
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Formation of lanthanide luminescent di-metallic helicates in solution using a bis-tridentate (1,2,3-triazol-4-yl)-picolinamide (tzpa) ligand†
Isabel N.
Hegarty
a,
Dawn E.
Barry
a,
Joseph P.
Byrne‡
 a,
Oxana
Kotova
ab and
Thorfinnur
Gunnlaugsson
*ab
a,
Oxana
Kotova
ab and
Thorfinnur
Gunnlaugsson
*ab
aSchool of Chemistry and Trinity Biomedical Sciences Institute (TBSI), Trinity College Dublin, The University of Dublin, Dublin 2, Ireland. E-mail: gunnlaut@tcd.ie
bAdvanced Materials and BioEngineering Research (AMBER) Centre, Trinity College Dublin, The University of Dublin, Dublin 2, Ireland
First published on 18th April 2023
Abstract
The chiral bis-tridentate (1,2,3-triazol-4-yl)-picolinamide (tzpa) ligand 1 was used in the formation of lanthanide di- and triple stranded di-metallic helicates in acetonitrile solution, where the changes in the ground and the Tb(III) excited state properties were used to monitor the formation of these supramolecular structures in situ under kinetic control.
The formation and the study of ordered structures and hierarchical supramolecular self-assembly has become an active area of research.1–3 The use of coordination chemistry to direct the synthesis of such structures is highly desirable.4–6 While many existing examples employ d-metal ions, the use of f-metals is less frequent in these systems.7,8 The f-metal ions have many unique physical properties and high coordination abilities opening new avenues for the construction of supramolecular systems.9,10 Recently, we11,12 and several other researchers13–16 have used lanthanide ions within these architectures. Supramolecular helicates are of particular interest as they are reminiscent of biomolecules such as the helices of polypeptides and DNA, representing the complex self-organisation found in nature. Using chiral ligands based on the 2,6-pyridine-dicarboxylic-amide (pda) structure, we have demonstrated the formation of chiral lanthanide luminescent complexes17 and triple-stranded di-metallic helicates, and their properties were then explored both in solution and the solid state.11a,18,19
With the view of extending the scope of using the f-metal ions in the coordination driven self-assemblies, we have also developed several examples of ligands based on the 2,6-bis(1,2,3-triazol-4-yl)pyridine (btp) motif,20 which included their applications in the formation of MOFs21 and lanthanide cross-linked hydrogels.22 Both pda and btp are versatile building blocks, which can form 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 (M:L) coordination complexes with the lanthanides completing their high coordination requirements (normally of nine), as each ligand is tridentate.23 With that in mind we recently combined these two coordination environments within a single ligand motif, the result being the formation of a new class of bis-tridentate (1,2,3-triazol-4-yl)-picolinamide (tzpa) ligands.24–26 Up until now, our effort has focused on exploring some of the coordination chemistry of the tzpa ligands with d-metal ions, which included the formation of chiral tetranuclear Cu(II) supramolecular assembly (M4L4) grids, using 1 (Fig. 1).26 However, no evidence for the formation of circular helicates was observed in the solid state using either Cu(II) or Fe(II) with tzpa ligands.25,26 We thus speculated that due to the coordination similarity with bis-tridentate pda ligands,18,19 the use of f-metal ions might yield such helical self-assemblies. Herein, we present the results from our investigation, and the first example of the use of tzpa ligands in the formation of lanthanide di-metallic helicates by using Tb(III) and 1 (and ligand 2, as a ‘half-helical’ model compound).
:3 (M:L) coordination complexes with the lanthanides completing their high coordination requirements (normally of nine), as each ligand is tridentate.23 With that in mind we recently combined these two coordination environments within a single ligand motif, the result being the formation of a new class of bis-tridentate (1,2,3-triazol-4-yl)-picolinamide (tzpa) ligands.24–26 Up until now, our effort has focused on exploring some of the coordination chemistry of the tzpa ligands with d-metal ions, which included the formation of chiral tetranuclear Cu(II) supramolecular assembly (M4L4) grids, using 1 (Fig. 1).26 However, no evidence for the formation of circular helicates was observed in the solid state using either Cu(II) or Fe(II) with tzpa ligands.25,26 We thus speculated that due to the coordination similarity with bis-tridentate pda ligands,18,19 the use of f-metal ions might yield such helical self-assemblies. Herein, we present the results from our investigation, and the first example of the use of tzpa ligands in the formation of lanthanide di-metallic helicates by using Tb(III) and 1 (and ligand 2, as a ‘half-helical’ model compound).
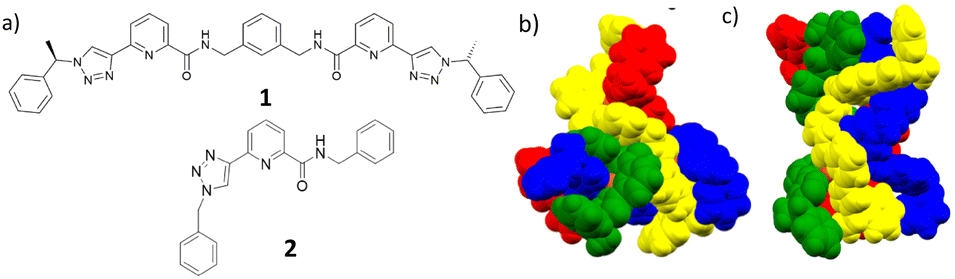 | ||
| Fig. 1 (a) The chiral bis-tridentate (1,2,3-triazol-4-yl)-picolinamide (tzpa) ligand 1 used in the current study, and the ‘half’ ligand 2, which was formed as a model ligand. The solid state structures of the tetranuclear [2 □× 2] square grids formed from 1 in our previous study using: (b) Cu(NO3)2·3 H2O and (c) [Cu(MeCN)4](PF6), respectively.26 | ||
The synthesis of 1 has previously been reported by us (see ESI† for characterisation).26 We foresaw that ligand 1 would be able to form a helicate (M2:L3) with lanthanide ions in polar solvents such as in CH3CN. Using chiral tzpa ligands (1 as the R,R enantiomer) would also aid in reducing the number of isomers in solution and possibly result in the formation of a single stereoisomer as in the case of the bis-tridentate pda ligands. Furthermore, in our work on btp ligands, we showed that Tb(III) complexes and self-assemblies of these triazole-containing systems are significantly more emissive than the corresponding Eu(III) assemblies.23c Hence, we selected Tb(III) for the current study.
Initially, compound 1 was reacted with Tb(CF3SO3)3 in a 2:3 M:L stoichiometry in CH3OH solution under microwave irradiation at 70 °C for 20 minutes. Initial visual observations indicated successful Tb(III) complex formation with 1, as both the solution and the isolated solid gave rise to Tb(III)'s characteristic green emission under UV irradiation (λex = 254 nm). However, attempts to characterise the paramagnetic complex by 1H NMR spectroscopy were unsuccessful. Furthermore, attempts to crystallise the product did not yield crystals of X-ray diffraction quality. Consequently, we decided to investigate the formation of the helical species in solution under kinetic control by monitoring the changes in the absorption and fluorescence of ligand 1 as well as the emerging delayed Tb(III) centred emission. These spectroscopic titrations were carried out at a ligand concentration of 1 × 10−5 M in CH3CN solution. At this concentration, the self-assembly between 1 and Tb(III) was fast and no substantial equilibration period was required.
The changes in the UV-visible absorption spectrum of 1 in CH3CN were monitored and are shown in Fig. 2 (left). The free ligand displayed two main bands, one centred at λ = 251 nm and the second band at λ = 291 nm (ε230 = 35400 cm−1·M−1). As can be seen from Fig. 2 (left), significant changes were observed upon addition of Tb(III), with the high energy band experiencing a hypochromic shift and broadening up to ca. 0.7 equivalents of Tb(III), after which the changes to this band reached a plateau at ca. 1 equivalent of Tb(III). Similar changes were observed for the λ = 291 nm band, which was also redshifted by 10 nm. These changes were accompanied by the appearance of two isosbestic points at 236 and 288 nm. Further additions of Tb(III) (1 → 3 equiv.) only resulted in minor changes, indicating the formation of stable species in solution.
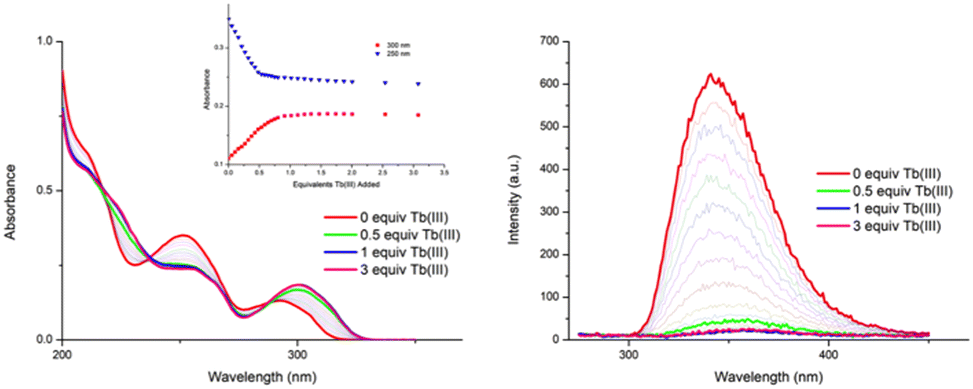 | ||
| Fig. 2 The overall changes in the (left) UV-visible absorption spectra and (right) fluorescence emission spectra (λ□□ = 260 nm) upon titrating 1 (1 × 10−5 M) against Tb(CF3SO3)3 (0 → 3 equiv.) in CH3CN at 22 °C. Inset: The binding isotherms of absorbance at λ = 300 and 250 nm, respectively. | ||
Concomitantly to monitoring the ground state properties, the changes in the ligand-centred fluorescence were recorded. Upon excitation of 1 at λ = 260 nm, ligand centred emission was observed at λ = 343 nm. As can be seen in Fig. 2(right), addition of Tb(III) resulted in almost full quenching of ligand-centred emission, with 93% reduction in the luminescence being observed at the addition of 0.5 equivalents of Tb(III). Such efficient quenching can be attributed to an energy transfer sensitisation process from the singlet excited state to the Tb(III) metal centre via the ligand triplet state.6,8a,18,19 Further additions of Tb(III) resulted in some further quenching, the overall process indicating a rapid population of the Tb(III) 5D4 excited state. This was indeed confirmed by the changes in the delayed Tb(III)-centred emission spectra upon excitation at λ = 260 nm; the overall changes and the corresponding binding isotherms can be seen in Fig. 3a and b, respectively. Here, the Tb(III)-centred transitions appearing at λ = 491, 545, 583, 622, 649, 668 and 679 nm correspond to deactivation of the 5D4 → 7FJ states (where J = 6 − 0) and were all clearly visible. As can be seen from Fig. 3b, a gradual enhancement was observed in all the transitions upon addition of Tb(III). In the case of ΔJ = 6 and 5, these were most significant up to the addition of ca. 0.6 equiv. of Tb(III) but thereafter, a gradual quenching was observed up to ca. 1 equiv. of Tb(III), with minor changes occurring between 0.5 → 3 equiv. of Tb(III). These changes would suggest the initial formation of emissive species in solution with a stoichiometry of 2:3 or 2:2/1:1 (i.e. Tb213, or a mixed Tb212/Tb111 species or both in solution). The gradual quenching of the Tb(III)-centred luminescence beyond the addition of one equivalent suggests the formation of a less emissive species, possibly 1:1.
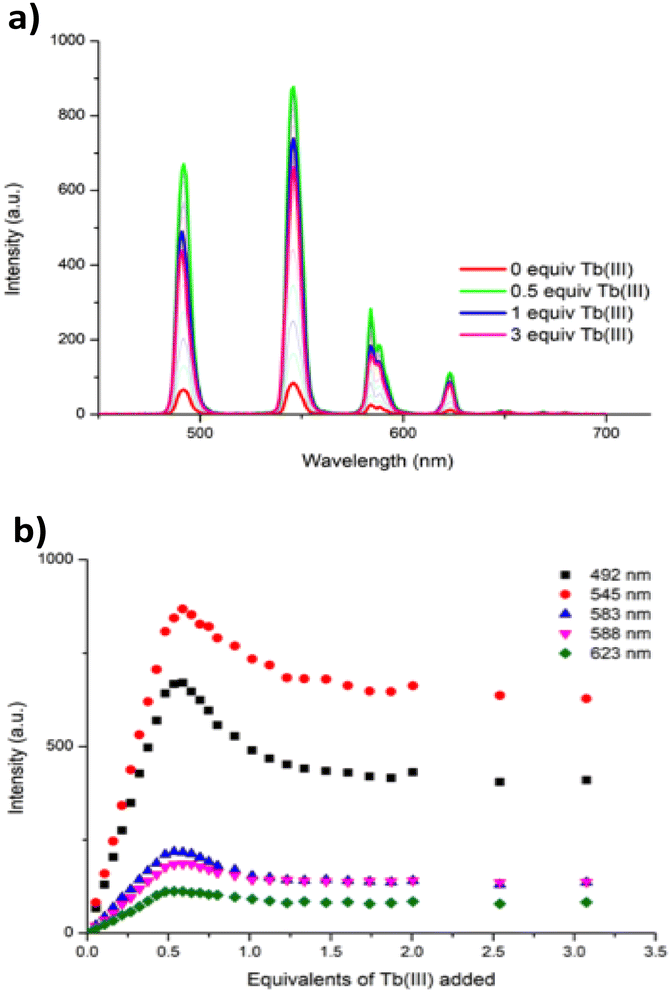 | ||
| Fig. 3 (a) The overall changes to the Tb(III)-centred phosphorescence spectra upon titrating 1 (1 × 10−5 M) against Tb(CF3SO3)3 (0 → 3 equiv.) in CH3CN at 22 °C. (b) The corresponding experimental binding isotherms of delayed Tb(III) emission at λ = 492, 545, 583, 588 and 623 nm. | ||
With the view of elucidating further the self-assembly processes in solution, the spectroscopic data obtained in the ground and the Tb(III) excited states were analysed using non-linear regression analysis, using the ReactLab EQUILIBRIA software.27 This allowed us to calculate stability constants for the proposed possible stoichiometries and analyse their speciation in solution at any given equivalent addition of Tb(III). By fitting the changes in these two windows, a better understanding of the various equilibrium processes could be obtained. Hence, we first analysed the changes in the ground state where a good fit to the experimental data was achieved, and the factor analysis suggested the formation of two main species in solution (see ESI†).
From this fitting, the binding constants (expressed as logβTb:L) were determined for each species as logβ□:□ = 24.1 ± 0.1 and logβ2:2 = 19.5 ± 0.1. These values are in excellent agreement with those (same stoichiometries) determined for the formation of triple and double stranded helicates reported by us using bis-tridentate pda ligands.18,19 The subsequently obtained speciation distribution diagram (See ESI†) showed that the M2L2 species appears most dominant in solution, reaching 100% abundance at 1.2 equivalents of Tb(III) (e.g. no free ligand exist). Surprisingly, the presence of M2L3 species reaches a maximum abundance of only 13% at approximately 0.5 equivalents of Tb(III) (as determined from the ground state) after which its concentration decreases to the benefit of the formation of the M2L2 species. We were also not able to distinguish the presence of the initial formation of the M1L1 species, or the same at high metal ion concentration, where such a stoichiometry can also exist. This speciation is consistent with the observation that the most emissive stoichiometry in solution was at ca. 0.5–0.6 equivalents of Tb(III). While the M3L2 species is likely the most emissive species due to exclusion of solvent from the inner coordination sphere of the Ln(III) ion, it does not appear to be the most stable kinetic product of the self-assembly formation in solution as determined from the ground state changes. Consequently, the changes in the delayed Tb(III)-centred emission were analysed in the same manner. As no Tb(III) emission exists prior to the self-assembly formation, the emission observed is direly associated with the step-wise formation of the Tb(III) self-assemblies in solution. The global changes to the Tb(III)-centred emission were analysed and confirmed the presence of two main species in solution, M2L3 and M2L2, Fig. 4. The binding constants were calculated for each species and M2L3 assembly was formed with logβ□:□ = 25.6 ± 0.1 while the second species (M2L2) had a calculated binding constant of logβ2:2 = 20.0 ± 0.1. Again, these calculated stability constants are comparable to those obtained from the changes in the absorption data (and that seen for the bis-tridentate pda ligands). Again the speciation distribution was calculated from the binding constant analysis and from 0 → 0.5 equivalents of Tb(III) the M2L3 species is most dominant in solution reaching ca. 46% abundance. However, upon further additions of Tb(III), the abundance of the M2L3 species decreases due to dissociation to the M2L2 species in the presence of excess metal. In fact, the formation of this stoichiometry only accounts for ca. 30% of the total speciation at ∼0.7 equivalents (e.g. when the M2L3 should be exclusively formed). This trend is in an agreement with the speciation estimated from the UV-visible absorption data (see ESI†). From these combined results it can be concluded that M2L3 is rapidly formed at low Tb(III) concentration. However, a mixture of both the M2L3 and M2L2 species exists in solution under kinetic control, with the latter being the dominant stoichiometry above one equivalent of Tb(III). While the binding constants determined herein are similar to those seen for the formation of related helicates of bis-tridentate pda ligands, then in comparison, the latter lead to almost exclusive formation of the M2L3 species at the same 0.68 Ln(III) equivalents.18,19 Hence, despite tzpa design being in part based on the pda ligand motif (where one of the hard oxygen donor atoms is replaced by a soft nitrogen donor atom), the coordination and the self-assembly properties of the bis-tridentate tzpa ligand in solution are strikingly different to that seen for analogues pda ligands. The latter form the M2L3 stoichiometry in high (>90%) yield under identical experimental conditions.
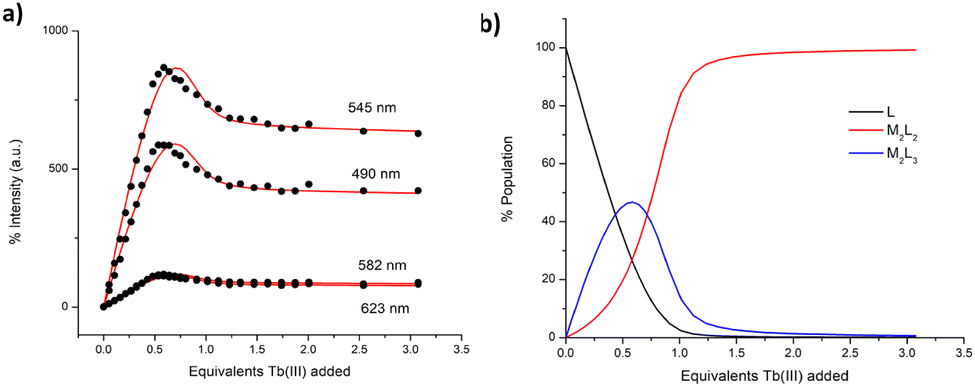 | ||
| Fig. 4 (a) The fit of the experimental binding isotherms using non-linear regression analysis software ReactLab.27 (b) The corresponding speciation distribution diagram obtained from the fit of the delayed luminescence titration data of ligand 1 against Tb(CF3SO3)3 in CH3CN. | ||
As alluded to above, we have also previously investigated the binding properties of (mono) pda ligands with lanthanides in comparison to that seen for the bis-tridentate pda analogues. These studies showed that in principle, the same coordination environment around a given lanthanide ion and binding constant was observed for both types of ligands.17a,b We decided to carry out similar comparative studies herein, and hence, we synthesised the achiral tzpa ligand 2, Fig. 1, which structurally mimics half of ligand 1.
The synthesis of 2, which was fully characterised, was achieved in three steps (See ESI†). As for 1 above, the Tb(III) titrations of 2 (c = 1 × 10−5 M) were carried out in CH3CN. The UV-visible absorption spectra displayed two main bands, centred at 250 nm and 290 nm (as in the case of 1). Both were affected by binding to Tb(III); the overall changes mirroring that seen for 1, occurring between 0 → 0.5 equivalents of Tb(III), suggesting the presence of the ML2 stoichiometry as the main species in solution (see ESI†). Concomitantly, the ligand fluorescence was quenched (∼90% at 0.5 equivalents), and the Tb(III) emission was ‘switched on’ (See ESI†). Global analysis of these results showed good agreement between the changes in the ground and the Tb(III) centred emission, with the ML2 stoichiometry being most prominent with logβ1:2 = 13.4 ± 0.1 (and formed in over 90% yield at 0.5 equivalents) and logβ1:1 = 6.5 ± 0.1 being determined from the ground state data (only forming beyond ca. 0.4 equiv., see ESI†), while fitting the changes in the Tb(III)-centred emission gave logβ□:□ = 14.5 ± 0.1 (∼85% at 0.5 equivalents) and logβ1:1 = 7.8 ± 0.1 (See ESI†). Hence, the results for 2 support the formation of the ML2 species over the ML3 species under kinetic control, and, as such, agree with what was seen for 1, where the M2L2 is the most abundant stoichiometry in solution. In the case of the self-assembly between 1 and Tb(III), this was further confirmed by HRMS analysis that showed two main species, the M2L2 and the ML2 for the Tb(III) self-assembly with 1, with isotopic distribution patterns matching the calculated ones (see ESI†).
In summary, we have demonstrated for the first time the use of a bis-tridentate tzpa ligand for the formation of luminescent di-metallic lanthanide helicates in solution. Here, the lanthanide emission properties were used to monitor and quantify the self-assembly processes. The results show that while the ‘expected’ M2L3 stoichiometry is initially formed, upon further Tb(III) additions, the M2L2 stoichiometry is the dominant one in solution. We are currently developing other analogues of 1 with the view to further examining the application of tzpa ligands in metal directed supramolecular self-assembly and material formations.
We thank Science Foundation Ireland (SFI PI Awards 13/IA/1865) and the SFI Amber Centre (SFI 12/RC/2278_P2) for financial support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) Z. Ashbridge, E. Kreidt, L. Pirvu, F. Schaufelberger, J. H. Stenlid, F. Abild-Pedersen and D. A. Leigh, Science, 2022, 375, 1035 CrossRef CAS PubMed; (b) J. Dong, Y. Liu and Y. Cui, Acc. Chem. Res., 2021, 54, 194 CrossRef CAS PubMed; (c) Z. Ashbridge, S. D. P. Fielden, D. A. Leigh, L. Pirvu, F. Schaufelberger and L. Zhang, Chem. Soc. Rev., 2022, 51, 7779 RSC.
- (a) J. Uchida, M. Yoshio and T. Kato, Chem. Sci., 2021, 12, 6091 RSC; (b) T. Gorai, J. I. Lovitt, D. Umadevi, G. McManus and T. Gunnlaugsson, Chem. Sci., 2022, 13, 7805 RSC.
- (a) A. B. Aletti, S. Blasco, S. J. Aramballi, P. E. Kruger and T. Gunnlaugsson, Chemistry, 2019, 5, 2617 CrossRef CAS; (b) A. J. Savyasachi, O. Kotova, S. Shanmugaraju, S. J. Bradberry, G. M. Ó’Máille and T. Gunnlaugsson, Chemistry, 2017, 3, 764 CrossRef CAS.
- (a) Q. V. C. van Hilst, N. R. Largesse, D. Preston and J. D. Crowley, Dalton Trans., 2018, 47, 997 RSC; (b) M. Cirulli, A. Kaur, J. E. M. Lewis, Z. Zhang, J. A. Kitchen, S. M. Goldup and M. M. Roessler, J. Am. Chem. Soc., 2019, 141, 879 CrossRef CAS PubMed; (c) D. Preston and P. E. Kruger, Chem. – Eur. J., 2019, 25, 178 CrossRef PubMed.
- D. A. Roberts, B. S. Pilgrim and J. R. Nitschke, Chem. Soc. Rev., 2018, 47, 626 RSC.
- (a) T. Gorai, W. Schmitt and T. Gunnlaugsson, Dalton Trans., 2021, 50, 770 RSC; (b) J. C. G. Bünzli, Acc. Chem. Res., 2006, 39, 53 CrossRef PubMed.
- X.-Z. Li, C.-B. Tian and Q.-F. Sun, Chem. Rev., 2022, 122, 6374 CrossRef CAS PubMed.
- (a) D. E. Barry, D. F. Caffrey and T. Gunnlaugsson, Chem. Soc. Rev., 2016, 45, 3244 RSC; (b) K.-H. Yim, C.-T. Yeung, M. R. Probert, W. T. K. Chan, L. E. Mackenzie, R. Pal, W.-T. Wong and G.-L. Law, Chem. Commun., 2021, 4, 116 CrossRef CAS PubMed.
- (a) J. A. Kitchen, Coord. Chem. Rev., 2017, 340, 232 CrossRef CAS; (b) S. J. Bradberry, A. J. Savyasachi, M. Martínez-Calvo and T. Gunnlaugsson, Coord. Chem. Rev., 2014, 273–274, 226 CrossRef CAS.
- (a) Y. B. Tan, M. Yamada, S. Katao, Y. Nishikawa, F. Asanoma, J. I. Yuasa and T. Kawai, Inorg. Chem., 2020, 59, 12867 CrossRef CAS PubMed; (b) L.-L. Yan, C.-H. Tan, G.-L. Zhang, L.-P. Zhou, J.-C. Bünzli and Q.-F. Sun, J. Am. Chem. Soc., 2015, 137, 8550 CrossRef CAS PubMed.
- (a) D. E. Barry, J. A. Kitchen, K. Pandurangan, A. J. Savyasachi, R. D. Peacock and T. Gunnlaugsson, Inorg. Chem., 2020, 59, 2646 CrossRef CAS PubMed; (b) S. J. Bradberry, A. J. Savyasachi, R. D. Peacock and T. Gunnlaugsson, Faraday Discuss., 2015, 185, 413 RSC.
- O. Kotova, C. O’Reilly, S. T. Barwich, L. E. Mackenzie, A. D. Lynes, A. J. Savyasachi, M. Ruether, R. Pal, M. E. Möbius and T. Gunnlaugsson, Chem, 2022, 8, 1395 CAS.
- (a) G. Gil-Ramírez, S. Hoekman, M. O. Kitching, D. A. Leigh, I. J. Vitorica-Yrezabal and G. Zhang, J. Am. Chem. Soc., 2016, 138, 13159 CrossRef PubMed; (b) K.-H. Yim, C.-T. Yeung, M. Y.-M. Wong, M. R. Probert and G.-L. Law, Chem. – Eur. J., 2022, e202201655 CAS; (c) C.-T. Yeung, K.-H. Yim, H.-Y. Wong, R. Pal, W.-S. Lo, S.-C. Yan, M. Y.-M. Wong, D. Yufit, D. E. Smiles, L. J. McCormick, S. J. Teat, D. K. Shuh, W.-T. Wong and G.-L. Law, Nat. Commun., 2017, 8, 1128 CrossRef PubMed; (d) L.-X. Cai, L.-L. Yan, S.-C. Li, L.-P. Zhou and Q.-F. Sun, Dalton Trans., 2018, 47, 14204 RSC; (e) X.-Z. Li, L.-P. Zhou, L.-L. Yan, D.-Q. Yuan, C.-S. Lin and Q.-F. Sun, J. Am. Chem. Soc., 2017, 139, 8237 CrossRef CAS PubMed.
- (a) Z. Wang, L. He, B. Liu, L.-P. Zhou, L.-X. Cai, S.-J. Hu, X.-Z. Li, Z. Li, T. Chen, X. Li and Q.-F. Sun, J. Am. Chem. Soc., 2020, 142, 16409 CrossRef CAS PubMed; (b) S.-Y. Wu, X.-Q. Guo, L.-P. Zhou and Q.-F. Sun, Inorg. Chem., 2019, 58, 7091 CrossRef CAS PubMed; (c) X.-Q. Guo, L.-P. Zhou, S.-J. Hu, L.-X. Cai, P.-M. Cheng and Q.-F. Sun, J. Am. Chem. Soc., 2021, 143, 6202 CrossRef CAS PubMed.
- (a) L.-L. Yan, C.-H. Tan, G.-L. Zhang, L.-P. Zhou, J.-C. Bünzli and Q.-F. Sun, J. Am. Chem. Soc., 2015, 137, 8550 CrossRef CAS PubMed; (b) Z. Ashbridge, O. M. Knapp, E. Kreidt, D. A. Leigh, L. Pirvu and F. Schaufelberger, J. Am. Chem. Soc., 2022, 144, 17232 CrossRef CAS PubMed.
- (a) Y. Wang, Y. Zhou, Z. Yao, W. Huang, T. Gao, P. Yan and H. Li, Dalton Trans., 2022, 51, 10973 RSC; (b) Y. B. Tan, M. Yamada, S. Katao, Y. Nishikawa, F. Asanoma, J. I. Yuasa and T. Kawai, Inorg. Chem., 2020, 59, 12867 CrossRef CAS PubMed; (c) C.-T. Yeung, K.-H. Yim, H.-Y. Wong, R. Pal, W.-S. Lo, S.-C. Yan, M. Yee-Man Wong, D. Yufit, D. E. Smiles, L. J. McCormick, S. J. Teat, D. K. Shuh, W.-T. Wong and G.-L. Law, Nat. Commun., 2017, 8, 1128 CrossRef PubMed; (d) M. Rancan, J. Tessarolo, A. Carlotto, S. Carlotto, M. Rando, L. Barchi, E. Bolognesi, R. Seraglia, G. Bottaro, M. Casarin, G. H. Clever and L. Armelao, Cell. Rep. Phys. Sci., 2022, 3, 100692 CrossRef CAS.
- (a) O. Kotova, B. Twamley, J. O’Brien, R. D. Peacock, S. Blasco, J. A. Kitchen, M. Martínez-Calvo and T. Gunnlaugsson, Chem. Sci., 2015, 6, 457 RSC; (b) C. Lincheneau, C. Destribats, D. E. Barry, J. A. Kitchen, R. D. Peacock and T. Gunnlaugsson, Dalton Trans., 2011, 40, 12056 RSC; (c) S. J. Bradberry, G. Dee, O. Kotova, C. P. McCoy and T. Gunnlaugsson, Chem. Commun., 2019, 55, 1754 RSC.
- (a) F. Stomeo, C. Lincheneau, J. P. Leonard, J. E. O’Brien, R. D. Peacock, C. P. McCoy and T. Gunnlaugsson, J. Am. Chem. Soc., 2009, 131, 9636 CrossRef CAS PubMed; (b) C. Lincheneau, R. D. Peacock and T. Gunnlaugsson, Chem. – Asian J., 2010, 5, 500 CrossRef CAS PubMed.
- (a) D. E. Barry, J. A. Kitchen, L. Mercs, R. D. Peacock, M. Albrecht and T. Gunnlaugsson, Dalton Trans., 2019, 48, 11317 RSC; (b) O. Kotova, S. Comby, K. Pandurangan, F. Stomeo, J. E. O’Brien, M. Feeney, R. D. Peacock, C. P. McCoy and T. Gunnlaugsson, Dalton Trans., 2018, 47, 12308 RSC.
- A. F. Henwood, I. N. Hegarty, E. P. McCarney, J. I. Lovitt, S. Donohoe and T. Gunnlaugsson, Coord. Chem. Rev., 2021, 449, 214206 CrossRef CAS.
- E. P. McCarney, C. S. Hawes, J. A. Kitchen, K. Byrne, W. Schmitt and T. Gunnlaugsson, Inorg. Chem., 2018, 57, 3920 CrossRef CAS PubMed.
- (a) I. N. Hegarty, S. J. Bradberry, J. I. Lovitt, J. M. Delente, N. Willis-Fox, R. Daly and T. Gunnlaugsson, Mater. Chem. Front., 2023, 7, 906 RSC; (b) I. N. Hegarty, A. F. Henwood, S. J. Bradberry and T. Gunnlaugsson, Org. Biomol. Chem., 2023, 21, 1549 RSC.
- (a) O. Kotova, J. A. Kitchen, C. Lincheneau, R. D. Peacock and T. Gunnlaugsson, Chem. – Eur. J., 2013, 19, 16181 CrossRef CAS PubMed; (b) J. P. Byrne, M. Martínez-Calvo, R. D. Peacock and T. Gunnlaugsson, Chem. – Eur. J., 2016, 22, 486 CrossRef CAS PubMed; (c) J. P. Byrne, J. A. Kitchen, J. E. O’Brien, R. D. Peacock and T. Gunnlaugsson, Inorg. Chem., 2015, 54, 1426 CrossRef CAS PubMed.
- D. E. Barry, C. S. Hawes, J. P. Byrne, B. la Cour Poulsen, M. Ruether, J. E. O’Brien and T. Gunnlaugsson, Dalton Trans., 2017, 46, 6464 RSC.
- I. N. Hegarty, H. L. Dalton, A. D. Lynes, B. Haffner, M. E. Möbius, C. S. Hawes and T. Gunnlaugsson, Dalton Trans., 2020, 49, 7364 RSC.
- I. N. Hegarty, H. L. Dalton, A. F. Henwood, C. S. Hawes and T. Gunnlaugsson, Chem. Commun., 2019, 55, 9523 RSC.
- (a) P. Gans, Data Fitting in the Chemical Sciences by the Method of Least Squares, Wiley, 1992 Search PubMed; (b) E. Joseph Billo, Excel for Chemists: A Comprehensive Guide, Wiley-VCH, 2nd edn, 2001 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and supporting figures. See DOI: https://doi.org/10.1039/d3cc01126g |
| ‡ Current address: School of Chemistry, University College Dublin, Dublin 4, Ireland. |
| This journal is © The Royal Society of Chemistry 2023 |