
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A water playground for peptide re-assembly from fibrils to plates†
Simone
Adorinni
 a,
Marina
Kurbasic
a,
Ana M.
Garcia
ab,
Slavko
Kralj
cd,
Ottavia
Bellotto
a,
Erica
Scarel
a,
Paolo
Pengo
a,
Rita
De Zorzi
a,
Michele
Melchionna
a,
Attilivio V.
Vargiu
*e and
Silvia
Marchesan
*a
a,
Marina
Kurbasic
a,
Ana M.
Garcia
ab,
Slavko
Kralj
cd,
Ottavia
Bellotto
a,
Erica
Scarel
a,
Paolo
Pengo
a,
Rita
De Zorzi
a,
Michele
Melchionna
a,
Attilivio V.
Vargiu
*e and
Silvia
Marchesan
*a
aDepartment of Chemical and Pharmaceutical Sciences, University of Trieste, Via. Giorgieri 1, 34127 Trieste, Italy. E-mail: smarchesan@units.it
bFacultad de Ciencias y Tecnologías Químicas, Instituto Regional de Investigación Científica Aplicada (IRICA), Universidad de Castilla-La Mancha, Ciudad Real 13071, Spain
cMaterials Synthesis Department, Jožef Stefan Institute, Jamova 39, 1000 Ljubljana, Slovenia
dDepartment of Pharmaceutical Technology, Faculty of Pharmacy, University of Ljubljana, 1000 Ljubljana, Slovenia
eDepartment of Physics, University of Cagliari, Cittadella Universitaria, S.P. 8 km. 0.7, 09042 Monserrato (CA), Italy. E-mail: vargiu@dsf.unica.it
First published on 29th October 2024
Abstract
Short-peptide amyloid assembly and disassembly play crucial roles in various research fields, which range from addressing pathologies that lack therapeutic solutions to the development of innovative soft (bio)materials. Hydrogels from short peptides typically show thermo-reversible gel-to-sol transition, whereby fibrils disassemble upon heating, and re-assemble upon cooling down to room temperature (rt). Despite ongoing intense research studies in this area, the majority focus on peptide–peptide interaction and neglect the structuring role of water in peptide supramolecular behavior. This study describes an unprotected tetrapeptide gelator that forms highly stable fibrils which, upon heating, re-organize into plates that persist upon cooling to rt. All-atom molecular dynamics (MD) simulations and experimental methods reveal water as a key player in the thermodynamics that accompany this irreversible morphological transition, and advance our understanding of supramolecular structures.
Introduction
Amyloid assembly and disassembly have attracted great interest in recent years, with Fmoc-derived gelators having gained wide popularity.1 By contrast, mastering the supramolecular behavior of short peptides devoid of synthetic appendages, which are more relevant to natural processes, is still very challenging. On one hand, amyloid disassembly could be of high value in a therapeutic context, yet fibril re-assembly is unavoidable when it is the thermodynamic product. On the other hand, general rules for the design of building blocks of three or four unprotected amino acids (aa) that self-organize into macroscopic – and ideally functional – hydrogels, as opposed to insoluble aggregates, are not yet fully elucidated,2 although integration of experiments with computational tools is enabling fast progress in the field.3It is well-established that amyloids have an amphipathic nature. Hydrophobic components segregate in steric zippers that exclude water and stabilize the structure.4 In particular, phenylalanine (Phe)-based zippers have been reported for minimalistic peptides.5 Yet, hydrophilic components must favorably interact with water to yield a bulk hydrogel. Gel-to-sol transition can be conveniently induced by heating, and usually the gel reforms upon cooling to rt. Reported examples of unprotected gelling peptides as short as three6 or four7 residues indeed display such thermo-reversible behavior.
For the design of self-assembling short peptides, the combination of D- and L-residues in heterochiral sequences is an attractive approach. Syndiotactic stereochemistry in cyclic peptides is well-known to permit the formation of nanotubes, with potential applications spanning from membrane transporters to hydrogels in confined droplets.8 We recently introduced D-aa in non-gelling, L-tripeptides as a convenient strategy to modulate self-assembly in linear sequences.6a Molecular dynamics (MD) simulations suggest that this approach may favor the segregation of hydrophobic and hydrophilic components on opposite faces of β-structures, thus yielding amphiphilic assemblies that favorably interact with water and gel.6aD-aa are advantageous for their known resistance to protease-mediated hydrolysis, and self-assembly of their derivatives has been used to develop new therapies.9 Besides, D-aa can interfere with amyloid fibrillation and hold therapeutic potential in related pathologies.10 There is thus scope to study self-assembling peptides with D-aa.
We report here the first unprotected D,L-tetrapeptide (Fig. 1) that assembles into a highly stable hydrogel at physiological pH and undergoes a thermally-induced irreversible re-organization from fibrils to plates. We support experimental data with all-atom MD simulations that focus not only on peptide–peptide but also on peptide–water interactions. Interestingly, the supramolecular re-organization is not dictated by a key conformational change of the peptide, but rather by an irreversible change in its hydration, revealing water as a key player in the process.
 | ||
| Fig. 1 (a) Tetrapeptide structure. (b) Zwitterion conformation resulting from MD simulations (aliphatic and aromatic side chains in grey and purple, respectively, with transparent molecular surfaces). (c) Tetrapeptide stack resulting from MD simulations. H-bonds between amides (dashed lines) display a pattern similar to β-sheets. (d) Ramachandran plot reporting the values of ϕ and ψ for the 2nd (D-Leu) and 3rd residues (L-Phe) of the tetrapeptide (the only ones for which both angles can be calculated). | ||
Results and discussion
The peptide was designed with two aliphatic D-aa at the N-terminus and the L-Phe-Phe self-assembling motif11 at the C-terminus, synthesized in solid phase, purified by HPLC and characterized by NMR and ESI-MS (Fig. 1a and ESI,† Section S1). In particular, the design included the elongated sequence of the reported gelator DLeu-Phe-Phe5b with D-Nle, whose linear sidechain favors intermolecular packing,6a and a heterochiral sequence to enable an amphipathic conformation, thanks to the segregation of aliphatic sidechains on the opposite side of the hydrophilic backbone.6a While design rules for self-assembling D,L-tripeptides are emerging,3b,6a to date there is limited understanding of the supramolecular behavior of unprotected D,L-tetrapeptides, and filling the gap answers an interesting and still open research question.12 It should be noted that even subtle chemical modifications can dramatically affect the supramolecular behavior. N-Acetylation of DLeu-Phe-Phe completely hindered self-assembly, and analogous results were obtained for the L-homochiral analog Nle-Leu-Phe-Phe (see ESI,† Section S8). These data confirmed the importance of both termini in establishing intermolecular salt bridges, as evident from the single-crystal XRD structure of a similar D,L-tripeptide, as well as heterochirality in hydrophobic short peptides to enable hydrogelation of amphipathic superstructures.6aMD simulations of DNle-DLeu-Phe-Phe in explicit water revealed that the most representative conformation of the zwitterion is a turn with all hydrophobic chains on the same side of the backbone (Fig. 1b). Such isotactic spatial arrangement was recently reported to be key for the self-assembly of hydrophobic D,L-tripeptides into stable hydrogels.6a In both cases, the backbone is kinked. Peptide molecules stack thanks to hydrogen-bonding between amides, with a pattern similar to β-sheets and the aromatic rings running up the stack in a helical arrangement (Fig. 1c). Dihedral angles of the peptide backbone were calculated for the 2nd (D-Leu) and the 3rd residue (L-Phe) and are shown in the Ramachandran plot (Fig. 1d).
1H-NMR spectra (see ESI,† Sections S1 and S2) revealed 3JNHCαH ∼ 4 Hz that was compatible with ϕ −60°, in contrast with the non-assembling homochiral L-tetrapeptide that displayed 3JNHCαH ∼ 8 Hz. The latter value is compatible with wider ϕ values and an extended conformation,13 as confirmed by MD simulations (see ESI,† Fig. S33). The presence of intramolecular H-bonds that could hold together the turn of the heterochiral tetrapeptide was verified by variable-temperature 1H-NMR spectroscopy, firstly in DMSO as a non-aggregating solvent (Fig. 2a–c), and then in the presence of water (Fig. 2d–f). 1H-NMR shifts of amide signals were visible and displayed a linear correlation with temperature from 298 K to 333 K, indicating no conformational loss. In particular, the NH chemical shifts of Leu and C-terminal Phe (Fig. 2a and c) displayed a temperature gradient Δδ/ΔT > −4.6 ppb K−1, which is a strong indication of involvement in H-bonding.14 Remarkably, the presence of water did not lead to loss of amide signals (Fig. 2d–f), indicating they were not exchanging with the solvent, although only the NH of Leu maintained a temperature coefficient within the expected range for H-bonds (Fig. 2d). Conversely, the same experiment carried out on the homochiral analog did not lead to any indication of H-bonds (see ESI,† Section S5).
 | ||
| Fig. 2 Intramolecular H-bonds in the heterochiral tetrapeptide. Variable-temperature 1H-NMR analysis of CONH signals in DMSO (a)–(c) and with 20% water (d)–(f). Note: higher amounts of water led to precipitation. | ||
In silico data were in qualitative agreement with experiments in solution. Detailed analyses were performed on the trajectories extracted from MD simulations of a single homo- or hetero-chiral peptide in explicit water. For each peptide, we performed a MD simulation of 1 μs in length at 298 K, followed by heating to 363 K in 20 ns, and finally an equilibrium simulation of 1 μs at this temperature. We analyzed the secondary structural content and the Ramachandran plot (reported only for the 2nd and 3rd residues of the tetrapeptide, that is those for which both angles can be calculated), the preferred conformations, the end-to-end intramolecular distances, the frequency of formation of intramolecular H-bonds, and the peptide hydration. The heterochiral peptide displayed an intrinsic propensity towards turns (Table S1, ESI†). Importantly, this feature seemed to be energy-driven and temperature-independent (i.e., there was no entropic gain with heating). The opposite was true for the homochiral analog, whose structural preference towards turns at rt was halved relative to the heterochiral isomer (Table S1, ESI†). The C1α-to-C4α distance (Fig. S34, ESI†) was significantly longer for the homochiral peptide (visiting extended conformations) relative to the heterochiral one (adopting turns). MD confirmed the formation of an intramolecular salt bridge between termini, and the engagement of Phe4 NH in intramolecular H-bonding, and of Phe3 and Leu NH to a lesser extent, in agreement with NMR data (Table S2, ESI†).
A turn stabilized by the intramolecular salt bridge between the charged termini was confirmed for the heterochiral peptide by single-crystal X-ray diffraction (XRD) (Fig. 3). Surprisingly, the NH of Leu and Phe3 were engaged in H-bonding, albeit intermolecularly with the CO of the same residues of an adjacent tetrapeptide molecule, as a distinctive feature of the solid phase, as opposed to the intramolecular H-bond of the peptide in solution. The dihedral angles of the crystal structures are compatible with one of the visited conformations observed by MD, albeit not the most representative one (Fig. S43, ESI†).
 | ||
| Fig. 3 Single-crystal XRD data (CCDC 2347110†) for the heterochiral tetrapeptide confirmed the turn conformation stabilized by the salt bridge between termini. The bent backbone is highlighted in yellow, while the hydrophobic sidechains of the central molecules in magenta. | ||
The gelator was first dissolved in alkaline phosphate buffer, thanks to repulsion between negative charges in its anionic form. Subsequent pH lowering to neutral triggered amyloid fibrillation of the zwitterion in samples as diluted as 0.050 wt%. When the concentration was increased to 0.67 wt% a self-supportive hydrogel was obtained (in the pH range 11.0–11.3), in contrast with the L-analog (see ESI,† Sections S7 and S8), confirming the key role played by the amino acid stereoconfiguration in the gelation process. For comparison, it is worth noting that two other reported unprotected tetrapeptide gelators had a minimum gelling concentration (mgc) of 17 wt% and 2.7 wt%,15 which are over 20- and 4-fold higher. Oscillatory rheology analyses (Fig. 4) showed the viscoelastic moduli reached a plateau within an hour, with a storage modulus G′ of 22.7 ± 0.147 kPa and a loss modulus G′′ of 2.06 ± 0.965 kPa (Fig. 4a). Stress sweep analyses showed a gel-to-sol transition at ca. 25 Pa (Fig. 4b), and frequency sweeps confirmed the hydrogel stability (Fig. 4c).
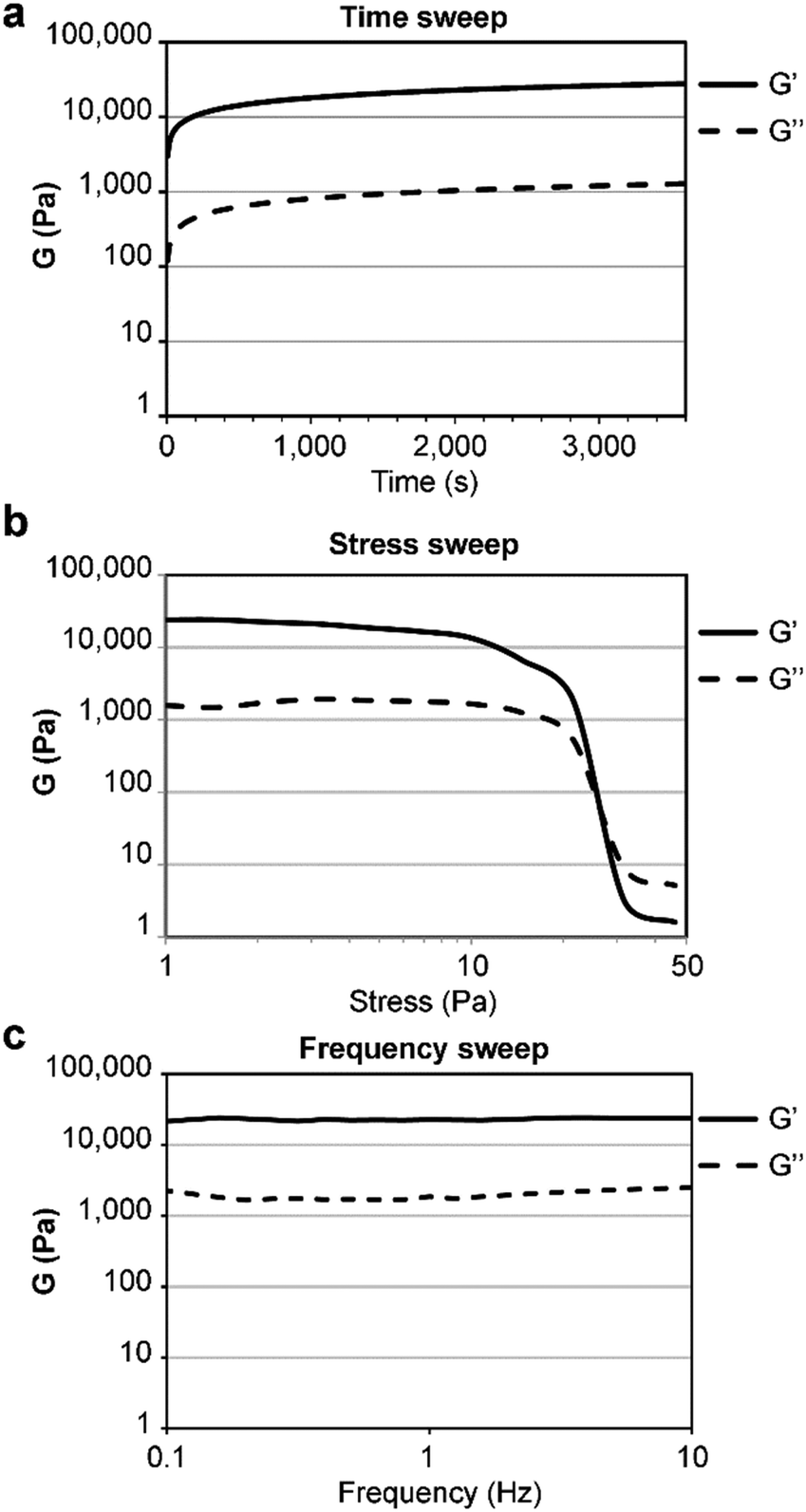 | ||
| Fig. 4 Oscillatory rheology of the heterochiral tetrapeptide hydrogel at 0.67 wt%. (a) Time sweep, (b) stress sweep, (c) frequency sweep. | ||
Interestingly, no cytotoxicity was found for the heterochiral peptide gel in live/dead assays on fibroblast cells (see ESI,† Section S9). Furthermore, the gel was highly resistant to protease digestion. Despite the presence of one natural peptide bond in the building block, the hydrogel was nearly unaltered after 24 h of treatment with a large excess of enzyme (see ESI,† Section S10). Interestingly, the amyloid assembly contributed to such resistance, since the majority of the tetrapeptide in solution was digested within a few hours. We infer that the amyloid structure displays dry regions of phenylalanine zippers5b,16 that reduce contact with water and thus provide protection against enzymatic hydrolysis.
Transmission electron microscopy (TEM) and atomic force microscopy (AFM) investigations revealed bundles of fibrils spanning the microscale in length (Fig. 5a and b). Heating up to 363 K was required to disassemble the stable supramolecular arrangement. Instead of dissolution, as typically observed for short peptide-based gels, an irreversible transition to plates occurred (Fig. 5c and d). We inferred that the partial loss of CD signal during heating could be ascribed to sedimentation of the plates (see ESI,† Fig. S40). Differential scanning calorimetry (DSC) confirmed the stability of the sample up to an endothermic transition at Tm = 362 K (see ESI,† Fig. S36).
 | ||
| Fig. 5 TEM (a) and (c) and AFM (b) and (d) micrographs of the heterochiral tetrapeptide forming fibrils at rt (a) and (b) which irreversibly converted into plates upon heating to 363 K (c) and (d). | ||
All-atom MD simulations of heterochiral peptides’ self-assembly revealed a general rigidification of the system after heating that persisted upon subsequent cooling to rt, with peptides sampling a significantly smaller range of conformations (see ESI,† Section S6). Initially (Fig. 6a), the supramolecular assemblies at rt gave rise to a 3D network containing water channels along three directions and compatible with the observed fibrillar hydrogel. During heating, solubility decreased, the hydrophobic peptides aggregated, and 5-to-9 water molecules per peptide molecule were released from solvation shells, leading to an increase in entropy and the formation of aggregates that are fully separated from each other along one direction (Fig. 6b). These aggregates could serve as seeds for the onset of plates, that is structures likely corresponding to a thermodynamic sink and thus leading to an irreversible transition. Re-assembly was driven by the entropic gain of the system due to the release of water molecules during the heating phase. Due to the temperature dependence of this term (TΔS) in the overall free energy balance of the process, this gain increases with increasing temperatures, leading the system to a deeper free energy minimum and rendering the process virtually irreversible.17 This picture is corroborated by several analyses including the calculation of the solvent accessible surface area (SASA), which decreases in the morphological transition, and the number of water molecules set free in the bulk phase, correlated with that reduction. The reduction in SASA (Table S4, ESI†), leading also to a reduction in the number of water–peptide H-bonds is, however, partly compensated by the increase in the number of such bonds involving only the peptides (Table S5, ESI†). Moreover, the overall number of bridging waters did not change before and after the heating/cooling steps. This is likely because virtually all the possible H-bonds of the termini and backbone were saturated in the MD simulation before the heating of the system, a picture consistent with the very minor changes in the nature of the surface exposed to the solvent (Fig. 6).
 | ||
| Fig. 6 Representative conformations extracted from MD simulations of heterochiral tetrapeptide assemblies at rt before (a) and after heating to 363 K and cooling to 298 K (b). Surfaces are colored by atom type (grey, white, blue, and red for C, H, N, and O respectively). | ||
Interestingly, the β-structure content was reduced, but no dramatic change to a different conformation was seen, as confirmed by MD simulations, FT-IR, circular dichroism (CD), and Thioflavin T fluorescence analyses (Fig. 7 and Fig. S40, ESI†). An increase of the temperature up to 363 K leads the peptide to sample different conformations, some corresponding to L-α-helices. However, this difference almost vanishes when simulating self-assembly of hundreds of tetrapeptides in explicit solvent (Fig. S35, ESI†), thus supporting the role of water thermodynamics in the process and the high stability of β-structures. Layered β-structures have been predicted to be the most stable secondary conformation for amyloid superstructures, due to the preference of backbones to engage in extended H-bonding to form sheets.18
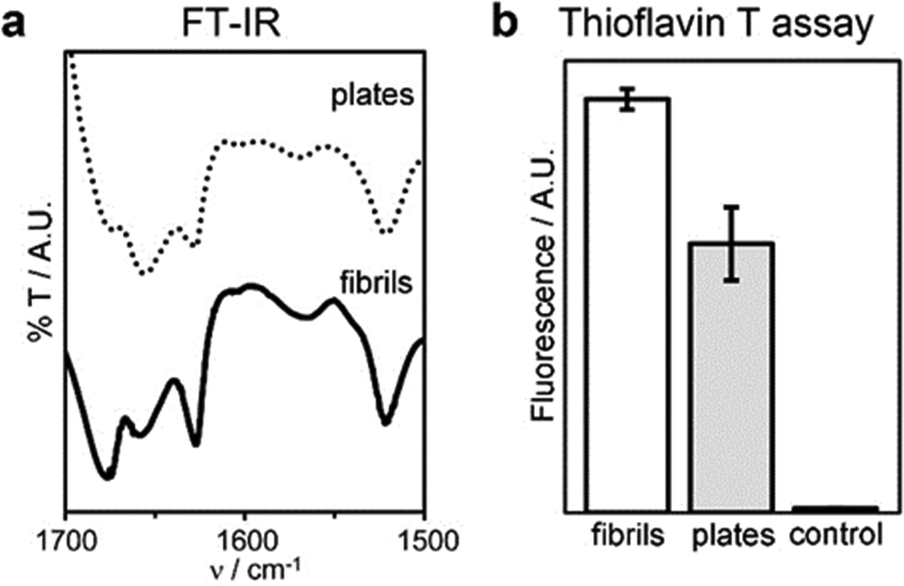 | ||
| Fig. 7 Conformation of fibrils and plates: (a) FT-IR spectra of the amide I–II regions, (b) thioflavin T fluorescence assay. | ||
Experimental section
Chemicals
2-Chlorotrytil resin, O-benzotriazole-N,N,N′,N′-tetramethyl-uronium-hexafluoro-phosphate (HBTU), 1-hydroxy-7-azabenzotriazole (HOAt), and Fmoc-protected amino acids were purchased from GL Biochem (Shanghai) Ltd. All solvents were purchased of analytical grade from Merck. All the other chemicals were from Sigma. High purity Milli-Q-water (MQ water) with a resistivity greater than 18 M Ω cm was obtained from an in-line Millipore RiOs/Origin system.Peptide synthesis and purification
Solid-phase peptide synthesis was performed as previously described.19 The crude peptide was too hydrophobic to be precipitated in cold ether, thus the majority of TFA was evaporated under argon flow, and the remaining oil was dissolved in a mixture of acetonitrile/water both with 0.1% of TFA and then purified by reverse-phase HPLC (Agilent Technologies). The HPLC Agilent 1260 Infinity system was equipped with a preparative gradient pump (1311B), semipreparative C-18 column (Kinetex, 5 microns, 100 Å, 250 × 10 mm, Phenomenex), autosampler (G1329B), photodiode array detector (G1315C). The gradient used consisted of acetonitrile (MeCN)/water with 0.1% TFA with the following program: t = 0–2 min. 30% MeCN; t = 12 min. 50% MeCN; t = 13 min. 95% MeCN; (heterochiral tetrapeptide tR = 11.7 min, homochiral tetrapeptide tR = 10.8 min, and acetylated tripeptide that was dissolved by adding 5% DMSO tR = 3.7 min); flow 3 ml/min. Analytical LC-MS traces are shown in the ESI† file to confirm purity. The compound was then freeze-dried to yield the corresponding peptide as a white fluffy powder. Peptide identity was verified by ESI-MS, 1H-NMR and 13C-NMR. 1H-NMR spectra were recorded at 400 MHz and 13C-NMR spectra were recorded at 101 MHz on a Varian Innova Instrument with chemical shift reported as ppm (in DMSO with tetramethylsilane as internal standard). ESI-MS spectra were recorded on an Agilent 6120 single quadrupole LC-MS system.Peptide self-assembly protocol
Each peptide was dissolved by 10 min of ultrasonication and heating in sodium phosphate buffer (0.1 M pH 12.0) to a final concentration of 30 mM and then an equal volume of sodium phosphate buffer (0.1 M pH 5.8) was added to get a final pH of 7.2 ± 0.1 and a final concentration of 15 mM.All-atom molecular dynamics (MD)
All-atom MD simulations and analyses were performed with the AMBER22 and AmberTools23 software packages.20 The parameters for D/L N-terminal norleucine were taken from the literature,21 and the ff19SB AMBER force field22 was employed together with an OPC model for water.23 Simulations of single peptides in solution. First, we performed MD simulations of a single homo-(Nle-Leu-Phe-Phe) and hetero-(DNle-DLeu-Phe-Phe) chiral peptide in explicit water as follows. The initial structure of the homochiral peptide was generated using the sequence command from the xleap tool of AMBER22 and relaxing the structure. The heterochiral peptide was built from the homochiral structure using VMD1.9.3.24 Starting from these structures, three consecutive restrained structural optimizations (up to 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 steps) were performed in the presence of harmonic restraints (k = 1 kcal mol−1 Å−1) applied to: (a) all non-hydrogenous atoms of the system; (b) backbone atoms; (c) Cα atoms. Reference structures at steps (b) and (c) were the final ones from the previous step. Next, up to 50000 cycles of unrestrained optimization were performed. Each system was then heated to 298 K in 1 ns via constant–pressure–temperature (NTP) MD simulations (using the isotropic Berendsen barostat and the Langevin thermostat) followed by an equilibration phase of 10 ns. Starting from the equilibrated structure, a MD simulation in the NVT ensemble of 1 μs in length was performed using a time step of 2 fs. Next, heating to 363 K in was simulated in 20 ns, followed by another equilibrium simulation of 1 μs in length at this temperature. Periodic boundary conditions were employed, and electrostatic interactions were estimated using the Particle Mesh Ewald scheme with a cutoff of 9.0 Å for the short-range evaluation in direct space and for Lennard–Jones interactions (with a continuum model correction for energy and pressure). Self-assembly. 5 independent MD simulations of the self-assembly of 512 heterochiral tetrapeptides in water solution were performed following published protocols.25 Briefly, the initial conformation of the assembling peptides was generated by placing their centers of mass on a 8 × 8 × 8 grid of 17.5 Å-spaced points. Initial orientations of peptides were randomized, and the systems were solvated with water molecules, for a total number of atoms around 235000. Initial structures of the 5 independent simulations were taken from the optimized structure of the corresponding peptides (simulation 1) and from the most populated conformations extracted (simulations 2 to 5) from a cluster analysis performed on the 1 μs-long MD simulation described in the previous paragraph. Hierarchical agglomerative clustering was performed, setting the number of clusters to 4 and using the average distance (average linkage keyword in cpptraj) and symmetric RMSD as metric (srmsd keyword). Each simulation was performed as described in the previous paragraph regarding the structural optimization and initial heating steps. Then, a MD simulation of 100 ns in length at 298 K (NVT ensemble) was performed, followed by: heating (in 20 ns) and equilibrium simulation (1 μs) at 363 K, cooling to 298 K in 100 ns, and finally another equilibrium simulation of 1 μs in length at this temperature.
000 steps) were performed in the presence of harmonic restraints (k = 1 kcal mol−1 Å−1) applied to: (a) all non-hydrogenous atoms of the system; (b) backbone atoms; (c) Cα atoms. Reference structures at steps (b) and (c) were the final ones from the previous step. Next, up to 50000 cycles of unrestrained optimization were performed. Each system was then heated to 298 K in 1 ns via constant–pressure–temperature (NTP) MD simulations (using the isotropic Berendsen barostat and the Langevin thermostat) followed by an equilibration phase of 10 ns. Starting from the equilibrated structure, a MD simulation in the NVT ensemble of 1 μs in length was performed using a time step of 2 fs. Next, heating to 363 K in was simulated in 20 ns, followed by another equilibrium simulation of 1 μs in length at this temperature. Periodic boundary conditions were employed, and electrostatic interactions were estimated using the Particle Mesh Ewald scheme with a cutoff of 9.0 Å for the short-range evaluation in direct space and for Lennard–Jones interactions (with a continuum model correction for energy and pressure). Self-assembly. 5 independent MD simulations of the self-assembly of 512 heterochiral tetrapeptides in water solution were performed following published protocols.25 Briefly, the initial conformation of the assembling peptides was generated by placing their centers of mass on a 8 × 8 × 8 grid of 17.5 Å-spaced points. Initial orientations of peptides were randomized, and the systems were solvated with water molecules, for a total number of atoms around 235000. Initial structures of the 5 independent simulations were taken from the optimized structure of the corresponding peptides (simulation 1) and from the most populated conformations extracted (simulations 2 to 5) from a cluster analysis performed on the 1 μs-long MD simulation described in the previous paragraph. Hierarchical agglomerative clustering was performed, setting the number of clusters to 4 and using the average distance (average linkage keyword in cpptraj) and symmetric RMSD as metric (srmsd keyword). Each simulation was performed as described in the previous paragraph regarding the structural optimization and initial heating steps. Then, a MD simulation of 100 ns in length at 298 K (NVT ensemble) was performed, followed by: heating (in 20 ns) and equilibrium simulation (1 μs) at 363 K, cooling to 298 K in 100 ns, and finally another equilibrium simulation of 1 μs in length at this temperature.
Differential scanning calorimetry (DSC)
DSC sample preparation and analysis was performed as previously described.26Rheometry
Dynamic time sweep rheological analysis was conducted on a Malvern Kinexus Ultra Plus Rheometer with a 20 mm stainless steel parallel plate geometry. The temperature was maintained at 25 °C using a Peltier temperature controller. Samples were prepared in situ and immediately analyzed with a gap of 1.00 mm. Time sweeps were recorded for 1 hour, using a frequency of 1.00 Hz and a controlled stress of 1.00 Pa. After 1 h, frequency sweeps were recorded using a controlled stress of 1.00 Pa and then stress sweeps were recorded using a frequency of 1.00 Hz.Transmission electron microscopy (TEM)
TEM micrographs were acquired on Jeol, JEM 2100, Japan, at 100 kV. TEM grids (copper-grid-supported lacey carbon film) were first exposed to the UV-ozone cleaner (UV-Ozone Procleaner Plus) for 45 min to make the grid surface more hydrophilic. Then, 24-hours-aged gels were precisely deposited on a TEM grid, dried for 15 min at RT, and contrasted by aqueous tungsten phosphate solution (pH 7.4).Atomic force microscopy (AFM)
Gels were prepared as described above and after 24 hours they were spread on a clean Si wafer surface. Atomic force microscopy (AFM) measurements were performed using a Nanoscope V microscope (Digital Instruments Metrology Group, model MMAFMLN) in tapping mode in air at room temperature, using standard μmasch® SPM probe (HQ:NSC15/AIBS) with tip height 12–18 μm, cone angle <40° (Resonant frequency 325 kHz, force constant of ∼40 N m−1). Image analysis has been performed with Gwyddion software.Fourier transformed infrared (FT-IR) spectroscopy
FT-IR spectra were registered using KBr-pellet method. The gel was prepared and after 24 h it was dried under vacuum. Then, it was mixed with KBr to make the pellet. Spectra were acquired at 1 cm−1 resolution and 128 accumulations on a PerkinElmer System 2000. Spectra were acquired also for the heated sample (see ESI†).Circular dichroism (CD)
A 0.1 mm quartz cell was used on a Jasco J815 Spectropolarimeter, with 1s integrations, 1 accumulation and a step size of 1nm with a bandwidth of 1nm over a range of wavelengths from 200 to 280 nm at 25 °C (Peltier). Samples were freshly prepared directly in the CD cell and the spectra immediately recorded. Averaged spectra at RT (n > 10) were plotted with Excel. After 1 hour of kinetics, a heating ramp was performed at 5 °C min−1 up to 85 °C.Thioflavin T fluorescence assay
Gel samples (100 μl total volume) were prepared in situ on wells of Greiner 96 flat clear-bottom black polystyrene. After 30 minutes, 20 μl of a solution of thioflavin T (23 mM in 20 mM glycine–NaOH pH 7.5, filtered with a 0.2 μm filter) were added in the wells. After 15 minutes, the fluorescence emission was analysed using a Tecan Infinite M1000 Pro, selecting an excitation wavelength of 446 nm and an emission wavelength of 490 nm, with a bandwidth of 20 nm. Average and standard deviations (n = 6) were calculated and plotted with Excel.Protease assay
The peptide (2.0 mg) was dissolved with 0.25 ml DMSO and 4.75 ml of 50 mM sodium phosphate buffer (pH 7.5) containing a large excess (5 mg) of proteinase K lyophilizate from Pichia pastoris (Roche – 03115879001 – 2 U mg−1) in 15 ml Falcon tubes. The tubes were incubated at 37 °C, 60 rpm, and at various timepoints 0.500 ml samples were taken and added to 0.200 ml NaOH 1M to stop the reaction, and analysed by HPLC (Abs 254 nm, same program as described above for peptide purification). The assay on gel was performed by preparing 2 mg-containing hydrogel (i.e., 248 μl total volume) in 15 ml-Falcon tubes. The gel was left to self-assemble overnight. The following morning, 4.75 ml of 50 mM sodium phosphate buffer (pH 7.5) containing a large excess (5 mg) of protease were added on top. The tubes were incubated at 37 °C, 60 rpm, and after 4 and after 24 hours, 2 ml of NaOH 1 M were added to completely dissolve the gel and slow-down protease activity prior to HPLC analysis as above. Average and standard deviation values (n = 3) were calculated and plotted with Excel.NMR analysis at different temperatures
Each sample was dissolved in DMSO-d6 (with or without 20% water) at concentration of 15 mM and analysed after 1 h with 1H-NMR at different temperatures (from 25 to 80 °C, every 5 °C). N. B. At each temperature sample was analysed after 10 min of NMR temperature stabilisation.Live/dead assay
The heterochiral tetrapeptide was dissolved in 0.1 M of sodium phosphate (pH 11.8) and by adding an equal volume of sodium phosphate (pH 5.8) the hydrogel was directly prepared (20 μl per well) in triplicate wells of an μ-slide Angiogenesis uncoated (Ibidi, Germany). Gelator solutions were sterilized by using filter 0.2 μm prior to use. The gel was kept at room temperature for 24 h. After that time, gels were covered/pre-treated with 30 μl of culture media (DMEM medium + 10% fetal bovine serum and 2 vol% penicillin/streptomycin from GIBCO) and left at rt for 1 h. The medium was removed and HaCaT cells (keratinocytes) were added to the gels (10000 cells per well in 30 μl media) and cultured in cell incubator (37 °C, 5% CO2) for 24 h by handling the slides according to the manufacturer's instructions. Cell viability was assessed by using acridine orange (5 μl per well of a 20 μM solution in 50 mM PBS) and propidium iodide (5 μl per well of a 30 μM solution in 50 mM PBS). Cells were imaged on an inverted microscope (Nikon Eclipse Ti–U) with green and red filters and 40× objective (N.A. 0.6). Controls consisted of untreated cells (no peptide). The homochiral tetrapeptide was tested under the same conditions of the heterochiral analog, although no gel was formed.
Conclusions
In conclusion, we describe the first uncapped D,L-tetrapeptide self-assembly into a hydrogel that, upon heating, undergoes an irreversible morphological transition from fibrils to plates, in marked contrast with all the short peptide assemblies described thus far. This investigation reveals that the peptide undergoes only minor conformational changes, while water plays a key role in the transition, whereby water–peptide interactions are replaced with peptide–peptide interactions, and the entropic gain of the water molecules that are set free locates the system in an energetic sink. These results shed new light on the modulation of peptide assemblies and their morphology.Author contributions
All authors have given approval to the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors acknowledge funding from the Italian Ministry of University and Research (MUR) through the PRIN program (grant SHAZAM n. 2022XEZK7K to S. M.), funded by the European Union – Next Generation EU. This research was also funded by the Slovenian Research and Innovation Agency (ARIS) through the core funding No. P2-0089 and ARIS projects: No. J2-3043, J2-3040, J2-3046, J3-3079, J7-4420. The authors thank the CENN Nanocenter (JSI, Slovenia) for TEM access. They acknowledge Elettra Sincrotrone Trieste for providing access to its synchrotron-radiation facilities and Dott. Fabio Hollan for HR-MS data acquisition. This research used the Savio computational cluster resource provided by the Berkeley Research Computing program at the University of California, Berkeley. A. V. V. acknowledges technical support from Andrea Bosin and Fabrizio C. Muredda (University of Cagliari).References
- K. Tao, A. Levin, L. Adler-Abramovich and E. Gazit, Chem. Soc. Rev., 2016, 45, 3935 RSC.
- (a) K. H. Chan, W. H. Lee, M. Ni, Y. Loo and C. A. E. Hauser, Sci. Rep., 2018, 8, 17127 CrossRef PubMed; (b) A. Brito, D. Dave, A. Lampel, V. I. B. Castro, D. Kroiss, R. L. Reis, T. Tuttle, R. V. Ulijn, R. A. Pires and I. Pashkuleva, J. Am. Chem. Soc., 2021, 143, 19703 CrossRef CAS.
- (a) P. W. Frederix, G. G. Scott, Y. M. Abul-Haija, D. Kalafatovic, C. G. Pappas, N. Javid, N. T. Hunt, R. V. Ulijn and T. Tuttle, Nat. Chem., 2015, 7, 30 CrossRef CAS; (b) M. Ramakrishnan, A. van Teijlingen, T. Tuttle and R. V. Ulijn, Angew. Chem., Int. Ed., 2023, 62, e202218067 CrossRef CAS.
- M. R. Sawaya, S. Sambashivan, R. Nelson, M. I. Ivanova, S. A. Sievers, M. I. Apostol, M. J. Thompson, M. Balbirnie, J. J. W. Wiltzius, H. T. McFarlane, A. O. Madsen, C. Riekel and D. Eisenberg, Nature, 2007, 447, 453 CrossRef CAS PubMed.
- (a) S. Mondal, L. Adler-Abramovich, A. Lampel, Y. Bram, S. Lipstman and E. Gazit, Nat. Commun., 2015, 6, 8615 CrossRef CAS; (b) S. Marchesan, L. Waddington, C. D. Easton, D. A. Winkler, L. Goodall, J. Forsythe and P. G. Hartley, Nanoscale, 2012, 4, 6752 RSC.
- (a) A. M. Garcia, D. Iglesias, E. Parisi, K. E. Styan, L. J. Waddington, C. Deganutti, R. De Zorzi, M. Grassi, M. Melchionna, A. V. Vargiu and S. Marchesan, Chem, 2018, 4, 1862 CrossRef CAS; (b) B. Adhikari, G. Palui and A. Banerjee, Soft Matter, 2009, 5, 3452 RSC; (c) S. Kumar, S. Bera, S. K. Nandi and D. Haldar, Soft Matter, 2021, 17, 113 RSC.
- (a) A. Boruah and A. Boy, Biomater. Sci., 2022, 10, 4694 RSC; (b) S. Kumar and A. Bajaj, Biomater. Sci., 2020, 8, 2055 RSC; (c) J. Naskar, G. Palui and A. Banerjee, J. Phys. Chem. B, 2009, 113, 11787 CrossRef CAS PubMed.
- (a) A. Méndez-Ardoy, J. R. Granja and J. Montenegro, Nanoscale Horiz., 2018, 3, 391 RSC; (b) M. R. Ghadiri, J. R. Granja, R. A. Milligan, D. E. McRee and N. Khazanovich, Nature, 1993, 366, 324 CrossRef CAS PubMed.
- J. Zhou, X. Du, N. Yamagata and B. Xu, J. Am. Chem. Soc., 2016, 138, 3813 CrossRef CAS.
- (a) V. Singh, R. K. Rai, A. Arora, N. Sinha and A. K. Thakur, Sci. Rep., 2014, 4, 3875 CrossRef; (b) A. Frydman-Marom, M. Rechter, I. Shefler, Y. Bram, D. E. Shalev and E. Gazit, Angew. Chem., Int. Ed., 2009, 48, 1981 CrossRef CAS.
- M. Reches and E. Gazit, Science, 2003, 300, 625 CrossRef CAS.
- J. Wang and B. Xu, Chemistry, 2018, 4, 1765–1767 CrossRef CAS.
- (a) M. Karplus, J. Chem. Phys., 1959, 30, 11 CrossRef CAS; (b) A. C. Wang and A. Bax, J. Am. Chem. Soc., 1996, 118, 2483 CrossRef CAS; (c) V. F. Bystrov, Prog. Nucl. Magn. Reson. Spectrosc., 1976, 10, 41 CrossRef.
- T. Cierpicki and J. Otlewski, J. Biomol. NMR, 2001, 21, 249 CrossRef CAS.
- J. Naskar, G. Palui and A. Banerjee, J. Phys. Chem. B, 2009, 113, 11787 CrossRef CAS.
- S. Mondal, L. Adler-Abramovich, A. Lampel, Y. Bram, S. Lipstman and E. Gazit, Nat. Commun., 2015, 6, 8615 CrossRef CAS PubMed.
- F. U. Hartl, A. Bracher and M. Hayer-Hartl, Nature, 2011, 475, 324 CrossRef CAS PubMed.
- A. Perczel, P. Hudáky and V. K. Pálfi, J. Am. Chem. Soc., 2007, 129, 14959 CrossRef CAS.
- D. Iglesias, M. Melle-Franco, M. Kurbasic, M. Melchionna, M. Abrami, M. Grassi, M. Prato and S. Marchesan, ACS Nano, 2018, 12(6), 5530–5538 CrossRef CAS PubMed.
- D. A. Case, H. M. Aktulga, K. Belfon, I. Y. Ben-Shalom, J. T. Berryman, S. R. Brozell, D. S. Cerutti, T. E. Cheatham, III, G. A. Cisneros, V. W. D. Cruzeiro, T. A. Darden, N. Forouzesh, G. Giambaşu, T. Giese, M. K. Gilson, H. Gohlke, A. W. Goetz, J. Harris, S. Izadi, S. A. Izmailov, K. Kasavajhala, M. C. Kaymak, E. King, A. Kovalenko, T. Kurtzman, T. S. Lee, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, M. Machado, V. Man, M. Manathunga, K. M. Merz, Y. Miao, O. Mikhailovskii, G. Monard, H. Nguyen, K. A. O’Hearn, A. Onufriev, F. Pan, S. Pantano, R. Qi, A. Rahnamoun, D. R. Roe, A. Roitberg, C. Sagui, S. Schott-Verdugo, A. Shajan, J. Shen, C. L. Simmerling, N. R. Skrynnikov, J. Smith, J. Swails, R. C. Walker, J. Wang, J. Wang, H. Wei, X. Wu, Y. Wu, Y. Xiong, Y. Xue, D. M. York, S. Zhao, Q. Zhu and P. A. Kollman, Amber 2023, University of California, San Francisco, 2023 Search PubMed.
- O. Bellotto, E. Scarel, G. Pierri, P. Rozhin, S. Kralj, M. Polentarutti, A. Bandiera, B. Rossi, A. V. Vargiu, C. Tedesco and S. Marchesan, Biomacromolecules, 2024, 25, 2476 CrossRef CAS.
- C. Tian, K. Kasavajhala, K. A. A. Belfon, L. Raguette, H. Huang, A. N. Migues, J. Bickel, Y. Wang, J. Pincay, Q. Wu and C. Simmerling, J. Chem. Theory Comput., 2020, 16, 528 CrossRef.
- S. Izadi, R. Anandakrishnan and A. V. Onufriev, J. Phys. Chem. Lett., 2014, 5, 3863 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graph., 1996, 14, 33 CrossRef CAS.
- S. Adorinni, S. Gentile, O. Bellotto, S. Kralj, E. Parisi, M. C. Cringoli, C. Deganutti, G. Malloci, F. Piccirilli, P. Pengo, L. Vaccari, S. Geremia, A. V. Vargiu, R. De Zorzi and S. Marchesan, ACS Nano, 2024, 18, 3011 CrossRef CAS.
- S. Marchesan, K. E. Styan, C. D. Easton, L. Waddington and A. V. Vargiu, J. Mater. Chem. B, 2015, 3, 8123 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2347110. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4tb01727g |
| This journal is © The Royal Society of Chemistry 2024 |