
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
CO2 switchable solvents for sustainable dissolution, modification, and processing of cellulose materials: a critical review
Peter McNeice
 * and
Ben L. Feringa
*
* and
Ben L. Feringa
*
Advanced Research Centre CBBC, Stratingh Institute for Chemistry, Faculty of Science and Engineering, University of Groningen, Nijenborgh 4, Groningen, 9747AG, The Netherlands. E-mail: pmcneice01@qub.ac.uk; b.l.feringa@rug.nl
First published on 6th November 2024
Abstract
Cellulose is a biopolymer with numerous applications ranging from food packaging to pharmaceutical formulations. However, the sustainability and use of cellulose materials is hindered by harsh processing conditions and toxic solvents. In recent years, a milder approach to cellulose processing and modification has emerged based on the use of CO2 switchable solvents. They facilitate the dissolution of cellulose through its activation with a base and CO2. Cellulose can then be regenerated in a range of morphologies (fibres, films, gels), or chemically modified in a controlled homogeneous manner. This not only avoids the need for toxic solvents, but prevents the waste associated with traditional heterogeneous cellulose modification. Based on the literature to date, we provide both a guide to the use of CO2 switchable solvents for cellulose dissolution and modification, and a critical analysis of these emerging methodologies for future applications of this important biobased material.
 Peter McNeice | Peter has always performed green chemistry, and his interests are in sustainable materials and catalysis. He conducted his PhD research titled “Base Stable and Basic ionic liquids for Catalysis” at Queen's University Belfast, supervised by Dr Andrew Marr, Dr Patricia Marr and Prof. Ken Seddon. He then performed postdoctoral research with Prof. Matthias Beller in the Leibniz Institute for Catalysis in Rostock, before moving to work for Prof. Ben Feringa at the University of Groningen. He is currently a postdoctoral fellow at the Max Planck Institute for Chemical Energy Conversion, in the group of Prof. Waltner Leitner and Dr Andreas Vorholt. |
 Ben L. Feringa | Ben L. Feringa a obtained his PhD in 1978 at the University of Groningen under Prof. Hans Wynberg, where he was appointed full professor in 1988, after working as a research scientist for Shell. He was named the Jacobus H. van't Hoff Professor of Molecular Sciences in 2004. His research interests include organic synthesis, asymmetric catalysis, nanotechnology, and molecular switches and motors. |
1. Introduction
1.1. Cellulose: potential and problems
The use of cellulose as a feedstock for bioplastics in order to reduce waste and the reliance on fossil feedstocks was highlighted as one of the most promising emerging technologies by the World Economic Forum in 2019.1 Cellulose (Fig. 1) is an obvious candidate for bioplastic production as it is the most abundant biorenewable and biodegradable resource on Earth, with 1.5 × 1012 tons estimated to grow every year.2 An added advantage of cellulose compared to other biomass sources is that being derived from waste,3,4 it does not compete with food production. The potential for cellulose to be used as a feedstock has been recognized in recent years, with several reviews showing its application to food packaging,5 water purification,6 pigments,7–9 polymer composites,10,11 and biomedical applications.12–14 | ||
| Fig. 1 The anhydroglucose unit (AGU) that makes up cellulose (left), and a photo of microcrystalline cellulose (right). Photo PMN, 2022. | ||
Despite its strong potential as a “green” feedstock, the preparation of materials from cellulose is currently not environmentally benign. Due to strong inter- and intra-molecular hydrogen bonding, alongside considerable hydrophobic interactions (Fig. 2), cellulose is highly crystalline, and insoluble in conventional solvents, compromising its processibility.2,15–17 As a result, industrial dissolution of cellulose is largely reliant on two processes, the viscose and Lyocell processes. The viscose process involves swelling cellulose in hot sodium hydroxide, treatment with carbon disulfide to give a xanthate derivative, and spinning fibres using aqueous sodium hydroxide.2,18 As well as the use of toxic carbon disulfide, there is significant metal waste produced when cellulose is precipitated from solution.2 The Lyocell process is more environmentally friendly, with cellulose being directly dissolved in N-methylmorpholine-N-oxide (NMMO), before regeneration by spinning. The use of NMMO is problematic however, as it is expensive and thermally unstable. This instability can lead to side-reactions and byproduct formation if a stabiliser is not used.19
 | ||
| Fig. 2 A representation of the bonding interactions in cellulose. Left is the inter- (red) and intramolecular (blue) hydrogen bonds, and right is a side on view showing the hydrophobic interactions of the axial hydrogen atoms. | ||
Due to the abovementioned problems with the solvents for cellulose processing, and in order to fully realise the potential of cellulose as a green source of polymers, there has been recent drive to develop more environmental systems to dissolve cellulose. Ionic liquids (ILs), and related deep eutectic solvents (DES) are examples of such systems. ILs are defined as materials consisting solely of ions, which can form liquids at the operating temperature.20,21 DES are low-melting mixtures of hydrogen bond donors and hydrogen bond acceptors, with choline chloride–urea being one of the most widely reported systems.22 It is generally accepted that ionic liquids dissolve cellulose via disruption of hydrogen bonds caused by both cations and anions.23 The dissolution mechanism in DES remains uncertain, although the interaction between cellulose and the counter anion of choline appears to be dominant.24,25 The use of these materials for cellulose dissolution and processing has been reported in multiple research articles and reviews.23,26–32 While some of these methods are entering the early stages of commercialisation,33 there is ongoing work to find more sustainable solvents for cellulose.
1.2. CO2 switchable solvents
An interesting development in this field is the use of CO2 switchable solvents. These were first reported by Jessop et al. and involve the conversion of a non-ionic mixture (an alcohol and an amine base) into an ionic liquid through the addition of CO2.35 The base abstracts a proton from the alcohol, which then forms a bicarbonate adduct with CO2.34 The subsequent addition of nitrogen gas returns the ionic liquid to its original neutral state (Fig. 3a).35 There is a drastic change in solvent polarity between the two states, which can be observed by a phase separation when a non-polar solvent such as decane is also present (Fig. 3b). Many applications of CO2 switchable systems have been reviewed.36–38 These include the use of switchable solvents for polymerisation reactions, as extraction media, for CO2 capture, to separate reaction products and catalysts, to switch the properties of materials and surfactants, or to turn catalysts “on” or “off”.39 | ||
| Fig. 3 A representation of a CO2 switchable solvent (a), and the use of the technique for separations (b). Adapted from Jessop et al.35 with permission from Springer Nature, 2024. | ||
Furthermore, there have been increasing reports on the use of CO2 switchable solvents for the dissolution and modification of cellulose. Herein, we provide a review on the use of CO2 switchable solvents for the dissolution and processing of cellulose. We begin with a guide to dissolving cellulose in CO2 switchable solvents (section 2.1), and the preparation of cellulose films, fibres, gel, etc. (section 2.2). Subsequently, modifications of cellulose in CO2 switchable solvents are discussed (section 3), and the preparation of functional materials (section 4), before concluding with future challenges in the field (section 5).
2. Cellulose dissolution in CO2 switchable solvents
Two alternate methods to dissolve cellulose using CO2 solvents were reported independently by Jérôme and co-workers,40 and Xie and co-workers.41 In the so-called “derivative method” cellulose is used as the alcohol for the switchable system and is incorporated into the solvent through the formation of an in situ carbonate anion (Scheme 1).40 The “non-derivative” method follows the original switchable solvent model (Fig. 3a) to create a polar solvent in which cellulose is dissolved.41 Both systems use DMSO as a co-solvent in order to assist the CO2 solubility. These were obviously important breakthroughs as they allowed cellulose to be dissolved at lower temperature, and in a shorter time (1 h, 40–60 °C) than was possible using conventional ionic liquids (8 h, 90 °C).40 Despite the environmental benefits presented by the use of CO2 switchable solvents which we discuss in sections 2.1 and 2.2, there are downsides such as the use of toxic DBU, and the difficulty in recycling the base. These drawbacks, and potential solutions are discussed in section 5.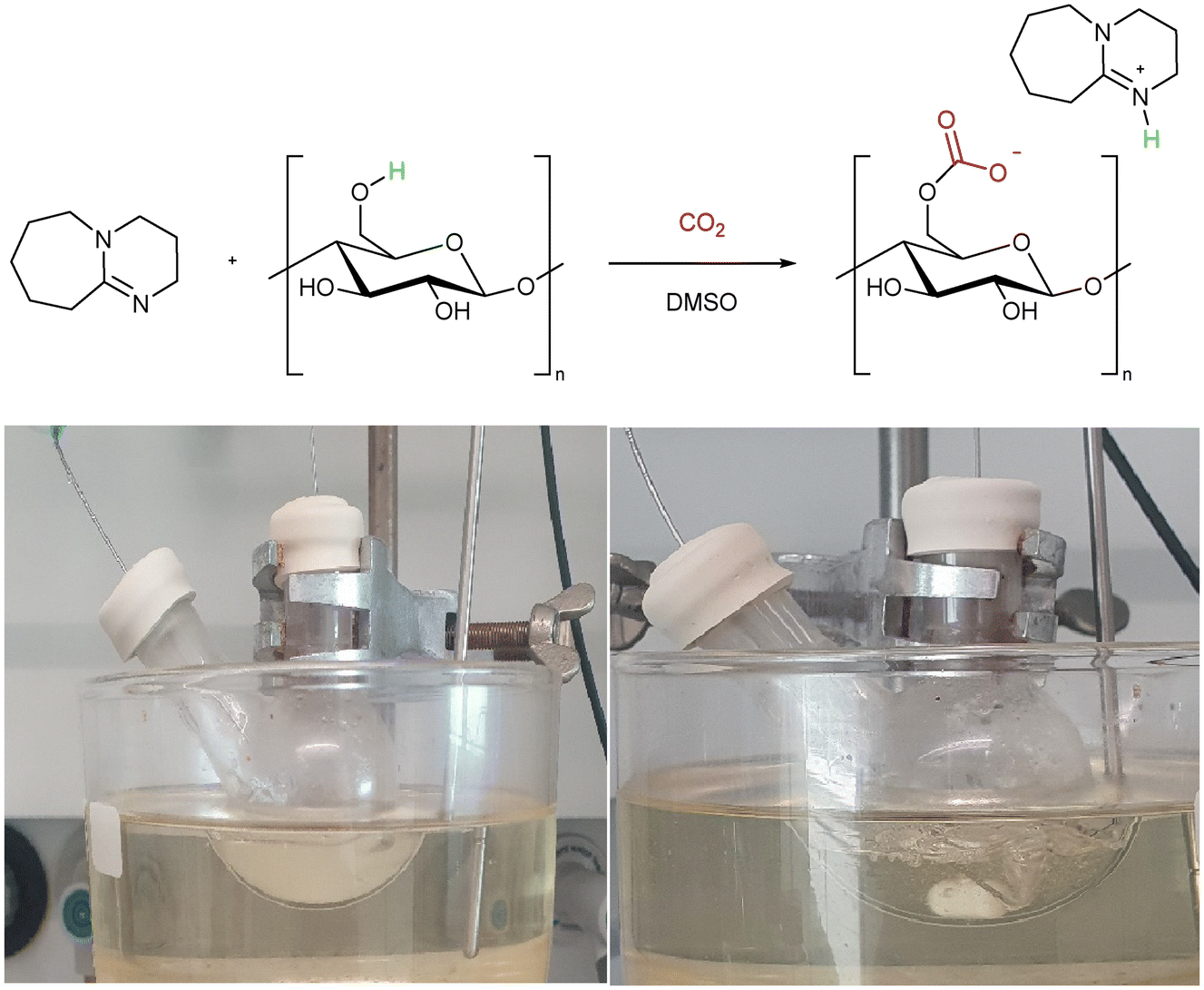 | ||
| Scheme 1 (Top) The “derivative” switchable solvent method to solubilise cellulose. (Bottom) The dissolution of cellulose in a DMSO/DBU/CO2 mixture. Photo PMN, 2022. | ||
2.1. Factors affecting dissolution
In this section we discuss the important parameters and conditions which determine the amount of cellulose that can be dissolved, and the speed at which it is dissolved. A summary is presented in Table 1. In the derivative process, the choice of base is important. A phosphazene base allowed 15 wt% cellulose dissolution, compared to 8 wt% using 1,3,3-tetramethylguanidine (TMG) and 4 wt% using 1,8-diazabicyclo-[5.4.0]undec-7-ene (DBU) (Fig. 4).40 As the dissolution ability does not follow a trend in pKa values (Fig. 4), the authors propose that the higher wt% of dissolved cellulose is due to a greater ability of the base to stabilise positive charge via delocalisation. This is more evident at higher cellulose loadings. Wang et al. reporting that 2-tert-butyl-1,1,3,3-tetramethylguanidine (BTMG), with its electron donating tert-butyl group, could stabilise the [BTMG − H]+ cation and thereby facilitate the dissolution of 10 wt% cellulose in 5 min at 22 °C. In contrast, DBU and TMG were unsuccessful after 2 h under the same conditions.42 Without the use of BTMG, Jérôme and co-workers reported that a minimum temperature of 40 °C and a minimum CO2 pressure of 2 bar were required to successfully dissolve microcrystalline cellulose.40 The cellulose was regenerated by heating the solution under vacuum at 30 °C for 30 min. The authors found no reduction in the degree of polymerisation of the cellulose, indicating no decomposition had occurred. However, the crystallinity index was decreased, and the cellulose was more prone to catalytic hydrolysis, thereby demonstrating a method to convert cellulose into glucose, and other commodity chemicals.40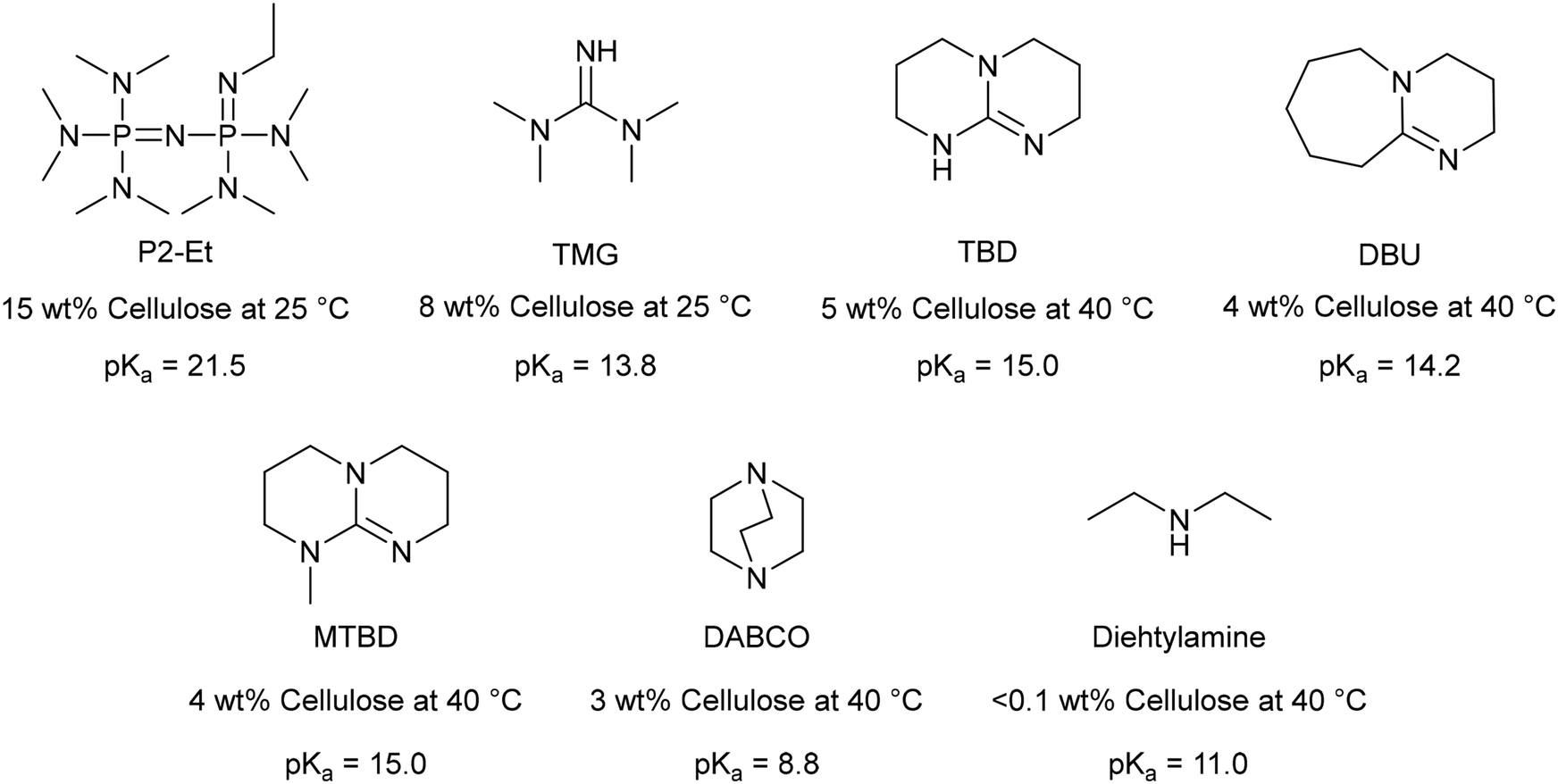 | ||
| Fig. 4 Structures, pKa values, and wt% of dissolved cellulose possible for a selection of superbases at 2 bar CO2.40 | ||
| Condition | Effect |
|---|---|
| Temperature | >30 °C can aid cellulose solubility for a given base,40,41 but reduces the cellulose carbonate anion stability43 |
| CO2 pressure | -Higher CO2 pressure increases solubility40,41,43 and improves solubility kinetics43 |
| Cellulose concentration | -Increased concentration increases viscosity (leading to reduced processability)43 |
| Cellulose species | Higher molecular weight cellulose is more difficult to dissolve41,45 |
| Co-solvent | -DMSO is essential |
| -Second co-solvent can aid solubility by improving CO2 uptake45 | |
| Time | -Maximum carbonate anion formation after 15 min |
| -A longer time does not improve dissolution43 | |
| Base species | -The ability to stabilise charge via delocalisation is required40–43,47 |
| -Delocalisation is more important than pKa |
Since its original use for cellulose dissolution, the derivative method has been studied further to analyse the dissolution mechanism, and optimise the process (see Table 1). Cramail, Meier, and co-workers altered various parameters during cellulose dissolution using the derivative method.43 They assessed the effect of the change in conditions by monitoring the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O FTIR absorbance at 1665 cm−1, concluding that a larger absorbance indicated an enhanced cellulose/CO2 interaction, and a more stable carbonate anion. Based on this analysis methodology, they established that temperature and CO2 pressure have the greatest effect on the in situ carbonate. At temperatures greater than 30 °C the carbonate absorbance was weaker, presumably due to Le Chatelier's Principle, as the reaction between a base and CO2 in the presence of alcohols is exothermic.44 Increasing the CO2 pressure from 5 to 40 bar resulted in increased carbonate formation. Furthermore, the kinetics of formation were faster above 20 bar, with maximum carbonate formation achieved in 10 min at 20 bar, but 5 min at 40 bar. Despite the increased pressure resulting in greater carbonate formation, the authors present their optimised conditions as 30 °C and 5 bar for 15 min, as they also took environmental considerations into account, and beyond 15 min there was no increase in carbonate formation. Employing a co-solvent has also been reported to have an effect on dissolution.45 The wt% of dissolved cellulose in a DMSO/base/CO2 mixture can be increased at a fixed temperature and pressure by adding a co-solvent with a low Henry's constant, such as propylene carbonate. The wt% of dissolved microcrystalline cellulose could be increased from 5 to 10, and for paper cellulose, which has a higher degree of polymerisation, it increased from 1 to 5 wt%. This is despite propylene carbonate being unable to dissolve cellulose when used as the sole solvent in a CO2 switchable system. The authors propose that the low Henry's Constant of propylene carbonate allows a larger amount of CO2 to be dissolved, thereby aiding cellulose solubility through increased cellulose carbonate formation.
O FTIR absorbance at 1665 cm−1, concluding that a larger absorbance indicated an enhanced cellulose/CO2 interaction, and a more stable carbonate anion. Based on this analysis methodology, they established that temperature and CO2 pressure have the greatest effect on the in situ carbonate. At temperatures greater than 30 °C the carbonate absorbance was weaker, presumably due to Le Chatelier's Principle, as the reaction between a base and CO2 in the presence of alcohols is exothermic.44 Increasing the CO2 pressure from 5 to 40 bar resulted in increased carbonate formation. Furthermore, the kinetics of formation were faster above 20 bar, with maximum carbonate formation achieved in 10 min at 20 bar, but 5 min at 40 bar. Despite the increased pressure resulting in greater carbonate formation, the authors present their optimised conditions as 30 °C and 5 bar for 15 min, as they also took environmental considerations into account, and beyond 15 min there was no increase in carbonate formation. Employing a co-solvent has also been reported to have an effect on dissolution.45 The wt% of dissolved cellulose in a DMSO/base/CO2 mixture can be increased at a fixed temperature and pressure by adding a co-solvent with a low Henry's constant, such as propylene carbonate. The wt% of dissolved microcrystalline cellulose could be increased from 5 to 10, and for paper cellulose, which has a higher degree of polymerisation, it increased from 1 to 5 wt%. This is despite propylene carbonate being unable to dissolve cellulose when used as the sole solvent in a CO2 switchable system. The authors propose that the low Henry's Constant of propylene carbonate allows a larger amount of CO2 to be dissolved, thereby aiding cellulose solubility through increased cellulose carbonate formation.
Despite the in situ formation of cellulose carbonate anion being widely reported, evidence of this was not provided until 2018. Indirect proof of the carbonate formation was obtained by trapping the in situ formed carbonate with benzyl bromide (Scheme 2).43 FTIR data showed the presence of a CO carbonic ester at 1740 cm−1, and a C–O bond, formed between the benzyl carbon and the carbonyl carbon, at 1256 cm−1. The cellulose benzyl carbonate was soluble in DMSO, allowing for NMR analysis. The benzylic CH2 protons were shifted downfield, whilst the carbonyl carbon was visible at 155 ppm, confirming that under these conditions, the cellulose reacted via the carbonate group and not the hydroxyl groups. This compared well with a test reaction between benzyl bromide and octanol (in place of cellulose) where 1H and 13C NMR, and a high-resolution mass spectrum were obtained of the predicted product. We discuss the mechanism of cellulose modification further in section 3.2.
 | ||
| Scheme 2 Trapping of the cellulose carbonate formed in situ during the derivative switchable method.43 | ||
Further evidence for the carbonate formation was later reported using both NMR and computational studies.46 Cellulose was first dissolved in mixtures of tetrabutylammonium acetate and DMSO, which does not require CO2 and does not alter the structure of cellulose. 13CO2 was then bubbled through the solution and the shift in the 13C NMR peaks monitored. These shifts were compared to those calculated for the model compound β-1,4-dimethyl-glucopyranoside (Scheme 3) using the gauge-independent atomic orbital (GIAO) method applied to MP2-level wave functions, and with the basis set pcSseg-1. The observed shift in the cellulose C6 carbon was within 1 ppm of the calculated 13C NMR value. Additionally, the presence of a C1 shift indicates the C2 alcohol is also functionalised. The authors propose that the primary C6 alcohol is substituted first, but at higher degrees of substitution, possibly caused by elevated CO2 pressure, the secondary alcohols can also react with CO2 to form carbonates.
 | ||
| Scheme 3 The model compounds used to calculate the 13C NMR shift of modified cellulose.46 | ||
In the non-derivative method, the switchable solvent is generally prepared in the absence of cellulose by using an alcohol (Fig. 3a), before this switchable solvent is used to solubilise the biopolymer. However, Xie and co-workers41 reported that DMSO as a co-solvent was still necessary to fully dissolve the cellulose. Of the range of alcohols tested (MeOH, EtOH, n-PrOH, n-BuOH, and ethylene glycol), only swelling of cellulose occurred, and not full dissolution, without DMSO. When combined with DMSO the authors report that shorter chain alcohols formed more polar solvent systems, as determined using Nile Red indicator, which reduced the crystallinity of regenerated cellulose to a greater extent than longer chain alcohols. Furthermore, replacing methanol with ethylene glycol allowed an increase from 5 to 10 wt% cellulose to be dissolved at 60 °C with 0.5 MPa CO2. NMR and IR analysis showed that a guanidinium ethylene di-carbonate formed (Scheme 4) and this was unaffected by the addition of cellulose, thereby resulting in the “non-derivative” name for this system. Whilst Jérôme and co-workers reported the pKa of the organic base to be unrelated to the cellulose dissolution,40 the bases tested all had pKa values greater than 18. Xie and co-workers demonstrated that the weaker bases dibutylamine (pKa = 11.3) and triethylamine (pKa = 10.8) did not form a solvent system capable of dissolving cellulose.41 However, as noted above, delocalisation of positive charge is also a factor in forming a switchable solvent system, and occurs to a greater extent in DBU, TMG, and phosphazene bases compared to aliphatic amines (see Fig. 4). It was shown that >99% of the ethylene glycol/DMSO/TMG mixture could be recovered by distillation after precipitation of cellulose via flushing with N2 at 70 °C.41 The derivative method was capable of reducing cellulose crystallinity, transforming it from cellulose I to the more easily processible cellulose II. This is a further demonstration of a green method to prepare cellulose for further transformation.
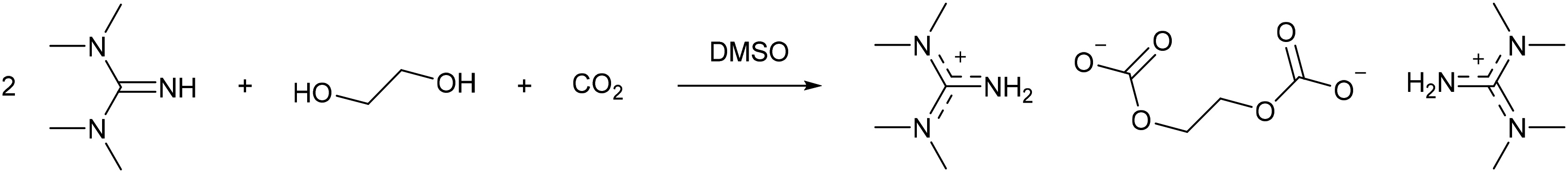 | ||
| Scheme 4 The formation of a guanidinium ethylene di-carbonate used for non-derivative cellulose dissolution.41 | ||
2.2. Cellulose dissolution and regeneration for materials
The facile methods to dissolve cellulose discussed above provide a key starting point for developing cellulose materials. In this section we discuss the production of fibres, films, and gels from cellulose solubilized in CO2 switchable systems. At the end of the section, we provide a summary of the factors which affect the quality of the materials produced.An efficient method to prepare fibres from a cellulose solution is essential for applications, and several methods have been reported for CO2 switchable solvents. Using the derivative method, Kirchberg and Meier injected 7 wt% cellulose solutions (DBU/DMSO/CO2) into a vigorously stirred anti-solvent.48 They reported water and acetone to give only particles, but MeOH, EtOH, and i-PrOH gave fibres. As the fibres formed in EtOH were the most stable, this method was selected for solvent recycling and optimisation. After removal of the fibres, EtOH was completely separated from the DBU/DMSO mixture via vacuum distillation (90 °C, 20 mbar, 30 min). The authors reported that the DBU/DMSO mixture could be re-used to dissolve cellulose, and fibres were formed using the recovered EtOH for five cycles. However, 22–58% of the DBU/DMSO mixture was lost during each cycle, with IR analysis showing that it was trapped in the cellulose fibres. The cellulose was then purified by washing 10 times with EtOH.
The concentration of the cellulose solution was found to be crucial for fibre formation. Homogeneous fibres were formed from 4, 5, and 6 wt% cellulose solutions, but using 2 or 3 wt% solutions led to uneven coiled fibres. The mechanical properties of fibres from a 4 wt% solution were analysed. Before washing, the fibre diameter was between 90–170 μm and elongation was 0.36–2.54 mm. The authors state that although the diameter is larger, and the elasticity lower than in the traditional Viscose process fibres, the fibres prepared here are comparable in terms of smoothness, which is an important industrial parameter.
Li et al. demonstrated that using a more systematic process during fibre formation could give greater control over the fibre properties.49 A self-made double diffusion wet spinning set-up was used to draw fibres out of a water, or 30 vol% DMSO in water, coagulation bath (Fig. 5). Fibres with diameters of 93–158 μm were obtained at 25 °C by varying the drawing ratio, with a higher ratio giving a larger diameter. Solvent recycling was carried out by distilling the contents of the coagulation bath to remove water, and 87% DMSO and 42% DBU were recovered. This is lower than in the case of the Kirchberg and Meier method where used EtOH was used,48 indicating that EtOH may be a more sustainable solvent for the coagulation bath. A main concern to be addressed, which we discuss in section 5, is the poor recovery of DBU, as this reduces the environmental benefits of these systems.
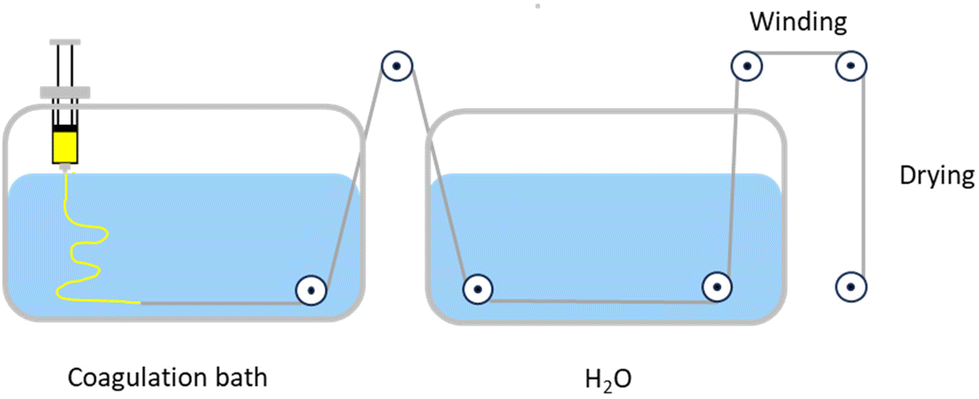 | ||
| Fig. 5 Double-diffusion wet spinning apparatus used to form cellulose fibres. Adapted from Li et al.49 with permission from American Chemical Society, 2024. | ||
The mechanical properties of the fibres; modulus (E), strength (σb), and elongation at break (εb), were measured as a function of the preparation conditions. The most important parameters were found to be the DMSO composition and temperature of the coagulation bath, and the fibre residence time in the bath. Increasing or decreasing the coagulation bath temperature from 25 °C gave fibres with poorer mechanical properties, as did extending the residence time or reducing the DMSO concentration from 30 vol%. An increased drawing ratio improved the mechanical properties of the fibres. The strongest fibres prepared by this method were 1.26 cN dtex−1, using a 30 min residence time in a 30 vol% DMSO, 25 °C bath. The authors anticipate that a more industrially feasible process is to use a shorter residence time and no DMSO, which gave fibres with 1.05 cN dtex−1. These are of poorer quality than Viscose fibres (1.8–2.5 cN dtex−1), but despite this drawback, almost no fibrillation was observed so the strength may not be a limiting factor in further development of this more sustainable process.
An alternative process to wet spinning is electrospinning, whereby charged fibres are drawn out of solution towards a metal collector (Fig. 6), allowing nanofibers to be prepared. This was used to prepare cellulose composite fibres.50 Both low molecular weight microcrystalline cellulose, and high molecular weight cellulose pulp were investigated. Solution viscosity and polymer molecular weight are amongst the most important factors for electrospinning of many materials. When using microcrystalline cellulose, fibres could not be produced using 1–10 wt% solutions in DBU/DMSO/CO2, regardless of the electrospinning conditions. Above 10 wt%, the solutions became unstable and resulted in cellulose precipitation. It was possible to form fibres when solutions of polyvinylalcohol (PVA) in DMSO were added to the cellulose solution. The ratio of cellulose to PVA required optimisation to prevent particle formation, with the optimum ratio determined to be 45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :55. This method produced beaded (i.e. non-uniform) fibres with a mean diameter below 100 nm. PVA was also required for cellulose pulp electrospinning, but only standing fibres with a mean diameter of 500 nm were produced. Standing fibres easily collapsed into larger, non-uniform fibres which are of limited applicability. Due to the beaded nature of the cellulose fibres produced here, this electrospinning method does not currently appear appropriate for preparing cellulose fibres from switchable solvent solutions. However, using different polymer additives may solve this problem, and allow electrospinning to become viable.
:55. This method produced beaded (i.e. non-uniform) fibres with a mean diameter below 100 nm. PVA was also required for cellulose pulp electrospinning, but only standing fibres with a mean diameter of 500 nm were produced. Standing fibres easily collapsed into larger, non-uniform fibres which are of limited applicability. Due to the beaded nature of the cellulose fibres produced here, this electrospinning method does not currently appear appropriate for preparing cellulose fibres from switchable solvent solutions. However, using different polymer additives may solve this problem, and allow electrospinning to become viable.
 | ||
| Fig. 6 An example of electrospinning equipment used to form cellulose nanofibers. | ||
Aside from making fibres, as in traditional cellulose processing, CO2 switchable solvent systems have been used for the preparation of films and aerogels. Investigations have shown that the properties of the final materials depend on the molecular weight of the cellulose source, and conditions used during cellulose regeneration.
Xie and co-workers51 used the derivative method (DMSO/DBU) to dissolve 5 wt% cellulose, with varied degree of polymerisation, in an autoclave using a CO2 pressure of 0.5 MPa at 50 °C. These solutions were cast onto glass plates and then submersed in a regeneration bath of either ethanol, methanol, aqueous sodium hydroxide (5 wt%), or aqueous sulfuric acid (5 wt%). Using scanning electron microscopy (SEM) and atom force microscopy (AFM) the authors found that smooth surfaces were obtained from the alcoholic baths, but rough surfaces formed when sodium hydroxide or sulfuric acid were used. Furthermore, the tensile strength of microcrystalline cellulose films regenerated using sodium hydroxide and sulfuric acid (25 MPa and 21 MPa, respectively) were lower than those of films regenerated in methanol and ethanol (42 and 55 MPa, respectively). Using this solvent regeneration method, forming amorphous films may be key to improved mechanical properties, as all films formed in alcoholic regeneration baths were amorphous, whilst those formed in the aqueous baths had cellulose II crystalline structures. It has previously been reported that aqueous conditions induce re-crystallisation of dissolved cellulose.16 Aside from regeneration using alcoholic baths, using cellulose sources with a higher degree of polymerisation (DP) improved the mechanical properties of cellulose. Tensile strength and elongation % at break increased on moving from microcrystalline cellulose (DP = 220) to cotton cellulose (DP = 350) to wood pulp cellulose (DP = 500). Furthermore, the films regenerated from ethanol had over 85% light transmittance in the visible region, demonstrating the potential of this method to prepare commercial cellulose films in a more environmentally friendly manner.
As an alternate method to solvent regeneration, Xie and co-workers52 report thermally induced CO2 release for film preparation. Here, a solution of 5 wt% cellulose in DBU/DMSO/CO2 was spread onto a plate and heated to 100 °C for 3 h to induce CO2 release, resulting in film formation. The authors also found that heating the solution to a higher temperature for a short time caused gelation/film formation. A lower gelation temperature (Tgel) was required for higher cellulose concentrations, or for higher molecular weight cellulose sources. As a contrast, the authors spread the cellulose solution on a plate before leaving it at room temperature for 3 h to absorb water, gradually forming a gel-like film. XRD patterns of the films show that the cellulose changed from a cellulose I crystalline structure to cellulose IVI when using the thermal treatment method, and becomes amorphous after the water absorption treatment. This indicates a rearrangement of hydrogen bonds during dissolution and regeneration. The mechanical properties of the cellulose films (tensile strength and elongation % at break) were greater for the thermally treated film compared to the water absorption film. Furthermore, the thermally treated film had a smoother surface, and better transparency (80% at 800 nm) compared to the water absorption film (<50% at 800 nm). The favourable properties of the crystalline cellulose films are in contrast to the solvent regeneration method discussed above,51 where amorphous films had better mechanical properties and higher transmittance. In both cases, using water to induce film formation led to poorer quality cellulose films.
Cramail, Meier, and co-workers investigated the effect of various reaction conditions in a derivative system on the properties of a cellulose aerogel.53 After forming a cellulose solution (5 wt% cellulose in DMSO, 3 equiv. DBU, 5 bar CO2, 30 °C, 15 min) and transferring it into a mould, the authors used an antisolvent to coagulate a wet cellulose gel over 24 h, and washed it to remove DMSO and DBU (Scheme 5). The DMSO and DBU could be recovered in 90 and 60% yield, respectively. Finally, the wet gel was freeze-dried to produce an aerogel (Scheme 5). Powder XRD and FTIR were used to demonstrate that the cellulose was chemically unmodified during aerogel formation. When cellulose solutions lower than 5 wt% were formed, aerogels could not be prepared due to the instability of the wet-gel stage (Scheme 5). As the cellulose wt% increased from 5–10%, there was an almost linear increase in aerogel density from 0.08 to 0.12 g cm−1. Despite a slight decrease in density when the coagulating solvent was changed from water to methanol, the increase in cellulose concentration had the largest effect on density. Changing methanol for ethanol or isopropanol, or changing DBU for DBN or TMG, had little effect on aerogel density. However, the pore size increased from 1.2 μm to 3.5 μm to 4.5 μm as the base was changed from DBU to DBN to TMG. Furthermore, the aerogels with lower densities had lower overall porosity. The authors do not provide an explanation for this, although it would appear to be due to differences in the structure of the wet gel. As discussed above for films, the gels were more resistant to breakage upon compression when higher molecular weight cellulose pulp was used compared to microcrystalline cellulose. Ultimately, the most difficult step in cellulose aerogel formation, dissolution of cellulose, was easily performed using a CO2 switchable solvent, and aerogels with densities of 0.05–1.12 g cm−1, and pore sizes 0.6–2.5 μm were prepared.
 | ||
| Scheme 5 Cellulose aerogel preparation using CO2 switchable solvents as demonstrated by Cramail, Meier, and co-workers.53 | ||
For the preparation of pure cellulose materials, the dissolution temperature, and CO2 pressure do not seem to influence the properties of the regenerated cellulose. The properties are affected by the wt% of dissolved cellulose and the molecular weight/degree of polymerisation of the dissolved cellulose. Better quality fibres were formed from higher concentration cellulose solutions,48 and stronger films and aerogels were formed from higher molecular weight cellulose sources.51–53 Aside from these chemical considerations, the mechanical processing method must be considered for fibre formation, with drawing49 and electrospinning50 allowing greater control than precipitation in a stirred antisolvent.48 In all cases, water has a negative effect on the formation and quality of both fibres and films. Derivatisation of cellulose can also lead to enhancements in the mechanical properties of the subsequent materials, which is discussed in sections 3.3 and 3.4.
3. CO2 switchable solvents for chemical modification of cellulose
3.1. Traditional modifications
Cellulose is not only used to prepare materials in its natural form (section 2.2) but can also be modified to alter its properties.2,54 The most common modifications involve the synthesis of cellulose ethers and esters via reaction of the hydroxyl groups. Many of these are commercially available.55–57 Cellulose ethers are largely water soluble, allowing them to find use as rheology modifiers,54–58 such as in the food industry where they give a creamier texture without adding fat, and also in the paint and coating, textile, and construction industries. Additionally, they have been used for tissue engineering54 and bio-sensing.54,58 Cellulose esters are generally insoluble and are therefore used to produce fibres, plastics, films, and coatings.54,59,60The synthetic strategies used for traditional cellulose modifications are described extensively by Heinze et al.54 As discussed previously (section 1), harmful solvents such as CS2, and dimethylacetamide/LiCl mixtures can be used to solubilise cellulose and allow it to react. However, a heterogeneous process, where the cellulose does not dissolve, is mainly employed in industry. This generally leads to uneven substitution along the chain which can result in irreproducible products. Therefore, a fully substituted form of cellulose can be synthesised, before partial hydrolysis to form the desired degree of substitution (DS), which leads to waste during synthesis. This is the standard process for the synthesis of cellulose acetate.54 Furthermore, strong acids or bases are required to catalyse the process, promoting side-reactions and cellulose decomposition which leads to reduced molecular weight and limits its applicability to polymer materials.
While analogous greener synthesis methods are beginning to be developed,61–63 such as preparing water soluble forms of cellulose for further processing,64 the use of switchable solvents to perform cellulose derivatisations with higher precision could be a major improvement as a soluble/dissolved reactive cellulose species can be produced under mild conditions using more environmentally friendly reagents (section 1.2). In the following sections, we will discuss the modification of cellulose in CO2 switchable solvents, focusing on the control over the DS, and the conditions which most affect the substitution. We summarise these conditions in section 3.5 (Table 2).
| Condition | Effect on DS |
|---|---|
| Substrate excess | Most important factor: DS increases as excess increases47,65–69,76,79,80 |
| Relative reactivity of substrate | DS decreases as substrate reactivity decreases42,66,69,71,72 |
| Time | -Little effect for a reactive substrate (e.g. acetic anhydride)66,67 |
| -Important for substrates with low reactivity (e.g. fatty acid esters)72 | |
| Temperature | -Increasing the temperature can improve DS66,67 |
| -Excess temperatures can cause hydrolysis of the newly formed bonds and reduce DS76,79 | |
| Heating method | -Microwave heating can reduce the reaction time, even for substrates with poor reactivity69,71 |
| Precipitation solvent | -MeOH and EtOH can reduce DS by transesterification68 |
| -iPrOH is more suitable68 | |
| Cellulose source | Higher molecular weight cellulose gives lower DS68 |
3.2. Reaction mechanism of cellulose modification in CO2 switchable solvents
In section 2.1 we discussed evidence for the in situ formation of a cellulose carbonate anion, which was trapped through a reaction with benzyl bromide to give cellulose benzyl carbonate (Scheme 2).43 There are other reports of alkyl halides reacting via the cellulose carbonate anion.42 Despite this, it is repeatedly reported that the products formed during cellulose modification in CO2 switchable solvents result from reaction of the cellulose hydroxyl groups (see sections 3.3–5). As an example, we will discuss the reaction between a cellulose solution and acetic anhydride. The reaction could proceed via one of two pathways, either reaction at the carbonate (Scheme 6A), or reaction at the hydroxyl moiety with loss of CO2 (Scheme 6B).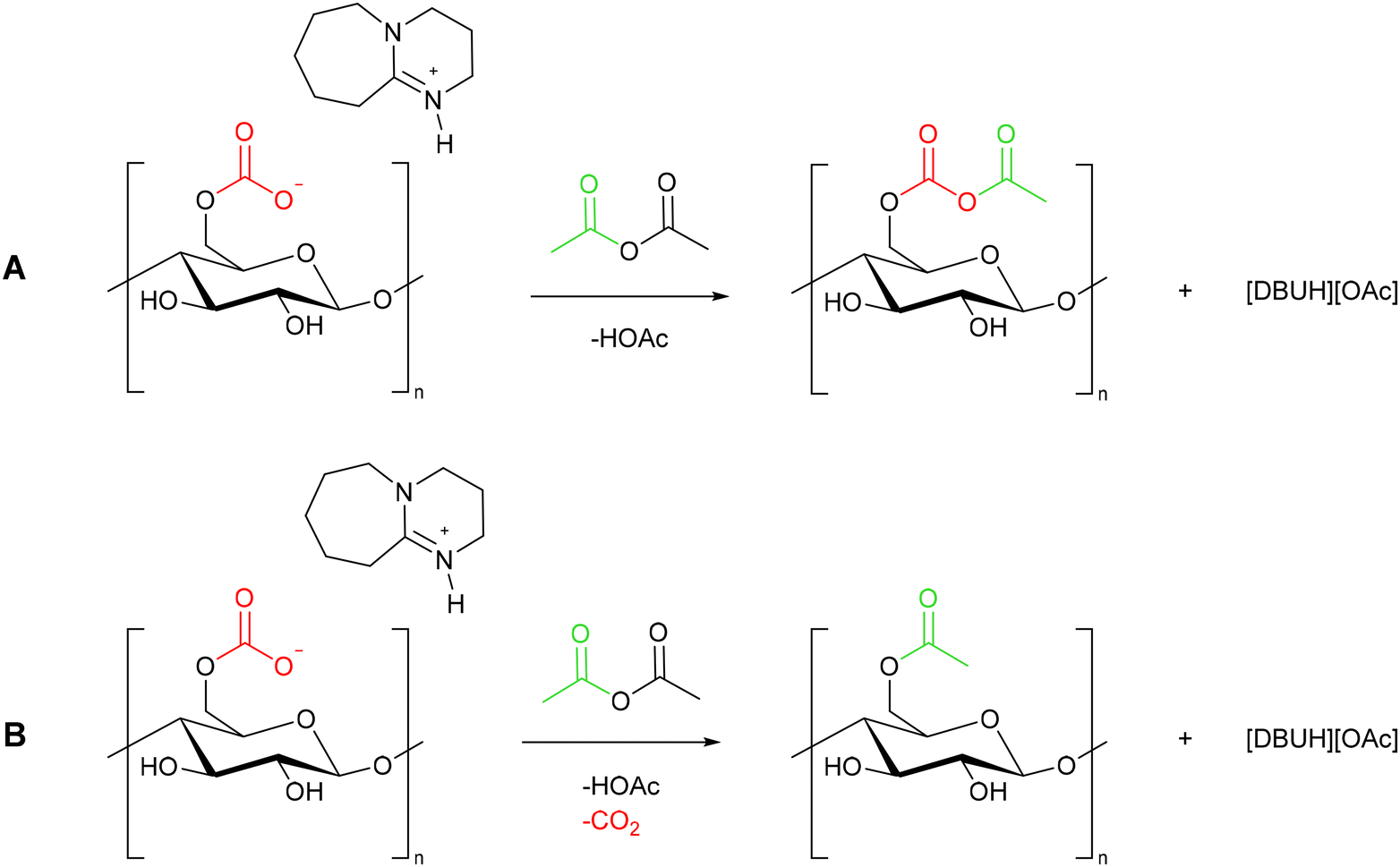 | ||
| Scheme 6 The possible reaction pathways between a cellulose solution and acetic anhydride via either the carbonate anion (A) or the cellulose hydroxyl moiety (B). | ||
It is difficult to differentiate between these products spectroscopically. FTIR will show a CO stretch regardless of the reaction pathway, while 13C NMR is also ambiguous. We expect the shift of the carbonyl carbons of the newly inserted group to overlap with a potential in situ formed carbonate. The majority of cellulose modification reactions performed in CO2 switchable solvents are aimed at the formation of esters (4.2–4.4), so the problem is repeated. Evidence for one mechanism rather than the other could be obtained by using a reactant without a carbonyl group. Such a reaction, between cellulose and acrylonitrile, was performed in a switchable solvent.65 However, the characterisation data provided is not clear enough to determine the reaction pathway. In the FTIR, the carbonyl stretch appears visible at 1600 cm−1, but the 13C NMR is not displayed to 150 ppm where the carbonyl carbon would be present.
To our knowledge, no study addresses this point specifically, other than as proof of in situ carbonate formation.43 We believe this is an area where further investigation is needed to unequivocally determine the mechanism of reactivity of cellulose in CO2 switchable solvents. Either mechanism, or both together are possible. For this reason, in the following sections we will draw the reaction product as it is represented in the original literature, which generally corresponds to Scheme 6B.
3.3. Preparation of cellulose derivatives, and their corresponding films
Cellulose acetate is the most widely produced cellulose derivative. Making use of a switchable solvent system allows the synthesis of cellulose monoacetate in a single step,66 overcoming the wasteful traditional process of complete derivatisation followed by partial hydrolysis (section 3.1). Furthermore, when the conditions are varied, controllable degree of substitution (DS) of between 1 and 3 can be achieved.66–69The non-derivative approach was first reported for this transformation, with acetic anhydride used as the derivatising agent.66 Microcrystalline cellulose (5 wt%) was first dissolved in a MeOH/DMSO/DBU mixture using 0.5 MPa CO2 at 60 °C for 3 h. The temperature was raised to 80 °C upon addition of acetic anhydride, and a DS of 2.41 was achieved after only 5 min when 3.6 equivalents of anhydride was used. It should be noted that each anhydroglucose unit from cellulose (Fig. 1) can react with 3 equivalents of anhydride. The reaction proceeded slowly after 5 min, with a 3 h reaction time required to increase the DS to 2.94. Decreasing the temperature, or the excess of anhydride reduced the DS. The industrially relevant DS of 0.98 was achieved using 2.4 equivalents of anhydride, with a 5 h reaction time required to compensate for the reduced amount of substrate. Cellulose propionate and butyrate could also be synthesised using this method, with a lower DS for cellulose butyrate indicating a relationship between the reactivity of the acylating agent and the extent of substitution. This is supported by an increased DS for the reaction between cellulose and alkyl halides containing electron withdrawing groups.42 A downside to the non-derivative process is seen through a side reaction, where the activated methanol goes on to form methyl acetate (Scheme 7). However, the authors do note that this is an important product in the chemical industry, and can be separated in 82% yield by distillation.
 | ||
| Scheme 7 The side reaction of the methyl carbonate anion to form methyl acetate when using the non-derivative method of cellulose dissolution.66 | ||
The side reaction of the alcohol in the non-derivative process (Scheme 7)66 can be avoided by using the derivative process, with similar DS achieved (Scheme 8).67 In this study, higher molecular weight cellulose pulp was substituted with acetic anhydride. The dissolution of this higher molecular weight cellulose required a longer reaction time, 3 h at 60 °C, which is consistent with observations made in the research into dissolution discussed earlier (section 2.2, Table 1). As with the non-derivative method, the main factor affecting the DS was the number of equivalents of anhydride. The majority of the reaction occurred within a short time (30 min in this particular case). A maximum DS of 2.89 was achieved after an 8 h reaction at 80 °C, using 5 equivalents of acetic anhydride.
 | ||
| Scheme 8 The derivative method for cellulose acetylation using acetic anhydride.67,69 | ||
These studies highlight the similarities in results between the derivative and non-derivative method for cellulose substitution. However, a direct comparison cannot be made between the reaction times because a higher molecular weight cellulose was used for the derivative process.
After cellulose has been dissolved in the derivative switchable system, the modification reaction time can be reduced through microwave heating.69 For instance, corncob cellulose could be reacted with acetic anhydride to achieve a DS between 1.83–2.91 with reaction times ranging from 3 to 15 min. A 10 min reaction time was sufficient to prepare cellulose esters of different chain lengths using propionic, butyric, valeric, and caproic anhydrides (Fig. 7). The DS decreased as the chain length increased due to the decreased reactivity of the anhydride, but it was still possible to form cellulose caproate with a DS of 2.20 after 10 min.
 | ||
| Fig. 7 Cellulose esters of different lengths, and their DS, prepared in a CO2 switchable solvent with 10 min microwave heating.69 | ||
In the above systems, methanol or water was added to precipitate the cellulose derivative, and up to 93% of the DMSO could be recovered by distillation.66,67,69 However, to recover DBU, the protonated [DBUH]+, which forms through a reaction with the in situ produced acetic acid (see Scheme 8), must first be neutralised with aqueous sodium acetate. An extraction with ethyl acetate allowed 91% of the DBU to be recovered. Despite recovery of sodium acetate being possible in a 55% yield,69 there is significant waste associated with this stage of the process.
The neutralisation and extraction steps can be avoided if vinyl acetate is used as acylating agent rather than acetic anhydride.68 Vinyl alcohol forms during the reaction rather than acetic acid. This bi-product tautomerises to acetaldehyde which evaporates, driving the equilibrium towards the products, and allowing for simpler recycling of DMSO and DBU (Scheme 9). After the reaction, the product is precipitated by adding i-PrOH before fractional distillation is used to recover i-PrOH (92%), DMSO (96%) and DBU (87%). An E-factor of 1.92 was calculated for the process. The authors also state that acetic acid catalysed de-acetylation of the product, which reduces the molecular weight and is known as vinegar syndrome,70 is prevented through the use of vinyl acetate as a reactant. The solvent used to precipitate the product was also noted to be important. When MeOH or EtOH were used, a lower DS was achieved. This may be due to transesterification catalysed by DBU, a factor that should be taken into account when designing new CO2 switchable solvent systems for cellulose. The DS of the obtained cellulose acetate could be tuned between 1 and 2.94, depending on the excess of vinyl acetate used, and even commercial filter paper could be modified with a DS of 2.66. The cellulose analogues with DS higher than 2 were used to prepare films which exhibited ultimate tensile strength of 20–43 MPa and Young's modulus of 747–2005 MPa.
 | ||
| Scheme 9 The reaction of a cellulose solution with vinyl acetate and the subsequent tautomerisation and evaporation of vinyl alcohol.68 | ||
This procedure was expanded to prepare cellulose esters with C2–C8 chains and mixed ester chains, facilitated via the use of microwave heating.71 In contrast to Yao et al.69 the increasing chain length did not reduce the DS, most likely due to the increased reactivity of vinyl esters compared to anhydrides. However, when mixed cellulose esters were prepared, the incorporation of the second ester group decreased as its chain length increased. This shows it is more difficult to add a second ester group, especially when the derivatising agent has reduced reactivity. DS between 2.0 and 2.3 were obtained using a reaction time of 10 min and 140 °C (300 W) microwave heating. The similar DS allowed a comparison of the mechanical properties of these materials in relation to the different modifying groups. As the chain length increased in the cellulose esters and mixed cellulose esters, there was a decrease in glass transition temperature, which would lead to enhanced processability of the final materials in an industrial setting. A linear increase in hydrophobicity, as determined by water contact angle, was also observed with increasing chain length. The Young's modulus decreased from 13.6 MPa to 7.51 MPa in the mixed cellulose esters, due to increased disruption of the inter- and intra-molecular bonding of the cellulose (see Fig. 2). This highlights the ease of preparing tailored cellulose materials using a CO2 switchable solvent system by varying the length of the ester group.
The mechanical properties of films formed from cellulose fatty acid esters can also be altered by using an unsaturated fatty acid rather than a saturated one. Meier, Cramail, and co-workers made use of sunflower oil as a sustainable esterification reagent (Fig. 8).72 Unlike many switchable solvent reactions, a two-phase mixture formed, which homogenised as the reaction proceeded. A relatively long reaction time of 24 h, at 115 °C, was used. As has been noted above, reactions are more difficult when the derivatising agent has low reactivity. The DS could be varied from 0.34 to 1.59, depending on the reaction time and the equivalents of fatty acid used. Cellulose analogues with DS above 1 were soluble in THF, and were used to prepare films by solvent casting. The films of cellulose analogues with lower DS were more flexible, which the authors attribute to a greater availability of free OH groups for hydrogen bonding. The Young's Modulus of films prepared from modifying microcrystalline cellulose with 1.5 or 3 equivalents of sunflower oil were 375 or 233 MPa, respectively. The Young's modulus could be increased to over 400 MPa by using higher molecular weight cellulose pulp, or filter paper, as a cellulose source. These are higher than fatty acid based films prepared by traditional heterogeneous cellulose processing (50 MPa).73 Furthermore, the maximum stress values of films from the CO2 switchable solvent method (16–22 MPa) are a considerable improvement on traditional processing (1.7–2.4 MPa).74
 | ||
| Fig. 8 Cellulose fatty acid esters prepared using sunflower oil in CO2 switchable solvents.72 | ||
Chemical modification of cellulose does not have to be a single reaction step. Modification in a switchable solvent can produce cellulose derivatives which are soluble in organic solvents, removing the main obstacle to its processability. Further reactions have led to the formation of cellulose carboxythiols,75 amides,76 and thiocarbamates.77
Meier and co-workers performed a thiol–ene reaction in DMSO using a cellulose fatty acid ester which had been prepared using a switchable solvent system (Scheme 10).75 Thiols of different lengths were reacted with the cellulose fatty acid ester which led to an increase in the molecular weight and thermal stability of the final material. In general, after addition of the thiol group, films prepared from the cellulose analogues became more elastic and more hydrophobic than their unmodified counterparts.
 | ||
| Scheme 10 An example of a thiol–ene reaction performed on a cellulose fatty acid derivative.75 | ||
Carboxylic acid moieties are an alternate functional group to modify. After dissolving filter paper in a CO2 switchable solvent system, Meier and co-workers reacted it with succinic acid, a bio-based feedstock, for 30 min at 25–80 °C to achieve DS of between 1.5 and 2.6.76 The DMSO and DBU could be recovered, but as with systems using acetic anhydride discussed in section 3.3, the [DBUH]+ salt needed to be neutralised using NaOH. Following the succinylation, the newly added carboxylic acid moiety was used to install amide groups via Ugi-4CR and Passerini-3CR reactions in DMSO (Scheme 11). Full conversion of the carboxylic acid groups was observed. The cellulose analogues were soluble in a range of organic solvents, and had glass transition temperatures below 100 °C, which would allow for homogeneous or thermal processing to produce cellulose materials.
 | ||
| Scheme 11 A general scheme for the Ugi-4CR (left) and Passerini-3CR (right) reactions performed on succinylated cellulose.76 | ||
An in situ tandem reaction removes the need for separating and washing the intermediate species, and can therefore be considered more environmental. This has been demonstrated for the formation of cellulose thiocarbamates, where cellulose dissolution is followed by modification with an in situ formed isothiocyanate (Scheme 12).77 As well as removing the harmful solvents used to dissolve cellulose by using a CO2 switchable solvent, this methodology benefits from avoiding the handling of toxic isothiocyanates, as this species forms in situ from a mixture of elemental sulfur and isocyanide, before reacting immediately. DBU has three roles in this process; assisting in the dissolution of cellulose, catalysing the reaction between sulfur and isocyanide, and catalysing the modification of cellulose with isothiocyanate. Due to the multiple roles of DBU, the authors report that the suspension of sulfur and isocyanide must be added dropwise to the cellulose solution to prevent too much DBU being removed from the cellulose–DBUH complex, as this results in precipitation of cellulose. The DS could be altered between 0.52 and 2.16, and was mainly dependant on the excess of isocyanide added, although increasing the cellulose concentration from 2 wt% to 5 wt% also increased the DS. Aside from the desired modification, a side reaction was possible, forming a cellulose oxocarbamate due to the presence of trace amounts of water. This reaction could not be completely supressed, but was limited by increasing the cellulose concentration and the excess of isocyanide. Upon completion of the reaction, the product was precipitated using iPrOH before fractional distillation was used to recover the DBU (79.1%), DMSO (91.5%), and iPrOH (95.6%). Overall the authors calculate the in situ process to have an E-factor of 2.95,77 whereas traditional process to modify cellulose can have E-factor values as higher than 10.62
 | ||
| Scheme 12 The in situ synthesis of isothiocyanates and the tandem synthesis of cellulose thiocarbamates using a CO2 switchable solvent.77 | ||
3.4. Synthesis of cellulose graft polymers in CO2 switchable solvents
Cellulose graft co-polymers, where the cellulose hydroxyl groups are used to grow other polymers directly on the cellulose surface, are used to reduce the hydrophilicity of cellulose in order to make it more compatible in composite materials.54 Previously this has been carried out heterogeneously due to the poor solubility of cellulose. This means that the reaction is controlled, and can be limited by, the supramolecular and morphological structure of the cellulose used.54 In particular, low grafting efficiency can be observed without thermal or chemical pre-treatment.78As seen in previous sections, dissolution of cellulose in a switchable solvent allows for control over the DS. This has been demonstrated for L-lactide79–81 and ε-caprolactone47 cellulose co-polymers, thereby producing fully bioderived composites (Scheme 13).
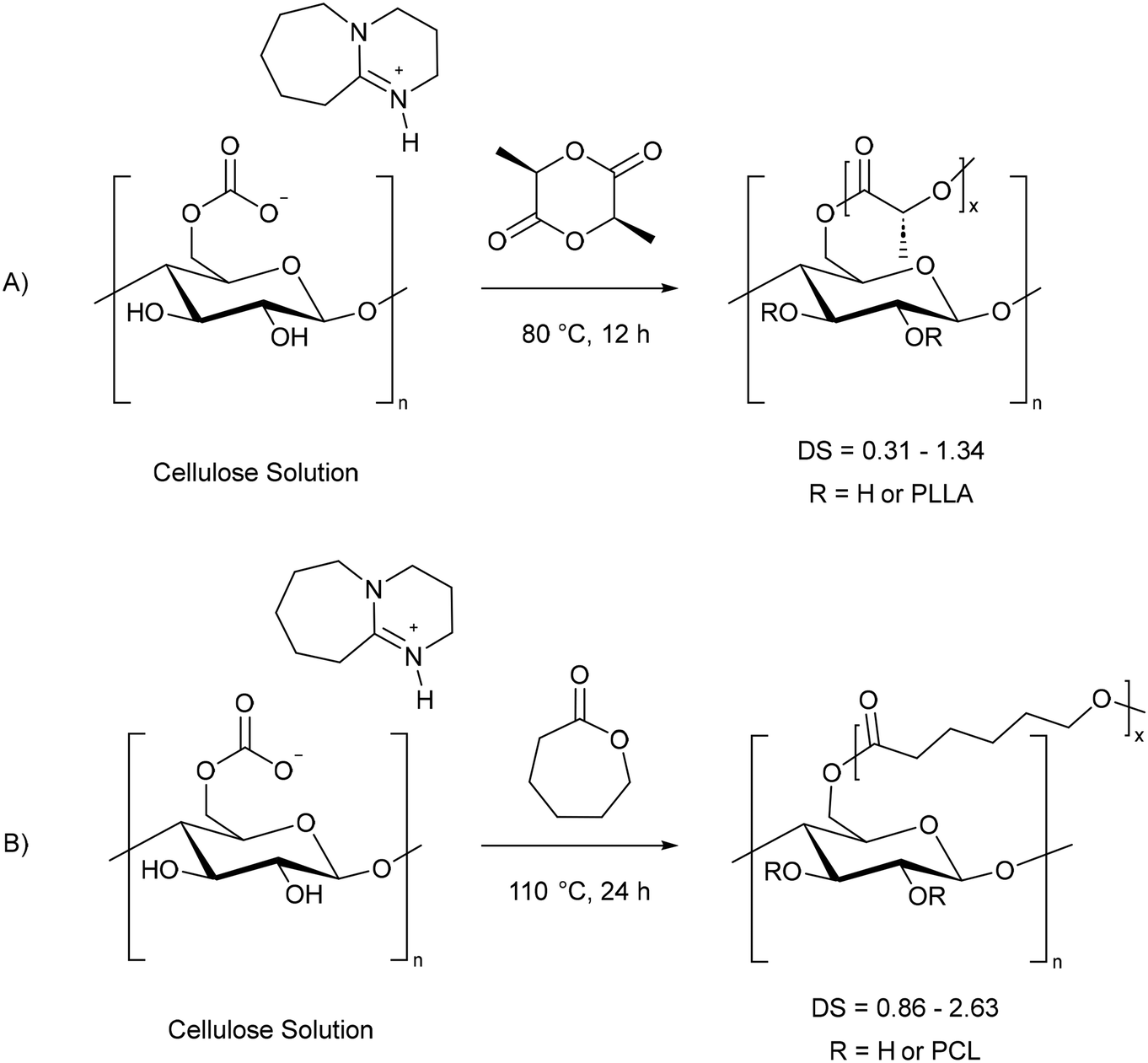 | ||
| Scheme 13 Grafting L-lactide (A)79 and ε-caprolactone (B)47 onto cellulose using CO2 switchable solvents. PLLA = polylactic acid, PCL = polycaprolactone. | ||
As the base used during cellulose dissolution can act as an in situ catalyst,43,69,77 this provides an additional advantage by removing the need to add an additional base catalyst, or harmful tin complex, which are often used for lactide ring opening polymerisation (ROP).82,83 As already noted, the degree of substitution can be controlled due to the homogeneous reaction mixture. The largest influence on the cellulose DS is through increasing the equivalents of L-lactide or ε-caprolactone. Song et al. report an increase in DS from 0.31 to 0.83 to 1.34 as the equivalents of L-lactide relative to cellulose are raised from 1 to 3 to 5.79 A maximum polylactic acid (PLLA) DS of 2.35 was achieved using 10 equivalents of L-lactide. In contrast, using melt co-polycondensation of cellulose and lactic acid catalysed by SnCl2, a limited DS range of 0.60–0.96 was achieved.84 Meanwhile the DS of polycaprolactone (PCL) grafted cellulose increased from 0.86 to 2.63 as the number of equivalents of ε-caprolactone increased from 5 to 15.47 This had a greater effect on the DS than increasing the reaction time or temperature.47 Grafting of PLLA can also be achieved using switchable solvents via transesterification of the in situ cellulose carbonate with PLLA.80 However, under the same conditions the degree of substitution was higher for the reaction of L-lactide compared to PLLA.
The molar substitution and total molecular weight of the substituted cellulose also increase with increasing DS, allowing the properties to be tuned. The glass transition temperature (Tg) of the cellulose polymers decreases as the DS increases,47,79 which would allow for thermoplastics with specific Tg to be synthesised. All the lactide and ε-caprolactone grafted cellulose analogues were soluble in a range of solvents, which depended on the DS. Both of these factors would allow the cellulose analogues to be processed further using traditional polymer methods.
Liu et al. demonstrate how this graft polymerisation can be implemented to enhance the properties of composite materials.81 They report that PLLA blended with cellulose had poor mechanical properties compared to unblended PLLA, with holes visible using SEM after tensile testing. However, blending L-lactide grafted cellulose (see Scheme 13A) with PLLA allowed better compatibility between the two polymers and prevented holes forming. Furthermore, as the amount of cellulose–PLLA polymer in the blend increased from 1 to 5 wt% the new composite material had impact strength of 772.3 kJ m−2, which is comparable to neat PLLA (793.9 kJ m−2). Similarly to the other lactide grafted polymers discussed above, the optimum blended polymer had a low Tg (58 °C) which would allow thermal processing. This demonstrates how cellulose can be used to improve the properties of PLLA whilst also reducing the cost through the incorporation of the biopolymer.
As the DS is easily controllable, and higher than that achieved using tin catalysts, CO2 switchable solvents are a promising method to prepare tuneable, biodegradable thermoplastics either directly from grafted cellulose, or by blending grafted cellulose with PLLA.
3.5. Summary of cellulose modification
We have discussed the modification of cellulose in CO2 switchable solvents in detail in section 3. A short guide to controlling the DS of these reactions is provided in Table 2. The DS of cellulose is largely controlled by the modifying substrate. Using a larger excess of substrate, or a more reactive species, produces a higher DS. Conditions such as temperature, time, etc. can be used to fine tune the DS, or promote a reaction where the substrate has a low reactivity.4. CO2 switchable solvents for the synthesis of functional cellulose materials
In the previous sections we have discussed strategies for the dissolution of cellulose in CO2 switchable solvents (2.1), the formation of simple cellulose materials (section 2.2), and the in situ modification of cellulose along with the important factors for controlling the degree of substitution (sections 3.3–3.5). In this section we look at examples where cellulose materials have been prepared for a specific purpose. These include water remediation,85–87 hydrogenation,88 electrochemical devices,89 and fluorescent materials and sensors.90–94Cellulose materials can be used as a support for nanoparticles, allowing them to have catalytic applications. Liang, Xie, and co-workers immobilised iron oxide nanoparticles in cellulose hydrogels (Fe3O4@CH) and carboxylate modified cellulose hydrogels (Fe3O4@CHC) (Scheme 14) before applying them to the oxidative degradation of Rhodamine B dye using H2O2.85
 | ||
| Scheme 14 A general scheme for the preparation of iron oxide nanoparticles entrapped in a modified cellulose gel (Fe3O4@CHC and Fe3O4@CH).85 | ||
After dissolving cellulose pulp in DMSO and tetramethylguanidine (1.5 MPa CO2, 50 °C, 3 h), excess CO2 was released and the solution mixed with 1 equivalent of succinic anhydride and varying amounts of pyromellitic dianhydride (PDMA) at room temperature to form a homogeneous solution (Scheme 15). This was placed in an oven at 80 °C for 5 h to form a gel, from which small discs were cut and suspended in water for 2 days to remove DMSO. The discs were suspended in solutions of FeCl3·6H2O and FeCl2·4H2O for 3 h, before being placed in a NaOH solution to form nanoparticles (Fe3O4@CHC and Fe3O4@CH), with the authors proposing that these are coordinated to the carboxylate groups of the modified cellulose (Fig. 9). Fe3O4@CH was prepared in a similar manner, without the addition of anhydride, and forming the gel at room temperature for 48 h rather than in an oven. FTIR, XRD, XPS, TEM and SEM were used to characterise the gels. It was found that both gels contained well-dispersed magnetite nanoparticles. The average crystallite size was smaller for Fe3O4@CHC (3.05 nm) than for Fe3O4@CH (6.02 nm). Fe3O4@CHC had a higher Fe loading, showing the importance of the carboxylate groups in controlling the particle size, and improving the immobilisation of nanoparticles within the gel.
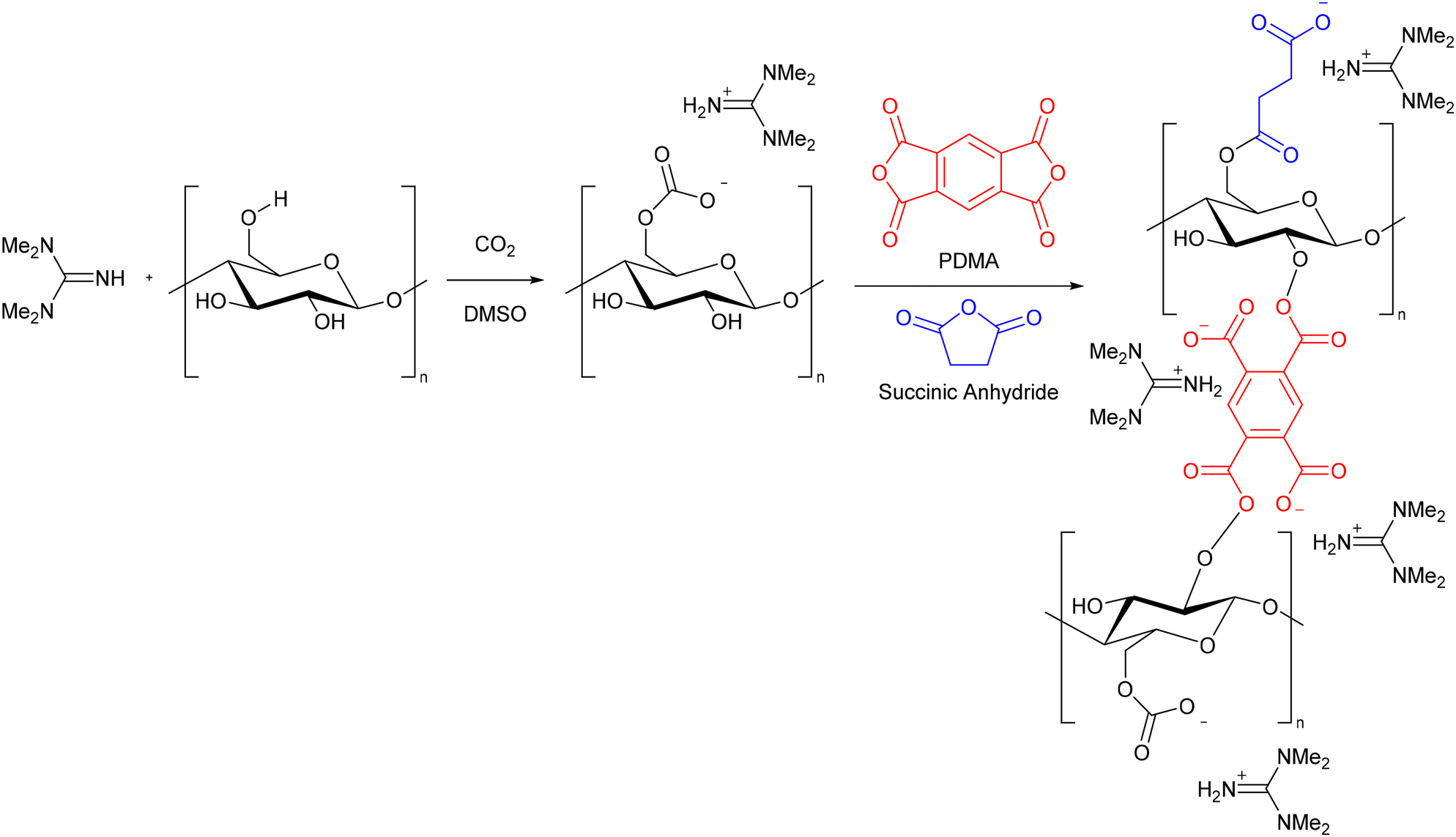 | ||
| Scheme 15 The synthesis of cellulose hydrogel precursors as proposed by Liang, Xie, and co-workers.85 Reproduced with permission from Elsevier, 2024. | ||
 | ||
| Fig. 9 The proposed interactions between Fe3O4 nanoparticles and the cellulose hydrogel.85 Reproduced with permission from Elsevier, 2024. | ||
Both gels (2.4 g L−1) were applied to the degradation of Rhodamine B (10 mg L−1) with H2O2 (1 mM) at pH 2. Fe3O4@CHC achieved 98% degradation after 3 h, whereas Fe3O4@CH and Fe3O4 nanoparticles achieved 92.9% and 71.8% degradation, respectively. The gels can absorb the dye to act as microreactors, thereby performing better than Fe3O4 nanoparticles, while the higher Fe loading of Fe3O4@CHC means it outperforms Fe3O4@CH. The gels were separated from the solution using a magnet, before re-use. Fe3O4@CHC maintained its degradation ability over 8 runs (ca. 98%), although recycling should be performed at a lower level to properly assess whether the catalyst is losing activity. Fe3O4@CH degradation decreased to 75% after 3 runs, and 44% after 5 runs. The better recycling performance of Fe3O4@CHC is attributed to lower Fe leaching (2.6 ppm) compared to Fe3O4@CH (5.45 ppm) due to the anchoring effect of the carboxylate groups. However, further work is needed to entirely prevent any leaching, as this would eventually lead to catalyst deactivation.
The same group also demonstrated reductive degradation of 4-nitrophenol using cellulose hydrogel supported palladium nanoparticles and NaBH4.86 The gels were prepared in a similar manner to that described above,85 except that as well as a carboxylate gel (Pd@CHC) and a cellulose gel (Pd@CH), the authors removed the NaOH washing step to leave the guanidinium ionic liquid in place (Pd@CHI). A NaBH4 solution was used to generate Pd nanoparticles from PdCl2. Pd@CHI had a higher Pd loading (3.34%) compared to Pd@CHC (0.11%) and Pd@CH (0.35%), showing the ionic liquid assists in anchoring the Pd nanoparticles. Unlike the hard Fe nanoparticles described above,85 carboxylate groups do not appear to coordinate soft Pd nanoparticles to such a great extent, leading to low Pd loading. The highest loading was accompanied by the greater degradation activity of 4-nitrophenol using Pd@CHI compared to the other gels. Pd@CHI had a higher rate of reaction, and a lower activation energy. After the degradation reaction, the gels were recovered via filtration. Whilst Pd@CHI maintained its activity over 10 runs, the other gels had decreased activity after each use. The true stability of the Pd@CHI gel is difficult to determine as the recycling reactions are all performed at 100% 4-nitrophenol degradation, however analysis by TEM did show only a slight aggregation and difference in size distribution of the nanoparticles after the reaction. The sustainability of this method is reduced by the use of NaBH4 during the water remediation reaction.
This gel formation process was further adapted to prepare cellulose gel catalysts with tailored hydrophobicity.88 Using a CO2 switchable solvent, cellulose was modified with pyromellitic dianhydride (PDMA) and varying ratios of succinic anhydride and 2-dodecen-1-ylsuccinic anhydride (DSA) (Scheme 16). The gels prepared in this manner were soaked in a PdCl2 solution, before nanoparticles were generated using NaBH4 in the same manner as described by Xie and co-workers.86 The hydrophobicity of the gels, as determined by the water contact angle, increased as the amount of DSA used during preparation increased. The gels were applied to the hydrogenation of styrene and cinnamaldehyde using an atmospheric pressure of hydrogen gas, and 4-nitrophenol using NaBH4 as reductant. The hydrophobicity of the catalyst affected the performance in the different reactions. For the hydrogenation of non-polar styrene and cinnamaldehyde, the rate of reaction and turnover frequency (TOF) increased as the gel catalyst became more hydrophobic. The authors propose that this is due to better uptake of the substrate. For cinnamaldehyde, the authors state that both 3-phenylpropanol and 3-phenylproponaldehyde were products, but do not comment on the selectivity. Hydrogenation of the more polar 4-nitrophenol was found to be faster when a more hydrophilic gel catalyst was used. This highlights the benefit of using CO2 switchable solvent systems to prepare cellulose functional materials as the properties can be easily controlled.
 | ||
| Scheme 16 The formation of gels with tailored hydrophobicity using CO2 switchable solvents.88 | ||
While further improvements to these gels would be useful, such as a simpler production process, they have been used to successfully demonstrate the potential of CO2 switchable solvent methods to develop cellulose materials to catalyse different classes of redox reactions. These could be applied not only water remediation, but to the synthesis of bulk and fine chemicals.95
The removal of oil from water is another important challenge, as it otherwise leads to environmental damage and harm to wildlife.96 For cellulose to be effective at absorbing oil from water it needs to be modified to become more hydrophobic. Mu and co-workers modified cellulose with tetraethylorthosilicate (TEOS) to form a gel which could be dispersed in solvent and spray coated onto a sponge, filter paper, and glass surfaces (Scheme 17).87 Before spray coating, the hydrophobicity could be improved by soaking the gel in 1,1,1,3,3,3-hexamethyldisilazane (HMDS). The water contact angle (WCA) of polyvinylidenefluoride filter paper before coating was 37°. After coating with the cellulose–silica coating it became 110°, whilst coating with the cellulose–silica–HMDS gel increased the WCA to 165°, allowing it to be classified as a super-hydrophobic coating. The contact angles of paper, sponge, and glass coated with the modified cellulose were maintained after rubbing 20 times with sandpaper, demonstrating the robustness of the materials. Furthermore, water drops across the entire pH range did not penetrate the cellulose–silica–HMDS coating, showing that it could be used in seawater, a key requirement for the remediation of oil spills. A sponge soaked in the super hydrophobic coating was able to remove over 90% of a variety of oils from water, with a maximum capacity of 89 g g−1 for n-decane. Despite being common for preparing hydrophobic materials, the use of HDMS is not ideal from a green perspective. As the authors state that cellulose aerogels have better absorption capacity, and as they can also be prepared using CO2 switchable solvents,53 they may be a better solution to preparing cellulose materials for oil absorption.
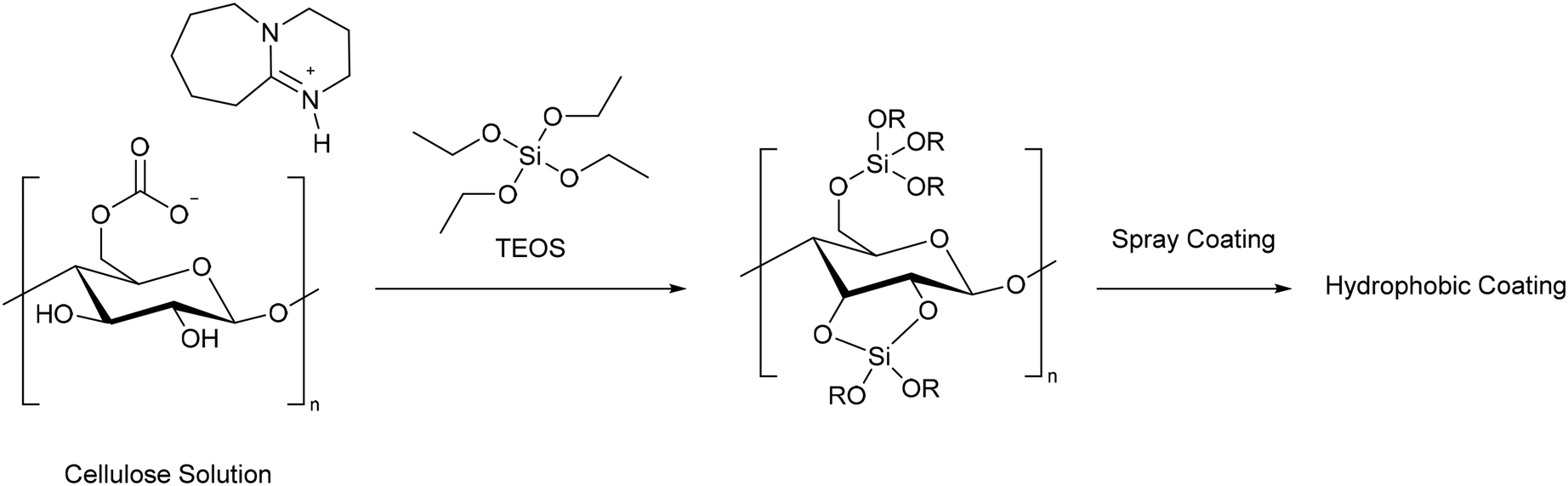 | ||
| Scheme 17 Preparation of a hydrophobic coating by reacting cellulose with tetraethylorthosilicate (TEOS) in a CO2 switchable solvent.87 | ||
Xie, Xu, and co-workers have prepared a cellulose electrolyte gel for a flexible supercapacitor using CO2 switchable solvents.89 Gel electrolytes have advantages over liquid electrolytes as they are less prone to leakage, and are easily shaped. Cellulose was dissolved in a switchable solvent using DMSO, 1.5 MPa CO2 and either DBU or TMG. These components were incorporated into the final electrolyte and contribute to the ionic conductance. The cellulose was modified with succinic anhydride, before polyvinyl alcohol (PVA) and epichlorohydrin (ECH) were added to the solution to form a cross-linked gel which was placed in a mould and dried at 60 °C for 2 h (Scheme 18). The authors tested the mechanical properties of electrolytes of different compositions. It was found that ECH was essential for gel formation, and that increasing the amount of PVA (0–200 wt% of cellulose) led to improved mechanical strength, with a maximum tensile strength of 197.6 kPa and elongation at break of 78.7%. Supercapacitors were fabricated by sandwiching the electrolyte between two electrode sheets prepared from a mixture of activated carbon, acetylene black, and polytetrafluoroethylene (PTFE) supported on a nickel foam. The ionic conductivity increased from 1.2–5.6 mS cm−1, as the amount of PVA in the electrolyte decreased due to the decreased viscosity and resistance to ion transport. The TMG-based gel had higher conductivity (4.1 mS cm−1) than the DBU-based gel (2.3 mS cm−1), probably due to its smaller size and better ion mobility. The TMG-based electrolyte with 1:1 cellulose to PVA was selected for further testing as it had a good balance between mechanical stability (tensile strength ca. 60 kPa) and conductivity (4.2 mS cm−1). This supercapacitor had an electrochemical window between −2.14 and 2.54 V. The maximum energy density at room temperature was 17.38 W h kg−1, and the capacitance was stable over 8000 charge–discharge cycles. An added advantage of using a gel electrolyte is its flexibility. The cyclic voltammograms of the cellulose polymer electrolyte remained consistent after 100 cycles when the supercapacitor was bent 120°. This demonstrates that CO2 switchable solvents contribute not only to processing cellulose, but can also be incorporated as functional components to enhance the properties of the final material.
 | ||
| Scheme 18 The formation of a cross-linked cellulose gel electrolyte as proposed by Xu, Xu, and co-workers.89 | ||
Several authors have used CO2 switchable solvents to prepare fluorescent cellulose derivatives. Cao et al. achieved this by adding different fluorescent pigments into a DMSO/DBU/CO2 cellulose solution and allowing them to react at 30 °C for 3 h.90 The emission wavelength was tuned based on the fluorescent derivative used (Fig. 10). All fluorescent cellulose analogues had good absorbance in the range 250–365 nm. The authors state that the fluorescent additive is bound to the cellulose via a carbonate moiety (see section 3.2) and that this enhances the fluorescent emission through a push–pull mechanism, leading to a large Stokes Shift, and distinct emission wavelengths. The fluorescent cellulose derivatives were used to prepare epoxy resin coatings. Despite using a CO2 switchable solvent for the first derivatisation, the authors activate cellulose by submerging it in a 2 M NaOH solution to react with a silane coupling agent. This further modified cellulose derivative is then mixed with diethylenetriamine and cured in a mould at 80 °C for 3 h. The epoxy coating exhibited uniform fluorescence which the authors claim is partly due the suppression of aggregation-caused quenching (ACQ), which is often observed for fluorescent pigments. Compared to pure epoxy coating, the fluorescent cellulose epoxy coating show improved hardness from H to 2H on the GB/T6739-2006 scale. The tensile strength is also increased from 56 MPa to 75–83 MPA due to the addition of fluorescent cellulose.
 | ||
| Fig. 10 The fluorescent cellulose derivatives prepared by Cao et al.90 | ||
Rather than preparing an epoxy resin, Zhang et al. grafted L-lactide to cellulose, to improve its processability, before adding a fluorescent tetraphenylethylene moiety to the cellulose backbone in a one-pot process (Scheme 19).91 The authors report that the process must be carried out in one pot, as isolating and purifying the L-lactide cellulose derivative before performing a second reaction in a new CO2 switchable solvent led to a low degree of substitution. We suggest this is due to the carbonate anion forming on the lactide rather than the cellulose, as would happen in the non-derivative process (section 2), preventing the reaction between cellulose and 1-[4-(bromomethyl)phenyl]-1,2,2-triphenylethene. The authors also comment that the fresh DBU would catalyse transesterification of polylactic acid, reducing the substitution on cellulose. The cellulose derivatives show aggregation-induced fluorescence. When in solution there is no fluorescence, but when the solvent evaporates, or an anti-solvent causes precipitation, fluorescence is observed. The absorbance peak occurs at 400 nm and the emission peak at 480 nm. The intensity of these peaks increases as the incorporation of fluorescent tetraphenylethylene increases. The L-lactide chain facilitates melt processing above 160 °C, whilst also allowing the cellulose derivative to dissolve in various organic solvents such as DMSO, THF, methanol and acetone, among others. This allowed the authors to prepare a fluorescent film using melt compress moulding, and fibres via electrospinning.
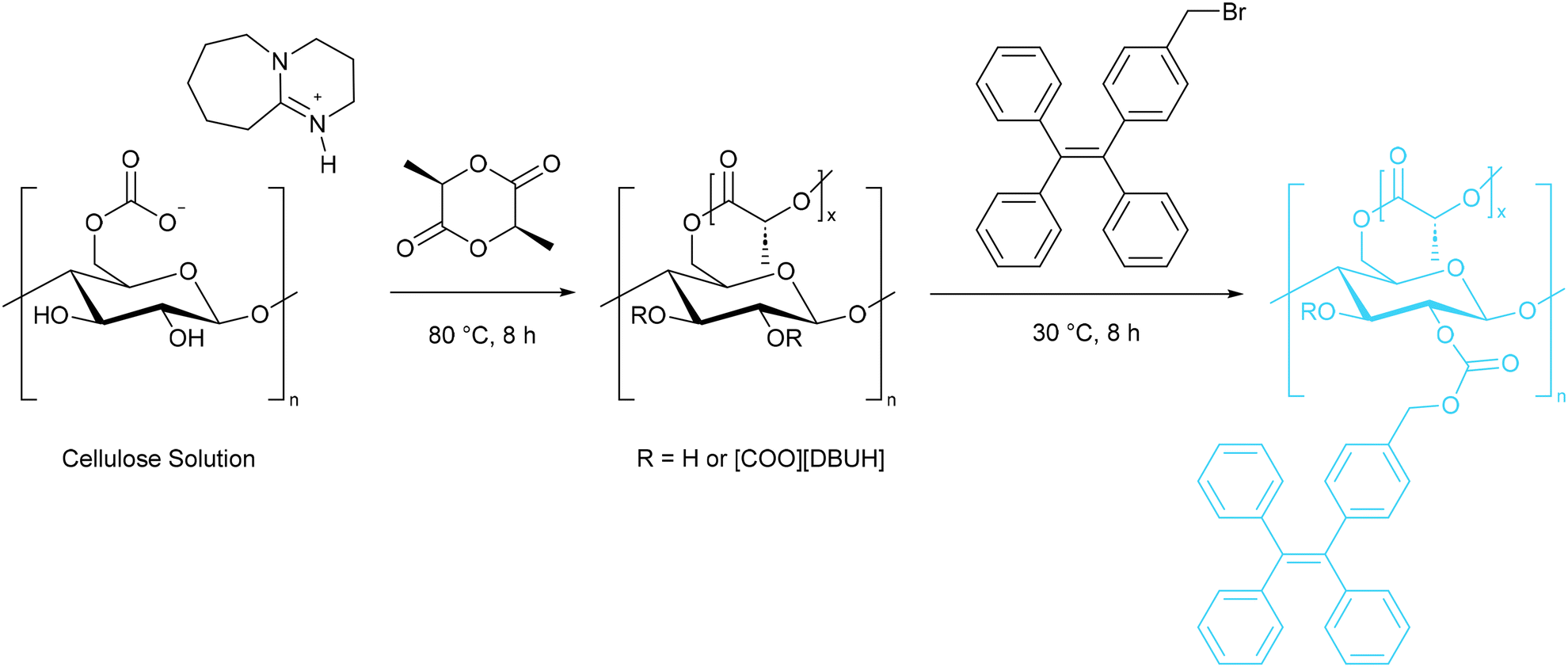 | ||
| Scheme 19 The preparation of a fluorescent cellulose derivative with a polylactic acid side chain.91 | ||
Cellulose materials with fluorescent quenching in the pH range 12–14 have been reported.92 These have been specifically applied to assessing the spoilage in shellfish.93 A CO2 switchable solvent was used to graft L-lactide, followed by 7-amino-4-(trifluoromethyl) coumarin (AFC) (Scheme 20).
 | ||
| Scheme 20 The synthesis of a fluorescent cellulose derivative for freshness monitoring of shellfish.93 | ||
The cellulose analogue could be excited at wavelengths ranging from 400 to 550 nm, with a maximum absorption peak at 450 nm, and emitted light between 400 and 600 nm, with maximum emission at 460 nm. AFC fluoresces at neutral pH, and is only slightly affected by acid. However, in the presence of base the colour changes from blue to cyan, and the intensity is greatly reduced (Scheme 21). Electrospinning was used to prepare a nano fibrous film, as these films have good absorbance of volatile gases. When the film was exposed to ammonia the fluorescence colour changed to cyan and the fluorescence was reduced. Heating the film could remove the ammonia and return the fluorescence to its original state, and this process could be repeated several times. The nanofibrous film was used to prepare a freshness analysis card. This was packaged with shrimp for 1–7 days at different temperatures (−18 °C, 5 °C, and 15 °C). The colour of the film was monitored and compared to the total volatile base nitrogen (TVBN), which is an industrial standard for freshness monitoring of aquatic products in the food industry. At 15 °C the film changed colour within 1 day, in agreement with the high TVBN level. At 5 °C, the film changed colour after 1 day, when the TVBN level was in the “borderline” range. At −18 °C, the film did not change, and the TVBN also indicated that the shrimp remained fresh. This demonstrated that the modified cellulose could function as a visual test for shellfish freshness, thereby contributing to a reduction in food waste using a biodegradable cellulose film.
 | ||
| Scheme 21 The change in colour of AFC modified cellulose when exposed to acid and base.93 | ||
The preparation of fluorescent on–off sensors for Fe3+ ions has been achieved using CO2 switchable solvents.94 The cellulose material was first prepared by grafting L-lactide, followed by methyl 6-bromomethyl coumarin-3-carboxylate. The fluorescent moiety (MCD) was formed in situ by a cyclocondensation reaction between DBU and methyl 6-bromomethyl coumarin-3-carboxylate (Scheme 22). The DS of both components depended on the excess in which they were added. The DS of PLLA was between 1.42 and 1.50 while the DS of MCD was 0.086–0.204.
 | ||
| Scheme 22 The preparation of a fluorescent sensor for Fe3+ in a CO2 switchable solvent.94 | ||
The absorption maximum occurred at 340 nm and the intensity increased with the DS of MCD. In contrast, the fluorescent maximum at 430 nm decreased with increasing DS of MCD, demonstrating that aggregation-caused quenching (ACQ) was taking place. Solutions of the cellulose derivative were prepared and a series of metal ions were added (Na+, K+, Ag+, Mg2+, Ca2+, Mn2+, Fe2+, Co2+, Ni2+, Cu2+, Zn2+, Cd2+, Ba2+, Hg2+, Al3+, Ga3+ and Fe3+). Only Fe3+ ions caused fluorescence quenching as determined visually and by the UV-vis absorption and fluorescent emission spectra. This quenching behaviour was still observed when Fe3+ and other metal ions were present at the same time, showing the sensor was selective for Fe3+. FTIR and XPS was used to analyse the binding of Fe3+ to the cellulose derivative, which showed that a carbonyl group was involved, and new Fe–O bonds were formed, but the PLLA did not take part. The authors therefore proposed that the formation of a complex between the carbonate linkage and Fe3+ was leading to non-radiative fluorescence quenching (Scheme 23). In solution, the limit of detection (LOD) for Fe3+ was calculated to be 3.74 × 10−6 M, and the quenching co-efficient (Ksv) was 5.639 × 103 M−1. A flat film of the fluorescent cellulose derivative was prepared using melt-compression moulding, and a nanofibrous film prepared by electrostatic spinning. The flat film was tested for Fe3+ sensing in flowing water. Under UV light, fluorescence was observed when water was dripped onto the film. This was immediately quenched when a 5 mM Fe3+ solution was run over the film. As water was again flowed over the film, the fluorescence was gradually restored (Scheme 23). The good sensitivity and selectivity, along with the reversible nature of this film are a demonstration of the potential for in-field water testing using cellulose materials prepared in CO2 switchable solvents.
 | ||
| Scheme 23 The mechanism of reversible fluorescence quenching proposed by Yu et al.94 | ||
5. Conclusions and future challenges
There are multiple uses of CO2 switchable solvents for cellulose processing. They can be applied to prepare unmodified cellulose materials such as fibres, films and gels. One can also gain control over cellulose functionalisation, with a tailored degree of substitution possible to allow materials with specific properties to be produced. This can lead to the preparation of functional materials for catalysis, sensing, and electrochemistry among other applications. The use of CO2 switchable solvents facilitates these processes without the use of the traditional harsh and wasteful conditions used for cellulose dissolution and modification. These conditions limit the environmental nature of materials formed from this biopolymer. The use of CO2 switchable solvents could dramatically improve the sustainability of industrial processes, and therefore the final materials, but there are still issues to be considered which we outline below.In our opinion, it is vitally important to conclusively determine the mechanism of reaction between cellulose and modifying groups (see section 3.2). While this does not affect the preparation of unmodified cellulose materials, the structure of modified cellulose must be known for the process to be industrially relevant. This is not a significant hurdle as many cellulose derivatives prepared by this method are soluble in common organic solvents, making their analysis relatively straightforward. If the reaction only proceeds via the in situ formed carbonate anion, then only cellulose esters can be prepared directly using this method, although the formed ester could be reduced to form the corresponding ether. Methods to perform this transformation would need to be developed. This would complicate the process and require extra materials and energy, thereby reducing the green advantages of CO2 switchable solvents. However, there are many catalytic approaches to hydrogenate esters,97–99 meaning this potential drawback would not prevent the use of CO2 switchable solvents for cellulose processing. Of course, it must be remembered that this may not be an issue at all, depending on the reaction mechanism.
Finally, although CO2 switchable solvents represent a more environmental approach to cellulose processing, further improvements are possible. These relate to the current use of DMSO co-solvent and a base. While DMSO and the solvent used for precipitation/coagulation can be recovered effectively, the base is often recovered in a lower yield. If an appropriate supported base could facilitate cellulose dissolution in CO2 switchable solvents, recovery might be simplified. Furthermore, the most commonly employed base, DBU, is toxic. Investigations into less harmful bases for this reaction would be beneficial, and would further contribute to the sustainability of these processes. For example, N-methylimidazole (NMI, Fig. 11 left) could be an ideal replacement for DBU. NMI fulfils the requirements for cellulose dissolution set out in section 2.1, namely a high pKa (ca. 19) and the ability to delocalise positive charge. Furthermore, NMI is non-toxic and has a lower boiling point than DBU which could facilitate its recovery by distillation after the reaction. Alternatively, imidazole could be supported on a resin (Fig. 11, right), which would simplify the recycling process.
 | ||
| Fig. 11 The structure of N-methylimidazole (left) and the method to support imidazole on resin (right). | ||
Similarly, the replacement of DMSO with a bio-based solvent would constitute a green improvement. According to the discussion in section 2.1, a new solvent should be polar to assist with charge delocalisation, and preferably aprotic to prevent it interfering with proton abstraction during the derivative process. Further research is required, but Cyrene (Fig. 12), which is derived from cellulose, is one possibility as it has similar polarity and boiling point when compared to DMSO.100 This could allow it to assist in cellulose dissolution, and be recovered by distillation after the reaction.
 | ||
| Fig. 12 The structure of Cyrene (dihydrolevoclucosenone). | ||
We have highlighted the immediate concerns which must be addressed for scale up of CO2 switchable solvents for cellulose. In addition, a full engineering, economic, and life cycle analysis has to be performed in the future to determine the feasibility of industrial use. Despite the challenges discussed here, CO2 switchable solvents have excellent potential to be industrially applicable for cellulose processing and modification in a more sustainable manner.
Data availability
This is a review article. All sources have been cited, allowing the authors of the original literature to be contacted.Conflicts of interest
There are no conflicts of interest to declare.References
- Top 10 Emerging Technologies 2019, World Economic Forum, 2019. https://www3.weforum.org/docs/WEF_Top_10_Emerging_Technologies_2019_Report.pdf (Accessed June 2023).
- D. Klemm, B. Heublein, H.-P. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 44, 3358–3393, DOI:10.1002/anie.200460587.
- R. Mori, RSC Sustainability, 2023, 1, 179–212, 10.1039/D2SU00014H.
- C. Campano, A. Balea, A. Blanco and C. Negro, Cellulose, 2016, 23, 57–91, DOI:10.1007/s10570-015-0802-0.
- Y. Liu, S. Ahmed, D. E. Sameen, Y. Wang, R. Lu, J. Dai, S. Li and W. Qin, Trends Food Sci. Technol., 2021, 112, 532–546, DOI:10.1016/j.tifs.2021.04.016.
- R. Das, T. Lindström, P. R. Sharma, K. Chi and B. S. Hsiao, Chem. Rev., 2022, 122, 8936–9031, DOI:10.1021/acs.chemrev.1c00683.
- B. Frka-Petesic and S. Vignolini, Nat. Photonics, 2019, 13, 365–367, DOI:10.1038/s41566-019-0448-9.
- R. M. Parker, T. H. Zhao, B. Frka-Petesic and S. Vignoli, Nat. Commun., 2022, 13, 3378, DOI:10.1038/s41467-022-31079-9.
- B. E. Droguet, H. L. Liang, B. Frka-Petesic, R. M. Parker, M. F. De Volder, J. J. Baumberg and S. Vignolini, Nat. Mater., 2022, 21, 352–358, DOI:10.1038/s41563-021-01135-8.
- A. Sharma, M. Thakur, M. Bhattacharya, T. Mandal and S. Goswami, Biotechnol. Rep., 2019, 21, e00316, DOI:10.1016/j.btre.2019.e00316.
- P. G. Gan, S. T. Sam, M. F. B. Abdullah and M. F. Omar, J. Appl. Polym. Sci., 2020, 137, 48544, DOI:10.1002/app.48544.
- J. Li, R. Cha, K. Mou, X. Zhao, K. Long, H. Luo, F. Zhou and X. Jiang, Adv. Healthcare Mater., 2018, 7, 1800334, DOI:10.1002/adhm.201800334.
- H. Seddiqi, E. Oliaei, H. Honarkar, J. Jin, L. C. Geonzon, R. G. Bacabac and J. Klein-Nulend, Cellulose, 2021, 28, 1893–1931, DOI:10.1007/s10570-020-03674-w.
- K. Heise, E. Kontturi, Y. Allahverdiyeva, T. Tammelin, M. B. Linder, Nonappa and O. Ikkala, Adv. Mater., 2021, 33, 2004349, DOI:10.1002/adma.202004349.
- B. Lindman and B. Medronho, BioResources, 2015, 10, 3811–3814, DOI:10.15376/biores.10.3.3811-3814.
- B. Medronho and B. Lindman, Adv. Colloid Interface Sci., 2015, 222, 502–508, DOI:10.1016/j.cis.2014.05.004.
- B. Medronho, H. Duarte, L. Alves, F. Antunes, A. Romano and B. Lindman, Nord. Pulp Pap. Res. J., 2015, 30, 58–66, DOI:10.3183/npprj-2015-30-01-p058-066.
- E. E. Treiber, in Cellulose Chemistry and its applications, ed. T. P. Nevell and S. H. Zeronian, Halsted Press, John Wiley, New York, 1985, ch. 18 Search PubMed.
- T. Rosenau, A. Potthast, H. Sixta and P. Kosma, Prog. Polym. Sci., 2001, 26, 1763–1837, DOI:10.1016/S0079-6700(01)00023-5.
- N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123–150, 10.1039/B006677J.
- J. P. Hallett and T. W. Welton, Chem. Rev., 2011, 111, 3508–3576, DOI:10.1021/cr1003248.
- E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060–11082, DOI:10.1021/cr300162p.
- Y. Li, J. Wang, X. Liu and S. Zhang, Chem. Sci., 2018, 9, 4027–4043, 10.1039/C7SC05392D.
- Y.-L. Chen, X. Zhang, T.-T. You and F. Xu, Cellulose, 2019, 26, 205–213, DOI:10.1007/s10570-018-2130-7.
- R. Häkkinen and A. Abbott, Green Chem., 2019, 21, 4673–4682, 10.1039/C9GC00559E.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975, DOI:10.1021/ja025790m.
- A. W. T. King, J. Asikkala, I. Mutikainen, P. Järvi and I. Kilpeläinen, Angew. Chem., Int. Ed., 2011, 50, 6301–6305, DOI:10.1002/anie.201100274.
- H. T. Vo, Y. J. Kim, E. H. Jeon, C. S. Kim, H. S. Kim and H. Lee, Chem. – Eur. J., 2012, 18, 9019–9023, DOI:10.1002/chem.201200982.
- S. Thiemann, S. J. Sachnov, F. Pettersson, R. Bollström, R. Österbacka, P. Wasserscheid and J. Zaumseil, Adv. Funct. Mater., 2014, 24, 625–634, DOI:10.1002/adfm.201302026.
- S. Elsayed, J. Helminen, S. Hellsten, C. Guizani, J. Witos, M. Rissanen, A. H. Rantamäki, P. Hyväri, P. Varis, S. K. Wiedmer, I. Kilpeläinen and H. Sixta, ACS Sustainable Chem. Eng., 2020, 8, 14217–14227, DOI:10.1021/acssuschemeng.0c05330.
- J. Zhang, J. Wu, J. Yu, X. Zhang, J. He and J. Zhang, Mater. Chem. Front., 2017, 1, 1273–1290, 10.1039/C6QM00348F.
- Z. Xia, J. Li, J. Zhang, X. Zhang, X. Zheng and J. Zhang, J. Bioresour. Bioprod., 2020, 5, 79–95, DOI:10.1016/j.jobab.2020.04.001.
- Ioncell, https://ioncell.fi/commercialization/, (accessed September 2023).
- D. J. Heldebrant, P. G. Jessop, C. A. Thomas, C. A. Eckert and C. L. Liotta, J. Org. Chem., 2005, 70, 5335–5338, DOI:10.1021/jo0503759.
- P. G. Jessop, D. J. Heldebrant, X. Li, C. A. Eckert and C. L. Liotta, Nature, 2005, 436, 1102, DOI:10.1038/4361102a.
- P. G. Jessop, S. M. Mercer and D. J. Heldebrant, Energy Environ. Sci., 2012, 5, 7240–7253, 10.1039/C2EE02912J.
- P. G. Jessop and M. F. Cunningham, CO2-Switchable Materials, Royal Society of Chemistry, Cambridge, 2020 Search PubMed.
- M. F. Cunningham and P. G. Jessop, Chem. Commun., 2023, 59, 13272–13288, 10.1039/D3CC03929C.
- A. Bordet, S. El Sayed, M. Sanger, K. J. Boniface, D. Kalsi, K. L. Luska, P. G. Jessop and W. Leitner, Nat. Chem., 2021, 13, 916–922, DOI:10.1038/s41557-021-00735-w.
- Q. Zhang, N. S. Oztekin, J. Barrault, K. De Oliveira Vigier and F. Jérôme, ChemSusChem, 2013, 6, 593–596, DOI:10.1002/cssc.201200815.
- H. Xie, X. Yu and Z. K. Zhao, Green Chem., 2014, 16, 2422–2427, 10.1039/C3GC42395F.
- C.-G. Wang, N. Li, G. Wu, T. T. Lin, A. M. X. Lee, S.-W. Yang, Z. Li and H.-K. Luo, Carbohydr. Polym. Technol. Appl., 2022, 3, 100186, DOI:10.1016/j.carpta.2022.100186.
- K. N. Onwukamike, T. Tassaing, S. Grelier, E. Grau, H. Cramail and M. A. R. Meier, ACS Sustainable Chem. Eng., 2018, 6, 1496–1503, DOI:10.1021/acssuschemeng.7b04053.
- D. J. Heldebrant, C. R. Yonker, P. J. Jessop and L. Phan, Energy Environ. Sci., 2008, 1, 487–493, 10.1039/B809533G.
- J. Zhang, S. Mi, F. Liu, Q. Qiao, H. Na and J. Zhu, Cellulose, 2022, 29, 6745–6758, DOI:10.1007/s10570-022-04670-y.
- M. Gunnarsson, D. Bernin, M. Hasani, M. Lund and E. Bialik, ACS Sustainable Chem. Eng., 2021, 9, 14006–14011, DOI:10.1021/acssuschemeng.1c05863.
- Q. Xu, L. Song, L. Zhang, G. Hu, J. Du, E. Liu, Q. Zheng, Y. Liu, N. Li and H. Xie, ChemistrySelect, 2017, 2, 7128–7134, DOI:10.1002/slct.201701639.
- A. Kirchberg and M. A. R. Meier, Macromol. Chem. Phys., 2021, 222, 2000433, DOI:10.1002/macp.202000433.
- J. Li, S. Lu, F. Liu, Q. Qiao, H. Na and J. Zhu, ACS Sustainable Chem. Eng., 2021, 9, 4744–4754, DOI:10.1021/acssuschemeng.0c08907.
- M. Heidari, K. N. Onwukamide, E. Grau, S. Grelier, H. Cramail, M. A. R. Meier and A. Greiner, Cellulose, 2021, 28, 6869–6880, DOI:10.1007/s10570-021-03967-8.
- L. Jin, J. Gan, G. Hu, L. Cai, Z. Li, L. Zhang, Q. Zheng and H. Xie, Polymers, 2019, 11, 994, DOI:10.3390/polym11060994.
- L. Zhang, W. Shi, H. Sheng, S. Feng, M. Yao, P. Chen, Q. Zheng and H. Xie, Green Chem., 2021, 23, 5856–5865, 10.1039/D1GC01771C.
- K. N. Onwukamike, L. Lapuyade, L. Maillé, S. Grelier, E. Grau, H. Cramail and M. A. R. Meier, ACS Sustainable Chem. Eng., 2019, 7, 3329–3338, DOI:10.1021/acssuschemeng.8b05427.
- T. Heinze, O. A. El Seoud and A. Koschella, Cellulose Derivatives: Synthesis, Structure, and Properties, Springer International Publishing AG, Switzerland, 2019 Search PubMed.
- Nouryon Cellulose Ethers, https://www.nouryon.com/products/cellulose-ethers/?_gl=1*a260kf*_up*MQ..&gclid=EAIaIQobChMI4bu715T-_QIVRg0GAB1q0AQnEAAYASAAEgIxlfD_BwE, (accessed November 2023).
- DOW cellulose ether, https://www.dow.com/en-us/search.html#q=carboxymethyl%20ether&t=All&sort=relevancy, (accessed November 2023).
- Ashland cellulosics, https://www.ashland.com/search?chemistry=cellulosics, (accessed November 2023).
- M. S. Rahman, M. S. Hasan, A. S. Nittai, S. Nam, A. K. Karmakar, M. S. Ahsan, M. J. A. Shiddiky and M. B. Ahmed, Polymers, 2021, 13, 1345, DOI:10.3390/polym13081345.
- K. J. Edgar, C. M. Buchanan, J. S. Debenham, P. A. Rundquist, B. D. Seiler, M. C. Shelton and D. Tindall, Prog. Polym. Sci., 2001, 26, 1605–1688, DOI:10.1016/S0079-6700(01)00027-2.
- V. Vatanpour, M. E. Pasaoglu, H. Barzeger, O. O. Teber, R. Kaya, M. Bastug, A. Khataee and I. Koyuncu, Chemosphere, 2022, 295, 133914, DOI:10.1016/j.chemosphere.2022.133914.
- A. Pettignano, A. Daunay, C. Moreau, B. Cathala, A. Charlot and E. Fleury, ACS Sustainable Chem. Eng., 2019, 7, 14685–14696, DOI:10.1021/acssuschemeng.9b02634.
- K. N. Onwukamike, S. Grelier, E. Grau, H. Cramail and M. A. R. Meier, ACS Sustainable Chem. Eng., 2019, 7, 1826–1840, DOI:10.1021/acssuschemeng.8b04990.
- W. Ge, J. Shuai, Y. Wang, Y. Zhou and X. Wang, Polym. Chem., 2022, 13, 359–372, 10.1039/D1PY00879J.
- P. McNeice, G. H. ten Brink, U. Gran, L. Karlson, R. Edvinsson and B. L. Feringa, RSC Sustainability, 2024, 2, 369–376, 10.1039/D3SU00317E.
- S. Mi, Z. Yao, F. Liu, Y. Li, J. Wang, H. Na and J. Zhu, Green Chem., 2022, 24, 8677–8684, 10.1039/D2GC02989H.
- Y. Yang, H. Xie and E. Liu, Green Chem., 2014, 16, 3018–3023, 10.1039/C4GC00199K.
- Y. Yang, L. Song, C. Peng, E. Liu and H. Xie, Green Chem., 2015, 17, 2758–2763, 10.1039/C5GC00115C.
- J. Wolfs and M. A. R. Meier, Green Chem., 2021, 23, 4410–4420, 10.1039/D1GC01508G.
- Z. Yao, S. Mi, B. Chen, F. Liu, H. Na and J. Zhu, ACS Sustainable Chem. Eng., 2022, 10, 17327–17335, DOI:10.1021/acssuschemeng.2c05872.
- N. S. Allen, M. Edge, J. H. Appleyard, T. S. Jewitt, C. V. Horie and D. Francis, Polym. Degrad. Stab., 1987, 19, 379–387, DOI:10.1016/0141-3910(87)90038-3.
- T. Sehn and M. A. R. Meier, Biomacromolecules, 2023, 24, 5255–5264, DOI:10.1021/acs.biomac.3c00762.
- K. N. Onwukamike, S. Grelier, E. Grau, H. Cramail and M. A. R. Meier, ACS Sustainable Chem. Eng., 2018, 6, 8826–8835, DOI:10.1021/acssuschemeng.8b01186.
- T. Kulomaa, J. Matikainen, P. Karhunen, M. Heikkilä, J. Fiskari and I. Kilpeläinen, RSC Adv., 2015, 5, 80702–80708, 10.1039/C5RA12671A.
- P. Wang and B. Y. Tao, J. Polym. Environ., 1995, 3, 115–119, DOI:10.1007/BF02067487.
- E. Esen, P. Hädinger and M. A. R. Meier, Biomacromolecules, 2021, 22, 586–593, DOI:10.1021/acs.biomac.0c01444.
- Z. Söyler, K. N. Onwukamike, S. Grelier, E. Grau, H. Cramail and M. A. R. Meier, Green Chem., 2018, 20, 214–224, 10.1039/C7GC02577G.
- J. Wolfs, R. Nickisch, L. Wanner and M. A. R. Meier, J. Am. Chem. Soc., 2021, 143, 18693–18702, DOI:10.1021/jacs.1c08783.
- H. Lönnberg, Q. Zhou, H. Brumer III, T. T. Teeri, E. Malmström and A. Hult, Biomacromolecules, 2006, 7, 2178–2185, DOI:10.1021/bm060178z.
- L. Song, Y. Yang, H. Xie and E. Liu, ChemSusChem, 2015, 8, 3217–3221, DOI:10.1002/cssc.201500378.
- S. Lu, J. Li, F. Liu, M. Chen, H. Na and J. Zhu, Polymer, 2021, 229, 124020, DOI:10.1016/j.polymer.2021.124020.
- F. Liu, S. Lu, W. Cao, J. Huang, Y. Sun, Y. Xu, M. Chen, H. Na and J. Zhu, Polymers, 2022, 14, 3449, DOI:10.3390/polym14173449.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176, DOI:10.1021/cr040002s.
- A. Carlmark, E. Larsson and E. Malmström, Eur. Polym. J., 2012, 48, 1646–1659, DOI:10.1016/j.eurpolymj.2012.06.013.
- S. Hua, F. Chen, Z.-Y. Liu, W. Yang and M.-B. Yang, RSC Adv., 2016, 6, 1973–1983, 10.1039/C5RA23182E.
- X. Zhu, L. Zhang, G. Zou, G. Chen, Y. Guo, S. Liang, L. Hu, M. North and H. Xie, Int. J. Biol. Macromol., 2021, 180, 792–803, DOI:10.1016/j.ijbiomac.2021.04.067.
- X. Li, F. Dong, L. Zhang, Q. Xu, X. Zhu, S. Liang, L. Hu and H. Xie, Chem. Eng. J., 2019, 372, 516–525, DOI:10.1016/j.cej.2019.04.123.
- J. Wang, Z. Xue, T. Yu, Z. Liu and T. Mu, Chem. – Asian J., 2017, 12, 1773–1779, DOI:10.1002/asia.201700335.
- H. Li, X. Zhu, T. He, C. Qu, Q. Tian, H. Xie, L. Hu, S. Liang, L. Zhang and J. Yuan, ACS Sustainable Chem. Eng., 2024, 12, 5390–5401, DOI:10.1021/acssuschemeng.3c03168.
- A. Zhu, J. Haibo, W. Yue, S. Qin, F. Zhang and Q. Xu, Chem. Eng. J., 2022, 446, 137032, DOI:10.1016/j.cej.2022.137032.
- Q. Cao, J. Dai, X. Bao, Z. Zhang, F. Liu, Y. Feng, H. Na and J. Zhu, Cellulose, 2021, 28, 10373–10384, DOI:10.1007/s10570-021-04184-z.
- W. Zhang, Y. Sun, Y. Xu, X. Yu, Y. Fang, F. Liu, H. Na and J. Zhu, Macromol. Chem. Phys., 2023, 224, 2300036, DOI:10.1002/macp.202300036.
- J. Liu, S. Lu, Q. Cao, J. Huang, F. Liu, H. Na, J. Zhu and Z. Jia, Carbohydr. Res., 2022, 520, 108630, DOI:10.1016/j.carres.2022.108630.
- Y. Sun, W. Zhang, S. Lu, W. Miao, M. Chen, F. Liu, H. Na and J. Zhu, Carbohydr. Polym., 2023, 301, 120346, DOI:10.1016/j.carbpol.2022.120346.
- X. Yu, Y. Xu, F. Liu, W. Zhang, Y. Sun, Y. Fang, L. Fang, X. He, H. Na and J. Zhu, J. Mater. Chem. A, 2023, 11, 23511–23522, 10.1039/d3ta05000a.
- J. Heveling, J. Chem. Educ., 2012, 89, 1530–1536, DOI:10.1021/ed200816g.
- B. L. Chivers, K. J. Morgan and B. J. White, Environ. Sci. Pollut. Res., 2021, 28, 754–762, DOI:10.1007/s11356-020-10538-0.
- S. Werkmeister, K. Junge and M. Beller, Org. Process Res. Dev., 2014, 18, 289–302, DOI:10.1021/op4003278.
- A. A. Strekalova, A. A. Shesterkina and L. M. Kustov, Catal. Sci. Technol., 2021, 11, 7229–7238, 10.1039/D1CY01603B.
- R. Qu, K. Junge and M. Beller, Chem. Rev., 2023, 123, 1103–1165, DOI:10.1021/acs.chemrev.2c00550.
- N. A. Stini, P. L. Gkizis and C. G. Kokotos, Green Chem., 2022, 24, 6435–6449, 10.1039/d2gc02332f.
| This journal is © The Royal Society of Chemistry 2024 |