 2.96 can be expected.
3.2 Starch cinnamyl ether synthesis and characterization
Starch-cinnamyl ethers with different DS (namely low, medium and high, samples CS1, CS2, and CS3 respectively) were synthesized, in order to tune the physico-chemical properties, and to check the readiness to undergo the photodimerization reaction. The availability of starch-cinnamyl ethers with different DS was considered as an opportunity to address several aspects: (i) the identification of diagnostic signals, robustness and sensitivity of the analytical techniques explored for the detailed characterization, such as solution and solid state NMR, UV-Vis, ATR-FTIR spectroscopies, (ii) solubility, thermal stability and optical behavior, being key issues for the design of sustainable material processing and features (i.e. the balance between water solubility for sustainable processing and hydrophobicity as a water vapor barrier is crucial for food packaging),48 (iii) the identification of the best DS for the subsequent photocrosslinking reaction.The synthesis of the cinnamyl-starch derivatives (CS1–3, Scheme 3) required first the complete dissolution of starch in DMSO at 80–90 °C; afterward the solution was reacted with powdered NaOH at room temperature for 1 h, and finally cinnamyl chloride was added.
 | ||
| Scheme 3 Starch etherification with cinnamyl moieties (CINN-Cl, cinnamyl chloride). Three different molar equivalents of NaOH and cinnamyl chloride were used, affording products CS1–CS3 with different degrees of substitution. | ||
In order to tune the substitution degree, different molar equivalents of NaOH and cinnamyl chloride in respect to the molar equivalents of AGU were tested (Table 2). Assuming that the maximum DS is very close to 3, reagent amounts correspond to CINN-Cl/AGU of 0.2, 1.0, 4.5 molar ratios, respectively.
The occurrence of the starch derivatization with the cinnamyl moieties was first assessed by FTIR-ATR spectroscopy (Fig. 2).
 | ||
| Fig. 2 FTIR-ATR of pristine yuca starch (PS), cinnamyl chloride (CINN-Cl), and cinnamyl starch samples CS1–3. | ||
The 1200–800 cm−1 region shows the characteristic band due to C–O vibrations in the COH and COC groups of starch. The spectrum is modified by the presence of cinnamyl moieties, in particular the appearance of the peaks at 691 cm−1, 966 cm−1 and 1650 cm−1 could be attributed to the double bond stretching (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) of the disubstituted alkene, and the peaks at 3000–3100 cm−1 to the C–H stretching of the alkene. The diagnostic signals of the cinnamyl units are visible only for the CS3 sample, with signal intensities as a function of the amount of reagents used, qualitatively indicating the presence of cinnamyl moieties in the starch derivatives.
C) of the disubstituted alkene, and the peaks at 3000–3100 cm−1 to the C–H stretching of the alkene. The diagnostic signals of the cinnamyl units are visible only for the CS3 sample, with signal intensities as a function of the amount of reagents used, qualitatively indicating the presence of cinnamyl moieties in the starch derivatives.
Since the FTIR sensitivity confirms the presence of the cinnamyl moiety only on the sample obtained with the highest amount of reagents, solution and solid phase NMR were used to further investigate the effectiveness of etherification reaction, demonstrating the presence of the covalent ether bond between the starch and the cinnamyl moieties. In addition, being quantitative, 1H-NMR allows the DS determination of the samples (Table 2). The spectra (Fig. 3) were recorded in the presence of deuterated trifluoroacetic acid, since it quickly exchanges the protons of the unsubstituted OH groups with deuterium atoms, thus eliminating disturbing signals overlapping with the proton peaks of the starch backbone, needed for an accurate DS quantification. The non-deuterated trifluoroacetic acid resulting from this equilibrium gives rise to a broad signal centered at around 7.5 ppm, out of the diagnostic region of interest.
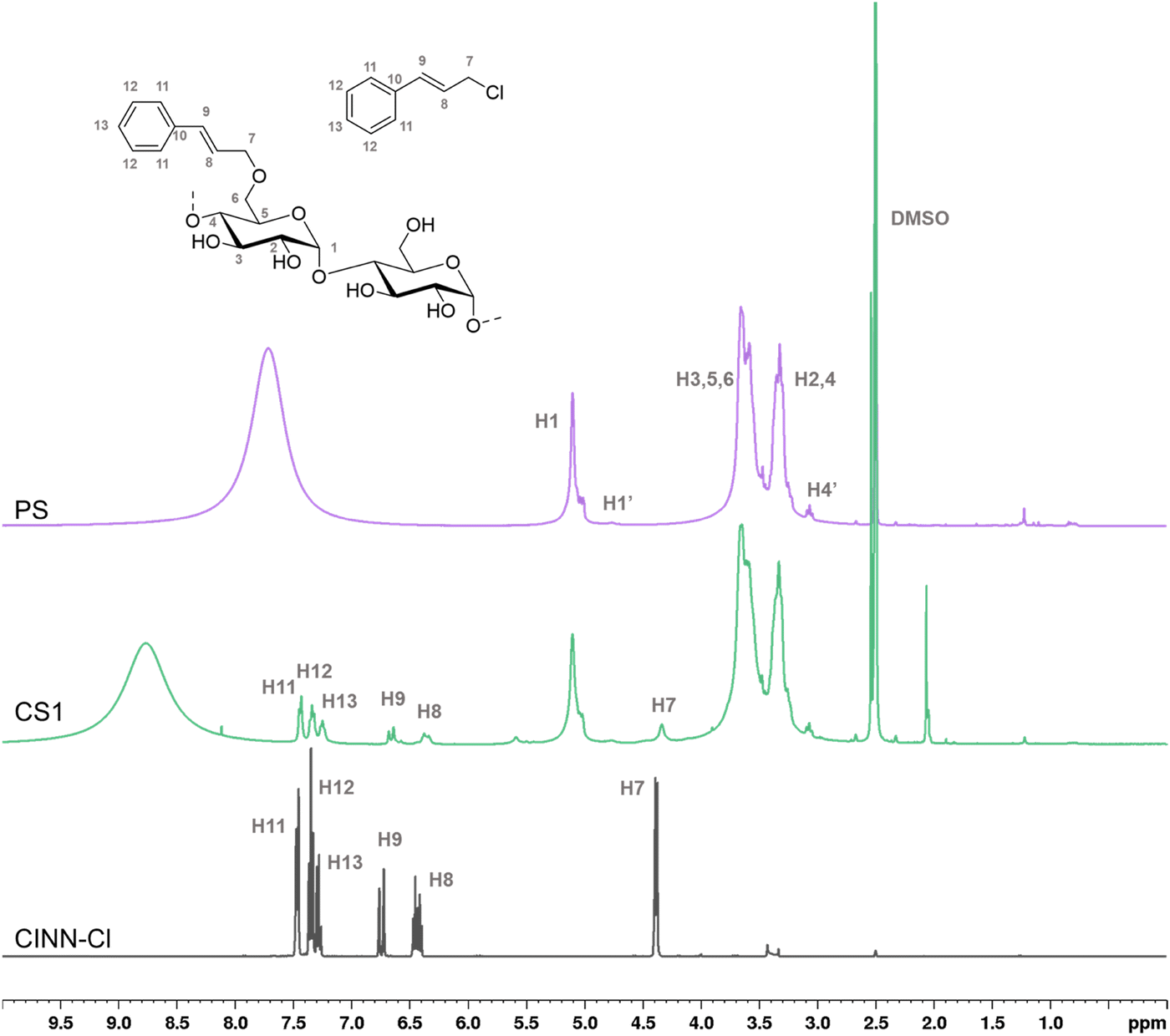 | ||
| Fig. 3 Comparison of 1H-NMR in DMSO-d6 of pristine starch (PS), CS1 (green) and cinnamyl chloride (black). For the sake of clarity, numbering of the cinnamyl moiety in cinnamyl chloride and in the starch derivatives has been arbitrarily assigned (not compliant with IUPAC nomenclature); H1 refers to the anomeric protons involved in the α(1,4) glycosidic linkage; H1′ refers to the anomeric protons involved in the α(1,6) glycosidic linkage; H4′ refers to the non-reducing terminal glucose units. | ||
It's worth noting that the appearance of the peaks related to the cinnamyl moieties can be observed even for the low substitution sample CS1, thus suggesting NMR as sensitive enough for the determination of etherification effectiveness. The aromatic protons (H-11–H-13) resonate in the range of 7.5–7.2 ppm, while the vinyl protons are detected at 6.65 ppm (H-9), and at 6.35 ppm (H-8). The allyl methylene protons (H-7) resonating at 4.34 ppm in the cinnamyl chloride are shifted to slightly higher fields (4.26 ppm) in the corresponding starch-cinnamyl ethers.
The broadening of the signals of the cinnamyl substituent in the functionalized samples if compared to the pure CINN-Cl is a consequence of the positional distribution of the group within the AGU moiety (C-2, C-3, and C-6), and of the three different starch components observed by GPC. The related 13C-NMR spectrum for the starch-cinnamyl ether (CS1) is reported in the ESI† (Fig. S1).
The anomeric protons α(1,4) and α(1,6) resonate at 5.10 ppm and 4.77 ppm, respectively; the integrals of these peaks in 1H-NMR spectra allow to quantitatively determine the DS (Table 2), as described by eqn (1).
Since it is expected that the photocross-linking reaction may afford insoluble products, solid state 13C CPMAS NMR spectra were recorded in order to obtain reference spectra before the photocatalytic process and fully characterize all cinnamyl-starch derivatives (Fig. S2, ESI†). Spectra of starch-cinnamyl ethers with 0.33 DS (CS2), pristine starch (PS), and cinnamyl chloride (CINN-Cl) are reported in Fig. 4. The resonances of starch carbons are found in the region of 94–105 ppm for C-1, 80–84 ppm for C-4, 68–77 ppm for C-2, C-3, C-5 and 58–65 ppm for C-6.13
 | ||
| Fig. 4 13C CPMAS NMR of (a) CINN-Cl, (b) pristine yuca starch (PS), (c) cinnamyl starch CS2, and (d) 13C CPPI NMR spectra of CS2 sample (cinnamyl methylene carbons involved in the ether linkage are highlighted). | ||
The vinyl carbons of CINN-Cl resonate at 134 and 125 ppm (C-9 and C-8, respectively, Fig. 4), while the phenyl ring resonances are located in the range 125–130 ppm (C11–13) and 136 ppm (C10). The presence of the corresponding peaks, despite being broader, in the substituted starch CS2 further confirms the presence of the cinnamyl moiety. The allylic carbon C-7 in the cinnamyl chloride resonates at 46 ppm (Fig. 4a), while it is not detected in the cinnamyl-starch samples (Fig. 4, and Fig. S2 in the ESI†). In agreement with the literature data on cinnamyl ethers49 the occurrence of the etherification reaction is expected to cause a shift of the allylic carbon C-7 to about 70 ppm, in the same spectral region of starch carbon atoms. In order to try to identify the C-7 signal and to definitely ascertain the formation of the covalent bond between starch and the cinnamyl moieties, a cross polarization with polarization inversion (CPPI) pulse sequence was applied to the solid-state NMR analysis of sample CS2. The parameters were optimized in order to obtain 13C spectra with CH2 resonances as negative peaks and suppressing all the other carbons resonances (e.g.: CH3, CH and quaternary).39 The 13C CPPI NMR spectrum of CS2 (Fig. 4d) shows three signals. The first centered at 61 ppm is due to the C-6 carbon of the glucose units. The other two located at about 68 and 71 ppm represent the cinnamyl allylic CH2 involved in the ether linkages (C-7), in agreement with the literature.49 The presence of two peaks may be due to different sites of grafting onto starch chains. Nevertheless, these signals, together with the absence of the peak at 46 ppm related to the allyl chloride, definitely prove the successful covalent grafting of cinnamyl moieties on yuca starch.
It is worth mentioning that the C-1 carbon signal is sensitive to starch crystalline conformations.13 As a matter of fact, the signal shape of the C-1 resonance of yuca starch is typical of crystalline starches, while the C-1 line shape of derivatized samples (CS1–3) indicates amorphization as a result of the processing, in agreement with XRD analyses (see Fig. S3, ESI†).
In summary, solution and solid-state NMR spectroscopy techniques definitively confirm the covalent ether bond of yuca starch with the cinnamyl moiety, while FTIR analysis qualitatively allows the identification of typical signals only with high DS samples.
It is expected that the solubility of starch derivatives could be influenced by the degree of substitution, since apolar groups derivatize the polar and hydrogen bond donors hydroxy groups. Hence, solubility behavior was studied on starch cinnamyl ethers CS1–3. Low substituted CS1 (DS = 0.09) was soluble in demineralized water; CS2 (DS = 0.33) was soluble in ethanol after a long mixing time at room temperature but not in water, whereas the CS3 derivative (DS > 1), was insoluble in polar solvents such as water, ethanol and acetone both at r.t. and under reflux, while it could be dissolved in toluene at 90 °C. Thus, tuning the degree of substitution allows starch to become water soluble when slightly functionalized, disturbing the strong intra- and intermolecular hydrogen bond network, while higher derivatization increases its solubility in less polar organic solvents, possibly influencing its applicability.
The ability to absorb light is at the basis for photoinduced reactions. In order to check this ability for the subsequent photo-crosslinking reaction, the optical properties of the starch-cinnamyl derivatives were investigated (Fig. S4, ESI†). Due to the solubility features in water of the derivatives, the UV-vis study was performed exclusively on the low substitution sample, namely CS1, and compared with cinnamyl chloride and pristine starch (Fig. S4a, ESI†). As expected, pristine yuca starch does not display any absorption; maximum absorption of CINN-Cl and CS1 was detected at 257 and 252 nm wavelength, respectively. The low hypsochromic shift in the absorption band for cinnamyl-starch may be reasonably related to the substitution of the allyl chloride with the ethereal oxygen, further confirming the occurrence of the etherification reaction, even in the low substitute sample. The optical properties of substituted derivative CS3, rendering it insoluble in water, were inspected by UV-DRS (Fig. S4b, ESI†). The diffuse reflectance spectrum of the CS3 powders revealed a remarkable depletion of the reflectance intensity, that is, an increase of the absorption ability, when compared to pristine starch; the observed behavior is an additional evidence of the cinnamyl chromophore grafting.
TGA analysis enables assessing both the thermal stability and the alteration of the degradation behavior of starch as a function of the substitution degree and distribution of cinnamyl units. These properties constitute an important attribute for its potential use in packaging products. Fig. 5 summarizes the TGA profiles of CS1, CS2 and CS3 samples in comparison to pristine yuca starch.
 | ||
| Fig. 5 TGA of pristine yuca starch (PS), compared to starch cinnamyl ethers CS1 (DS = 0.09), CS2 (DS = 0.33) and CS3 (DS = 1.24). | ||
The thermal degradation of pristine starch (PS) under a nitrogen atmosphere resulted in a two-step process, starting with a dehydration step between 35 °C and 144 °C corresponding to a weight loss of about 10%, followed by the decomposition of the polysaccharide initiating at 270 °C with a maximum weight loss at 297 °C. In the case of starch ethers with a lower DS (CS1 and CS2), a multi-step decomposition was observed, indicating the presence of non-functionalized and functionalized fractions of the polymer.46 As long as the substitution degree increases (i.e. CS3 sample), a single step profile is detected as well as a slight enhancement of the thermal stability, indicating a more homogeneous derivatization of polymer chains. These changes are better appreciated in the first derivative function (Fig. 5, inset).
3.3 Starch cinnamyl ether photodimerization
Cinnamyl moieties are known to give photocatalyzed reactions, based on a [2+2] pericyclic mechanism, upon exposure to irradiation at a suitable wavelength. Competing reactions have been observed by researchers, such as thermal cross-linking through a radical mechanism,50 and double bond isomerization (Scheme 4). The starch-cinnamyl ether CS1 (DS 0.09) was subjected to photoinduced dimerization by exposure at 365 nm (Scheme 4). The wavelength selection was based on the UV-Vis spectrum, showing a maximum absorption at ∼250 nm, featuring an extended tail terminating at ∼370 nm (ESI,† Fig. S4a). The study was performed exclusively on the low substitution sample due to its water solubility. | ||
| Scheme 4 Cross-linking products expected by (a) photoinduced reaction and (b) thermal radical reaction; inset: double bond isomerization by-product. New bonds are highlighted in red (Ph = phenyl). | ||
The evaluation of the absorbance in the UV range is a commonly used method to monitor cycloaddition reactions in cinnamyl derivatives,47 where the decrease in the peak intensity of the C–C double bond is considered diagnostic of the cycloaddition reaction. Thus, the photo-cross-linking ability of the cinnamyl-substituted starch was investigated by UV-Vis spectroscopy after irradiation at λ ∼ 365 nm and, subsequently, at λ = 254 nm (Fig. 6).
 | ||
| Fig. 6 (a) UV-Vis spectra of CS1 sample after exposure at λ ∼ 365 nm and, subsequently, at λ = 254 nm and (b) maximum absorption plotted against UV irradiation time at λ = 254 nm. | ||
A progressive decrease of the absorption band at ∼250 nm was detected, especially upon irradiation at λ = 254 nm; this observation can be ascribed to the conversion of double bonds into a saturated cyclobutane ring, thus losing p-conjugation with the benzene ring (Fig. 6a).51 The variation of the maximum absorbance (λ ∼ 250 nm) plotted against the irradiation time (Fig. 6b), showed an exponential decay until a plateau was reached after 140 min of irradiation. These preliminary results confirm that the double bonds in cinnamyl ether starch are photoreactive. However, UV-Vis spectroscopy does not give any insight into the reaction products (and related mechanism), i.e. photodimerization vs. isomerization. Here, for the first time on such complex derivatives, the photodimerization products were further studied through 1H-NMR analysis (Fig. 7 and Fig. S5, S6 in the ESI†). In order to overcome solubility and sensitivity problems in the NMR analysis, photodimerization was performed on the CS2 starch ether sample in DMSO-d6 and irradiated at λ ∼ 365 nm for 24 h. The cyclobutane signals, resonating between 3 ppm and 4.5 ppm, are usually considered a proof of the photodimerization reaction.52–56 However, these diagnostic peaks are expected in the crowded region of the starch signals. Upon UV exposure, NMR spectra showed changes in the aromatic and vinyl signals (shaded areas) and new peaks (*) at ∼5.9 and ∼6.9 ppm (Fig. 7). The new signals at ∼5.9 and ∼6.9 ppm can be ascribed to H-8 and H-9 in a Z configuration, as a result of photoinduced isomerization. Indeed, in the Z configuration, vinyl hydrogens resonate slightly more upfield.51 Even so, the decrease of the integral value of vinyl protons with respect to the aromatic H11–H13 may suggest partial photodimerization. This observation is an interesting hint deserving further investigation. Taken together, data indicate that, under the used experimental conditions, a competition between the two processes occurs, as widely reported for cinnamyl derivatives in several literature studies.53,54
 | ||
| Fig. 7 1H-NMR spectra of the CS2 starch ether in DMSO-d6 before and after 24 h UV exposure at λ ∼ 365 nm. Shaded areas highlight changes in the spectra and * indicates new peaks; relevant integrals are indicated (I). | ||
In summary, UV-Vis and NMR investigations support a partial but successful cross-linking reaction through the [2+2] cycloaddition of the double bonds of the starch cinnamyl units. Further investigations are needed in order to study the best degree of substitution and conditions (i.e. solvent) in order to reduce the isomerization side-reaction.
3.4 Cinnamyl starch film
To provide proof of the potential applicability of the novel bio-based polymer, films were produced by the solvent-casting technique. CS1 derivatives were used, due to their water solubility (a biocompatible solvent).The obtained film appears homogeneous and transparent (Fig. 8a). The observation of the films under a TLC UV lamp at λ = 254 nm or λ ∼ 365 nm shows emission in the Vis range as a qualitative confirmation of the surface exposure of the cinnamyl chromophoric units (Fig. 8b and c). Control films made by pristine starch are used as control (Fig. 8d–f).
 | ||
| Fig. 8 (a) Drop-cast film of cinnamyl ether CS1; film under UV lamp at (b) 254 nm and (c) at 365 nm wavelengths; (d) drop-cast film of pristine starch; film under UV lamp at (e) 254 nm and (f) at 365 nm wavelengths. | ||
4. Conclusions
An innovative bio-based polymer based on starch grafted with cinnamyl moieties was synthesized. FTIR, solution and solid-state NMR definitively demonstrate the effectiveness of the grafting process through a covalent ether bond. The degree of substitution can be tuned by varying the reagent ratio. Interestingly, starch is converted into a water soluble bio-inspired polymer when the degree of substitution is low, allowing film formation under biocompatible conditions.The crosslinking ability of the cinnamyl moieties was effective at λ ≥ 254 nm as demonstrated by UV-Vis spectra and suggested by solution-phase 1H-NMR analysis. NMR studies provided a more detailed understanding of the photochemical process, revealing a double bond isomerization competing with the cycloaddition and cyclobutane ring formation.
These outcomes appear encouraging and demonstrate the efficacy of the methodological approach adopted in supplying a bio-based light responsive system potentially applicable to packaging products.
Author contributions
SP, SFO, DB, EC, and LZ: conceptualization, investigation, visualization, data curation, writing – original draft, writing – review and editing; LR, SDV, AV, AS, and PM: investigation; SD, RB, MD, and LC: conceptualization, resources, writing – review and editing, supervision, funding acquisition, and project administration.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
The authors would like to acknowledge Fondazione Cariplo for funding the project Biostar-Pack (2020-0993) and funds by the European Union - Next Generation EU, Mission 4 Component 2 - CUP H53D23004450006. The authors would like to thank Dr Giorgio Emanuele Patriarca for the 1H, 13C, APT, and HSQC NMR spectra.References
- H. Xu, H. Canisag, B. Mu and Y. Yang, Robust and Flexible Films from 100% Starch Cross-Linked by Biobased Disaccharide Derivative, ACS Sustainable Chem. Eng., 2015, 3(11), 2631–2639, DOI:10.1021/acssuschemeng.5b00353.
- Z. Li, J. Wang, L. Cheng, Z. Gu, Y. Hong and A. Kowalczyk, Improving the Performance of Starch-Based Wood Adhesive by Using Sodium Dodecyl Sulfate, Carbohydr. Polym., 2014, 99, 579–583, DOI:10.1016/j.carbpol.2013.08.062.
- H. Ibrahim, M. Farag, H. Megahed and S. Mehanny, Characteristics of Starch-Based Biodegradable Composites Reinforced with Date Palm and Flax Fibers, Carbohydr. Polym., 2014, 101, 11–19, DOI:10.1016/j.carbpol.2013.08.051.
- J. J. G. Van Soest, S. H. D. Hulleman, D. De Wit and J. F. G. Vliegenthart, Crystallinity in Starch Bioplastics, Ind. Crops Prod., 1996, 5(1), 11–22, DOI:10.1016/0926-6690(95)00048-8.
- T. Ghosh Dastidar and A. Netravali, Cross-Linked Waxy Maize Starch-Based “Green” Composites, ACS Sustainable Chem. Eng., 2013, 1(12), 1537–1544, DOI:10.1021/sc400113a.
- M. K. Marichelvam, M. Jawaid and M. Asim, Corn and Rice Starch-Based Bio-Plastics as Alternative Packaging Materials, Fibers, 2019, 7(4), 32, DOI:10.3390/fib7040032.
- M. K. Marichelvam, P. Manimaran, M. R. Sanjay, S. Siengchin, M. Geetha, K. Kandakodeeswaran, P. Boonyasopon and S. Gorbatyuk, Extraction and Development of Starch-Based Bioplastics from Prosopis Juliflora Plant: Eco-Friendly and Sustainability Aspects, Curr. Res. Green Sustainable Chem., 2022, 5, 100296, DOI:10.1016/j.crgsc.2022.100296.
- K. J. Falua, A. Pokharel, A. Babaei-Ghazvini, Y. Ai and B. Acharya, Valorization of Starch to Biobased Materials: A Review, Polymers, 2022, 14(11), 2215, DOI:10.3390/polym14112215.
- S. H. Tay, S. C. Pang and S. F. Chin, A Facile Approach for Controlled Synthesis of Hydrophilic Starch-based Nanoparticles from Native Sago Starch, Starch/Staerke, 2012, 64(12), 984–990, DOI:10.1002/star.201200056.
- D. Le Corre and H. Angellier-Coussy, Preparation and Application of Starch Nanoparticles for Nanocomposites: A Review, React. Funct. Polym., 2014, 85, 97–120, DOI:10.1016/j.reactfunctpolym.2014.09.020.
- R. Santayanon and J. Wootthikanokkhan, Modification of Cassava Starch by Using Propionic Anhydride and Properties of the Starch-Blended Polyester Polyurethane, Carbohydr. Polym., 2003, 51(1), 17–24, DOI:10.1016/S0144-8617(02)00109-1.
- S. Garg and A. K. Jana, Characterization and Evaluation of Acylated Starch with Different Acyl Groups and Degrees of Substitution, Carbohydr. Polym., 2011, 83(4), 1623–1630, DOI:10.1016/j.carbpol.2010.10.015.
- D. Bugnotti, S. Dalle Vacche, L. H. Esposito, E. Callone, S. F. Orsini, R. Ceccato, M. D’Arienzo, R. Bongiovanni, S. Dirè and A. Vitale, Structure of Starch–Sepiolite Bio-Nanocomposites: Effect of Processing and Matrix–Filler Interactions, Polymers, 2023, 15(5), 1207, DOI:10.3390/polym15051207.
- F. Garavand, M. Rouhi, S. H. Razavi, I. Cacciotti and R. Mohammadi, Improving the Integrity of Natural Biopolymer Films Used in Food Packaging by Crosslinking Approach: A Review, Int. J. Biol. Macromol., 2017, 104, 687–707, DOI:10.1016/j.ijbiomac.2017.06.093.
- J. Delville, C. Joly, P. Dole and C. Bliard, Solid State Photocrosslinked Starch Based Films: A New Family of Homogeneous Modified Starches, Carbohydr. Polym., 2002, 49(1), 71–81, DOI:10.1016/S0144-8617(01)00302-2.
- Z. Zhang, G. Sèbe, X. Wang and K. C. Tam, UV-Absorbing Cellulose Nanocrystals as Functional Reinforcing Fillers in Poly(Vinyl Chloride) Films, ACS Appl. Nano Mater., 2018, 1(2), 632–641, DOI:10.1021/acsanm.7b00126.
- L. García-Guzmán, G. Cabrera-Barjas, C. G. Soria-Hernández, J. Castaño, A. Y. Guadarrama-Lezama and S. Rodríguez Llamazares, Progress in Starch-Based Materials for Food Packaging Applications, Polysaccharides, 2022, 3(1), 136–177, DOI:10.3390/polysaccharides3010007.
- J. D. Guzman, Natural Cinnamic Acids, Synthetic Derivatives and Hybrids with Antimicrobial Activity, Molecules, 2014, 19(12), 19292–19349, DOI:10.3390/molecules191219292.
- A. Mustafa, Dimerization Reactions in Sunlight, Chem. Rev., 1952, 51(1), 1–23, DOI:10.1021/cr60158a001.
- G. M. J. Schmidt, Photodimerization in the Solid State, Pure Appl. Chem., 1971, 27(4), 647–678, DOI:10.1351/pac197127040647.
- S. H. Moon, H. J. Hwang, H. R. Jeon, S. J. Park, I. S. Bae and Y. J. Yang, Photocrosslinkable Natural Polymers in Tissue Engineering, Front. Bioeng. Biotechnol., 2023, 11, 1127757, DOI:10.3389/fbioe.2023.1127757.
- N. Kawatsuki, K. Matsuyoshi, M. Hayashi, H. Takatsuka and T. Yamamoto, Photoreaction of Photo-Cross-Linkable Methacrylate Polymer Films Comprising 2-Cinnamoyloxyethoxybiphenyl Side Group by Linearly Polarized Ultraviolet Light and Liquid Crystal Alignment on the Resultant Films, Chem. Mater., 2000, 12(6), 1549–1555, DOI:10.1021/cm000066t.
- D. J. Adams, S. Chappellet, F. Lincker, M. Ibn-Elhaj, B. Watts, M. Iannuzzi, D. Šišak Jung, C. A. Pignedoli and D. Passerone, Identifying Photoreaction Products in Cinnamate-Based Photoalignment Materials, J. Phys. Chem. C, 2014, 118(28), 15422–15433, DOI:10.1021/jp504765f.
- Z. Wang, X. Zhuang, Y. Chen, B. Wang, J. Yu, W. Huang, T. J. Marks and A. Facchetti, Cinnamate-Functionalized Natural Carbohydrates as Photopatternable Gate Dielectrics for Organic Transistors, Chem. Mater., 2019, 31(18), 7608–7617, DOI:10.1021/acs.chemmater.9b02413.
- R. Sepehrifar, R. I. Boysen, B. Danylec, Y. Yang, K. Saito and M. T. W. Hearn, Design, Synthesis and Application of a New Class of Stimuli-Responsive Separation Materials, Anal. Chim. Acta, 2017, 963, 153–163, DOI:10.1016/j.aca.2017.01.061.
- M. Abdallh, C. Yoshikawa, M. T. W. Hearn, G. P. Simon and K. Saito, Photoreversible Smart Polymers Based on 2π + 2π Cycloaddition Reactions: Nanofilms to Self-Healing Films, Macromolecules, 2019, 52(6), 2446–2455, DOI:10.1021/acs.macromol.8b01729.
- A. Bazin, A. Duval, L. Avérous and E. Pollet, Synthesis of Bio-Based Photo-Cross-Linkable Polyesters Based on Caffeic Acid through Selective Lipase-Catalyzed Polymerization, Macromolecules, 2022, 55(11), 4256–4267, DOI:10.1021/acs.macromol.2c00499.
- L. Du, Z.-Y. Xu, C.-L. Huang, F.-Y. Zhao, C.-J. Fan, J. Dai, K.-K. Yang and Y.-Z. Wang, From a Body Temperature-Triggered Reversible Shape-Memory Material to High-Sensitive Bionic Soft Actuators, Appl. Mater. Today, 2020, 18, 100463, DOI:10.1016/j.apmt.2019.100463.
- Q. Hu, Y. Zhang, T. Wang, W. Sun and Z. Tong, pH Responsive Strong Polyion Complex Shape Memory Hydrogel with Spontaneous Shape Changing and Information Encryption, Macromol. Rapid Commun., 2021, 42(9), 2000747, DOI:10.1002/marc.202000747.
- M. Sangermano, I. Roppolo and A. Chiappone, New Horizons in Cationic Photopolymerization, Polymers, 2018, 10(2), 136, DOI:10.3390/polym10020136.
- M. Sangermano, N. Razza and J. V. Crivello, Cationic UV-Curing: Technology and Applications: Cationic UV-Curing: Technology and Applications, Macromol. Mater. Eng., 2014, 299(7), 775–793, DOI:10.1002/mame.201300349.
- R. Bongiovanni, S. D. Vacche and A. Vitale, Photoinduced Processes as a Way to Sustainable Polymers and Innovation in Polymeric Materials, Polymers, 2021, 13(14), 2293, DOI:10.3390/polym13142293.
- G. Yılmaz and Y. YagciMacromolecular Engineering by Photochemical Routes, Macromolecular Engineering, John Wiley & Sons, Ltd, 2022, pp. 1–18 DOI:10.1002/9783527815562.mme0054.
- H. Li, Y. Ma, X. Gao, G. Chen and Z. Wang, Probing the Structure-Antioxidant Activity Relationships of Four Cinnamic Acids Porous Starch Esters, Carbohydr. Polym., 2021, 256, 117428, DOI:10.1016/j.carbpol.2020.117428.
- Y. Du, L. Niu, X. Song, J. Niu, C. Zhang and K. Zhi, Dual-Modified Starch as Particulate Emulsifier for Pickering Emulsion: Structure, Safety Properties, and Application for Encapsulating Curcumin, Int. J. Biol. Macromol., 2024, 266, 131206, DOI:10.1016/j.ijbiomac.2024.131206.
- R. Zhang, F. Chu, Y. Hu, H. Hu, Y. Hu, H. Liu, C. Huo and H. Wang, Preparation of Photo-Crosslinking Starch Colloidal Particles, Starch/Staerke, 2020, 72(5–6), 1900175, DOI:10.1002/star.201900175.
- A. Rasouli, Y. Jamali, E. Tajkhorshid, O. Bavi and H. N. Pishkenari, Mechanical Properties of Ester- and Ether-DPhPC Bilayers: A Molecular Dynamics Study, J. Mech. Behav. Biomed. Mater., 2021, 117, 104386, DOI:10.1016/j.jmbbm.2021.104386.
- M. J. Tizzotti, M. C. Sweedman, D. Tang, C. Schaefer and R. G. Gilbert, New 1H NMR Procedure for the Characterization of Native and Modified Food-Grade Starches, J. Agric. Food Chem., 2011, 59(13), 6913–6919, DOI:10.1021/jf201209z.
- X. L. Wu and K. W. Zilm, Complete Spectral Editing in CPMAS NMR, J. Magn. Reson., Ser. A, 1993, 102(2), 205–213, DOI:10.1006/jmra.1993.1092.
- V. Tabaglio, A. Fiorini, V. Ndayisenga, A. Ndereyimana, A. Minuti, R. Nyembo Nyembo, D. Nyembo Ngoy and G. Bertoni, Sustainable Intensification of Cassava Production towards Food Security in the Lomami Province (DR Congo): Role of Planting Method and Landrace, Agronomy, 2023, 13(1), 228, DOI:10.3390/agronomy13010228.
- N. Charoenkul, D. Uttapap, W. Pathipanawat and Y. Takeda, Simultaneous Determination of Amylose Content & Unit Chain Distribution of Amylopectins of Cassava Starches by Fluorescent Labeling/HPSEC, Carbohydr. Polym., 2006, 65(1), 102–108, DOI:10.1016/j.carbpol.2005.12.030.
- V. Guillard, S. Gaucel, C. Fornaciari, H. Angellier-Coussy, P. Buche and N. Gontard, The Next Generation of Sustainable Food Packaging to Preserve Our Environment in a Circular Economy Context, Front. Nutr., 2018, 5 DOI:10.3389/fnut.2018.00121.
- L. Zoia, A. W. T. King and D. S. Argyropoulos, Molecular Weight Distributions and Linkages in Lignocellulosic Materials Derivatized from Ionic Liquid Media, J. Agric. Food Chem., 2011, 59(3), 829–838, DOI:10.1021/jf103615e.
- Z. Zhou, D. L. Topping, M. K. Morell and A. R. Bird, Changes in Starch Physical Characteristics Following Digestion of Foods in the Human Small Intestine, Br. J. Nutr., 2010, 104(4), 573–581, DOI:10.1017/S0007114510000875.
- R. Hoover, Composition, Molecular Structure, and Physicochemical Properties of Tuber and Root Starches: A Review, Carbohydr. Polym., 2001, 45(3), 253–267, DOI:10.1016/S0144-8617(00)00260-5.
- R. F. Tester, J. Karkalas and X. Qi, Starch—Composition, Fine Structure and Architecture, J. Cereal Sci., 2004, 39(2), 151–165, DOI:10.1016/j.jcs.2003.12.001.
- Y.-C. Shi, T. Capitani, P. Trzasko and R. Jeffcoat, Molecular Structure of a Low-Amylopectin Starch and Other High-Amylose Maize Starches, J. Cereal Sci., 1998, 27(3), 289–299, DOI:10.1006/jcrs.1997.9998.
- B. F. Bergel, S. Dias Osorio, L. M. Da Luz and R. M. C. Santana, Effects of Hydrophobized Starches on Thermoplastic Starch Foams Made from Potato Starch, Carbohydr. Polym., 2018, 200, 106–114, DOI:10.1016/j.carbpol.2018.07.047.
- Y. Kasashima, A. Uzawa, K. Hashimoto, T. Nishida, K. Murakami, T. Mino, M. Sakamoto and T. Fujita, Synthesis of Cinnamyl Ethers from α-Vinylbenzyl Alcohol Using Iodine as Catalyst, J. Oleo Sci., 2010, 59(10), 549–555, DOI:10.5650/jos.59.549.
- S.-J. Sung, K.-Y. Cho, H. Hah, J. Lee, H.-K. Shim and J.-K. Park, Two Different Reaction Mechanisms of Cinnamate Side Groups Attached to the Various Polymer Backbones, Polymer, 2006, 47(7), 2314–2321, DOI:10.1016/j.polymer.2006.02.003.
- P. L. Egerton, E. Pitts and A. Reiser, Photocycloaddition in Solid Poly(Vinyl Cinnamate). The Photoreactive Polymer Matrix as an Ensemble of Chromophore Sites, Macromolecules, 1981, 14(1), 95–100, DOI:10.1021/ma50002a019.
- M. D. Cohen, G. M. J. Schmidt and F. I. Sonntag, 384. Topochemistry. Part II. The Photochemistry of Trans-Cinnamic Acids, J. Chem. Soc., 1964, 2000, 10.1039/jr9640002000.
- G. M. J. Schmidt, 385. Topochemistry. Part III. The Crystal Chemistry of Some Trans-Cinnamic Acids, J. Chem. Soc., 1964, 2014, 10.1039/jr9640002014.
- P. L. Egerton, E. M. Hyde, J. Trigg, A. Payne, P. Beynon, M. V. Mijovic and A. Reiser, Photocycloaddition in Liquid Ethyl Cinnamate and in Ethyl Cinnamate Glasses. The Photoreaction as a Probe into the Micromorphology of the Solid, J. Am. Chem. Soc., 1981, 103(13), 3859–3863, DOI:10.1021/ja00403a039.
- H. Amjaour, Z. Wang, M. Mabin, J. Puttkammer, S. Busch and Q. R. Chu, Scalable Preparation and Property Investigation of a Cis-Cyclobutane-1,2-Dicarboxylic Acid from β-Trans-Cinnamic Acid, Chem. Commun., 2018, 55(2), 214–217, 10.1039/C8CC08017H.
- I. Fonseca, S. E. Hayes, B. Blümich and M. Bertmer, Temperature Stability and Photodimerization Kinetics of β-Cinnamic Acid and Comparison to Its α-Polymorph as Studied by Solid-State NMR Spectroscopy Techniques and DFT Calculations, Phys. Chem. Chem. Phys., 2008, 10(38), 5898, 10.1039/b806861e.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb01406e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |