DOI:
10.1039/D2TC02238A
(Paper)
J. Mater. Chem. C, 2022,
10, 14532-14541
Coordination-driven opto-electroactive molecular thin films in electronic circuits†
Received
29th May 2022
, Accepted 1st September 2022
First published on 2nd September 2022
Abstract
Surface engineering using controlled molecular structures, compositions, thicknesses, packing, and orientations is highly desirable for understanding nanoscale interfacial phenomena. The preparation of coordination-compound-based oligomer films with well-defined chemical structures, compositions, orientations, and thicknesses is crucial for ‘on-surface’ optoelectronic and electrical applications. We take advantage of the simple-yet-classic technique of layer-by-layer (LBL) construction to fabricate both homostructured Fe(II)/Co(II)-bis-terpyridyl (bis-tpy) oligomers and heterostructures. We exploit the electrochemical reduction method to prepare covalent template layers on which different transition metal ions and bis-terpyridine ligands are systemically assembled. Molecular thin films fabricated on transparent conducting oxides such as ITO substrates are utilized for optical, electrochemical, electrical, and electrical impedance spectroscopy studies to explore the possibilities of molecular-prototype devices. A gel electrolyte was placed between ITO/tpy-[Fe(bis-tpy)]5 and ITO/tpy-[Co(bis-tpy)]5 to mimic a molecular electronic device configuration to measure the current–voltage (I–V) response in a facile, cheap, fast, scalable, and clean-room-free approach. Electrical impedance spectroscopy was used to experimentally deduce charge-transfer resistance, contact resistance, and capacitance values, followed by circuit modeling. The circuit model was further validated via building a real electronic circuit using individual electrical components. Near-vertically-aligned molecular thin films could be suitable for various applications in optoelectronics and electrochromic and molecular electronics.
Introduction
Herbert Kroemer in his Nobel lecture in 2000 made a celebrated statement, ‘Interface is the devices’, appreciating the remarkable development of microelectronics for optoelectronic applications.1 Microelectronics has engendered many other sub-fields, and surface chemistry is one of them. The surface chemistry of nanoscale molecular thin films has been an intriguing research domain since its inception. This realm provides an enormous opportunity not only to fabricate atomic to molecular thick layers on diverse substrates but is also fruitful for applications in optics, semiconductors, spintronics, sensors, catalysis, energy storage, anticorrosion, and nanotechnology, including daily life, to mention but a few.2–11 So far, many effective and advanced platforms have been successfully introduced to engineer electrode surfaces using diverse molecules, so that it is generally believed that functional (organic) molecules would be the powerhouse of future optoelectronic devices.12–16 It has been well established that molecules are able to mimic electronic functions, and are thus effective in the fabrication of organic to molecular electronic and spintronic devices for understanding stimuli-responsive charge and spin transport phenomena in nanoscale molecular layers.17–21 Molecules offer many advantages over inorganic materials due to tunable physicochemical properties, ease of synthesis, solution processability, scalability, and compatibility with CMSO-technology.22–26 However, suitable surface modification methods are desirable to create chemically and thermally stable interfaces between electrode surfaces and molecules. In this regard, self-assembled monolayers (SAMs) formed between noble metals (Au, Ag) and thiolated or dithiolated organic molecules or phosphate and siloxane based on-surface molecular assemblies have served as a popular platform in understanding interfaces and on-surface phenomena, such switching, spin crossover, supramolecular assembly, sensing, catalysis, and molecular electronic and spintronic devices.27–30 Despite the many advantages that SAMs offer, there are some disadvantages as well. For instance, SAMs are not so great in terms of interfacial stability, as thiols are quite reactive; the process is limited to noble metals; it requires a long time to assemble; Au–S is not a pure covalent bond; and the thickness of the molecular layers cannot be varied systematically.31–35 Siloxane and phosphate linked on-surface molecular assemblies also require a longer reaction time (56–72 h) to provide a good surface coverage. In recent years, an electrochemical (e-chem) reduction method to make covalent bonds between electrode and molecules has captured attention and revealed excellent application potential in molecular electronics, supercapacitors, and on-surface molecular assembly for molecular-scale applications.36–41 The e-chem reduction method produces excellent durability, longer stability, and wider substrate scope.41–44 This e-chem reduction method is an extremely powerful technique to yield mixed molecular layers on different electrode surfaces whose electronic, optical and redox properties can be tuned to a greater magnitude. In this regard, the Gooding and Ciampi group has made significant contributions to growing molecular layers via the e-chem reduction of aryl diazonium salts that have been used in electrochemical studies, as single molecule junctions for current–voltage (I–V) traces and biomolecular sensing applications.45–49 In addition, the group has also succeeded in preparing organophosphonic acid monolayers on ITO via covalent attachment chemistry.50
In this work, we use an electrochemical grafting method performed on ITO as a working electrode that forms covalent bonds with a carbon of the terpyridine (tpy) moiety that has three binding centers. Covalent interfaces between ITO and tpy serve as template layers on which transition metal ions (M = Fe2+ or Co2+) and bis-terpyridine (bis-tpy, six binding centers) are assembled using a layer-by-layer (LbL) approach. Coordination-driven self-assembly facilitates a nexus between metal ions and the tpy ligand, forming covalent bonds (tpy-M-tpy), where metal ions experience octahedral geometry.51–54 Thanks to the layer-by-layer (LbL) method, we can extend the assembly to at least the 10th deposition unit (each unit = metal ions + bis-tpy ligand) to prepare either ITO/tpy-[Fe(bis-tpy)]10 or ITO/tpy-[Co(bis-tpy]10 or heterostructures ITO/tpy-[Fe(bis-tpy)]5-[Co(bis-tpy)5]. LbL deposited ‘on-surface’ coordination complexes were characterized by X-ray photoelectron spectra (XPS), UV-vis spectroscopy, and electrochemical studies. In addition, two modified ITO were sandwiched in a molecular electronic device configuration, ITO/tpy-[Fe(bis-tpy)]5/gel electrolyte/[(bis-tpy)5Co]5-tpy/ITO, to measure the current–voltage (I–V) characteristics. To deduce the individual electrical components of solid-state molecular junctions, electrical impedance spectroscopy was performed and experimentally observed data were fitted to an electrical circuit for further validation.
Results and discussion
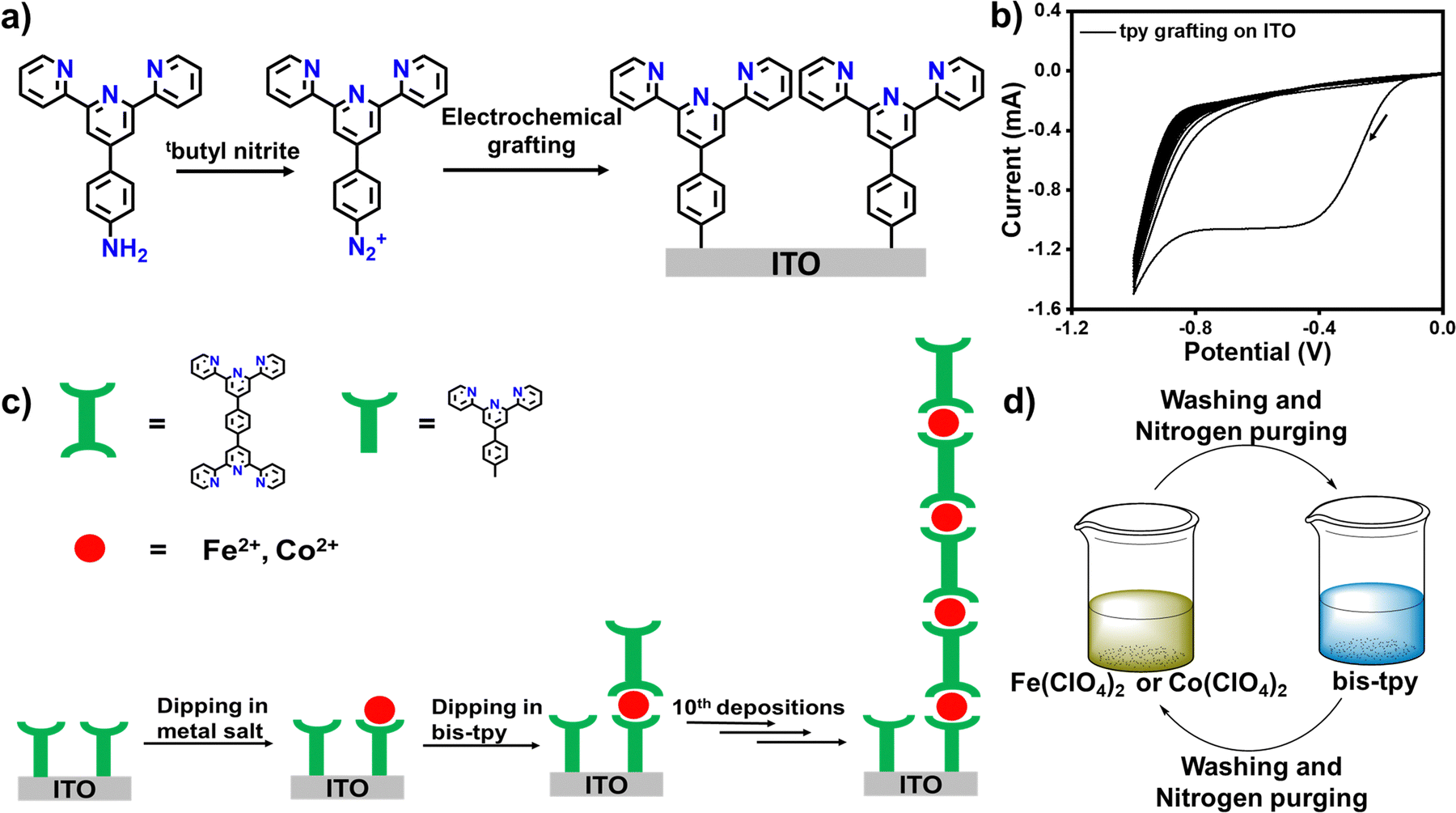
Metal–polypyridyl complexes have been well investigated due to their rich coordination chemistry, excellent optical properties, and reversible electrochemical charge transfer properties.55–60 In this work, we chose the bis-terpyridyl ligand (bis-tpy) that shows the structural advantages of having a ‘back-to-back’ metal ion receptor and unidirectionality, forming a chelated ring, providing extra stability to the in situ formed on-surface coordination complexes. 4′-Aminophenyl terpyridine (4′-NH2-tpy, Fig. 1a) was converted to the corresponding diazonium salts (the synthesis and characterization are discussed in ESI,† Fig. S1–S4) for electrochemical grafting onto transparent conductive oxide, ITO, as a working electrode in a conventional three-electrode electrochemical setup (see experimental section). A cathodic peak observed at −0.38 V (vs. Ag/AgCl) in the first cycle manifests the reduction of aryl diazonium salts and the formation of reactive radicals which immediately graft onto the ITO substrate (Fig. 1b). The CV scans were continued up to the 10th repeated CV scan, but no significant cathodic signals were observed after the 1st cathodic wave, indicating that the template layers grafted onto the ITO electrode surface inhibit further growth of molecular layers.41 Before dipping into the solution of metal salt precursor or ligand, the template layer was thoroughly washed with acetonitrile to remove any physiosorbed materials. For instance, the modified ITO was dipped into a solution containing Fe(ClO4)2 followed by dipping into a solution of bis-tpy ligand (Fig. 1c and d). Before proceeding with further dipping, the ITO was cleaned and dried under nitrogen using a Schlenk line. Similarly, Co(ClO4)2 was used instead of Fe(ClO4)2 and such metal ion/ligand alternative depositions were performed up to the 10th deposition step to deduce the relationship between absorbance and faradaic current as a function of deposition steps. In another deposition, a mixture of both metal ions (Fe2+ and Co2+) was used for 5 deposition cycles each. The present strategy not only creates a covalent bond at electrode/tpy but also helps to avoid the formation of ex situ coordination compounds for fabricating on-surface assemblies that have the desired chemical compositions and to perform optical, electrical, and frequency response measurements. Although the electrochemical grafted method offers many advantages, the possibility of the formation of non-uniform molecular layers cannot be ruled out. As we consider here, the diazonium salt of the terpyridine ligand does not contain any bulky group, so its thickness cannot be controlled to the monolayer scale, but it produces multilayers in irregularly branching fashions.
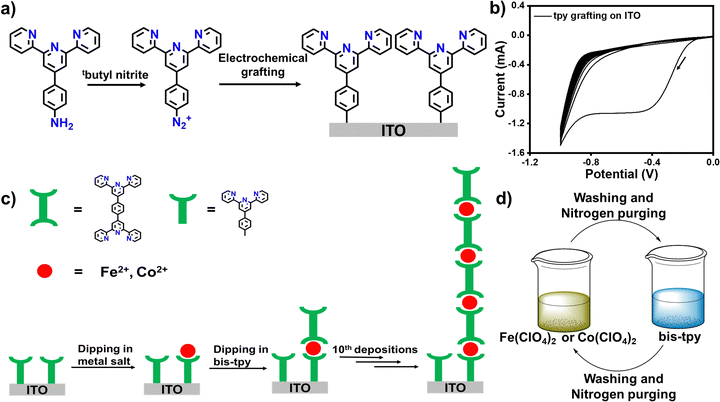 |
| | Fig. 1 (a) A schematic description of the in situ preparation of diazonium salts of 4′-NH2-phtpy followed by electrochemical grafting onto an ITO substrate (the formation of the template layer). (b) Cyclic voltammograms to monitor the electrochemical grafting of the tpy layers. ITO, Ag/AgCl, and a Pt wire are used as the working, reference, and counter electrodes, respectively. (c) The stepwise coordination process on the ITO surface with free template layers without coordinating metal ions. (d) A schematic diagram of the 10 deposition steps performed alternatively with either iron perchlorate or cobalt perchlorate and bis-tpy ligand via a layer-by-layer approach. | |
X-Ray photoelectron spectroscopy (XPS) analysis
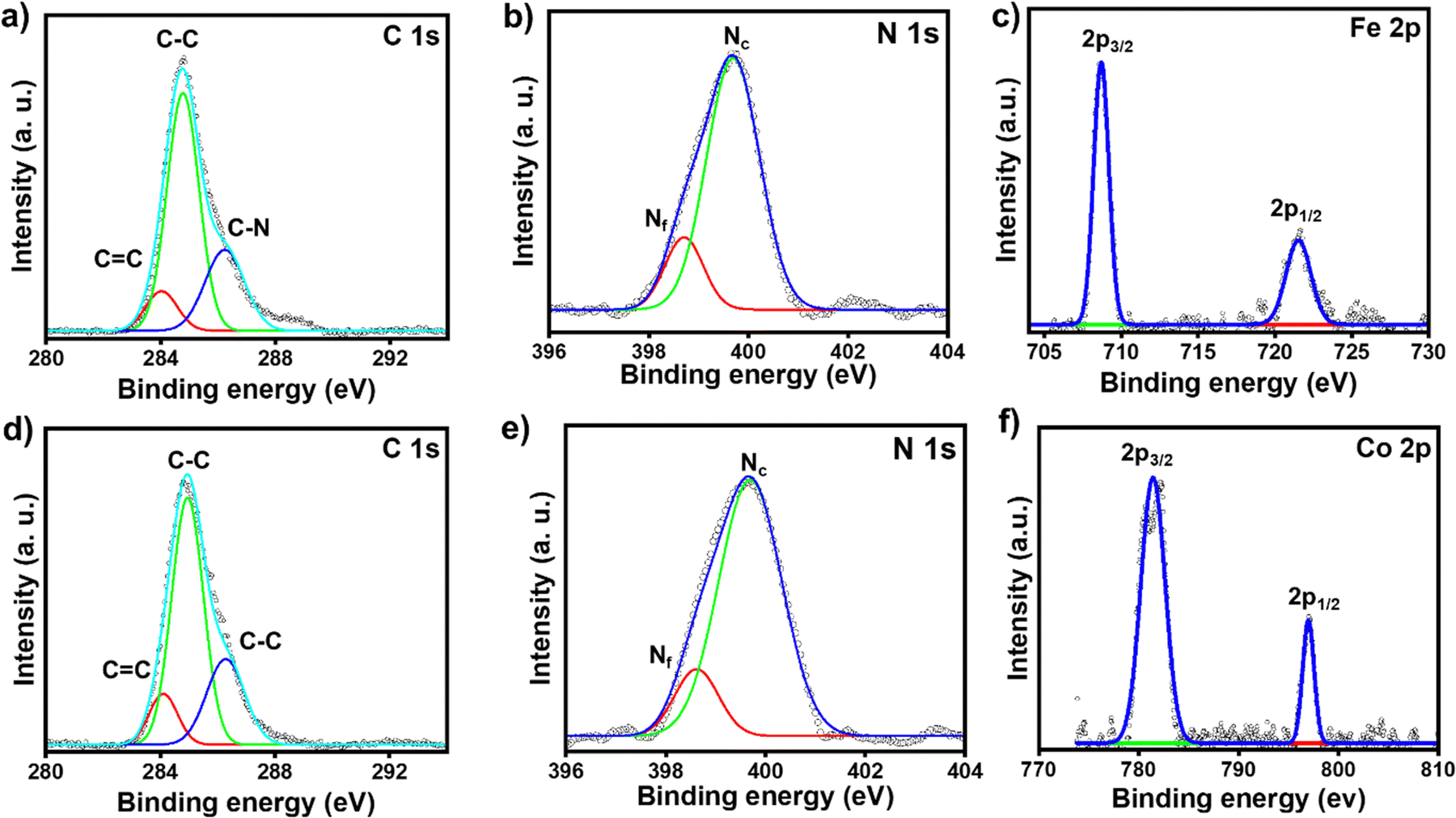
The chemical compositions of the respective molecular thin films deposited on the ITO electrode surfaces were characterized by XPS spectroscopy. The full survey scans of ITO/tpy-[Fe(bis-tpy)]5 and ITO/tpy-[Co(bis-tpy)]5 are shown in Fig. S5a and b (ESI†). The XPS data confirm the presence of elements such as C 1s, N 1s, Fe 2p and Co 2p in the respective surface-confined terpyridyl-based molecular assemblies (Fig. 2). For instance, the C 1s XPS spectrum of ITO/tpy-[Fe(bis-tpy)]5 was deconvoluted into three peaks centred at binding energies (BE) of 284 eV, 284.8 eV and 286.2 eV (Fig. 2a). The peak at 284 eV, which is a relatively low binding energy, corresponds to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C peak originating from the terpyridyl ligand, which is the central building block to form multi-component on-surface molecular assemblies. The binding energy with strong intensity appearing at 284.8 eV represents C–C bonds in the aromatic rings, while the peak at 286.2 eV, a high binding energy, corresponds to a carbon atom attached to an electronegative element such as nitrogen (CN). The C 1s spectrum did not show any asymmetric signals, indicating no shift in the binding energy of carbon atoms present in the ortho, meta, and para positions of the individual pyridine rings. The XPS spectrum of N 1s was fitted into two peaks centred at 399.6 eV and 398.7 eV corresponding to Fe2+ coordinated nitrogen in the terpyridine rings (Nc) and free terpyridine rings (Nf), respectively (Fig. 2b). The binding energy of free nitrogen (Nf) was cross-checked by recording the XPS spectra (C 1s, N 1s) of the as-prepared template layers (Fig. S5c and d, ESI†). The binding energy of N 1s in the template layer is observed at 398.8 eV which is 0.8 eV lower than that of metal-ion coordinated nitrogen (Nc), ensuring the coordination of terpyridyl nitrogen to Fe2+ ions. The respective intensity ratio of inner-core pyridine (coordinated nitrogen) and free pyridine units [I(Nc)/I(Nf)] was estimated at ∼4.9, indicating that nearly 83% of nitrogen are coordinated with the respective metal ions, while the remainder (∼17%) are free. This 17% of free nitrogen (Nf) is not surprising, as it originates not only from the template layers but also from the top layer of bis-tpy, as three pyridine rings remain free after the 10th LbL deposition step. The XPS peaks of Fe 2p in ITO/tpy-[Fe(bis-tpy)]5 appear as a doublet due to spin–orbit coupling. Two characteristic peaks of Fe 2p were observed at 708.7 eV and 721.6 eV corresponding to Fe 2p3/2 and Fe 2p1/2, respectively (Fig. 2c).61 ITO/tpy-[Co(bis-tpy)]5 shows a similar XPS signal at 781.4 eV which is the characteristics peak of Co 2p3/2, while the peak at 797 eV represents Co 2p1/2, indicating the in situ formation of an on-surface Co(II) terpyridine coordination compound (Fig. 2d and f). Overall, the XPS result confirms the formation of on-surface octahedral compounds and that both the metal ions are in 2+ oxidation states.62,63
C peak originating from the terpyridyl ligand, which is the central building block to form multi-component on-surface molecular assemblies. The binding energy with strong intensity appearing at 284.8 eV represents C–C bonds in the aromatic rings, while the peak at 286.2 eV, a high binding energy, corresponds to a carbon atom attached to an electronegative element such as nitrogen (CN). The C 1s spectrum did not show any asymmetric signals, indicating no shift in the binding energy of carbon atoms present in the ortho, meta, and para positions of the individual pyridine rings. The XPS spectrum of N 1s was fitted into two peaks centred at 399.6 eV and 398.7 eV corresponding to Fe2+ coordinated nitrogen in the terpyridine rings (Nc) and free terpyridine rings (Nf), respectively (Fig. 2b). The binding energy of free nitrogen (Nf) was cross-checked by recording the XPS spectra (C 1s, N 1s) of the as-prepared template layers (Fig. S5c and d, ESI†). The binding energy of N 1s in the template layer is observed at 398.8 eV which is 0.8 eV lower than that of metal-ion coordinated nitrogen (Nc), ensuring the coordination of terpyridyl nitrogen to Fe2+ ions. The respective intensity ratio of inner-core pyridine (coordinated nitrogen) and free pyridine units [I(Nc)/I(Nf)] was estimated at ∼4.9, indicating that nearly 83% of nitrogen are coordinated with the respective metal ions, while the remainder (∼17%) are free. This 17% of free nitrogen (Nf) is not surprising, as it originates not only from the template layers but also from the top layer of bis-tpy, as three pyridine rings remain free after the 10th LbL deposition step. The XPS peaks of Fe 2p in ITO/tpy-[Fe(bis-tpy)]5 appear as a doublet due to spin–orbit coupling. Two characteristic peaks of Fe 2p were observed at 708.7 eV and 721.6 eV corresponding to Fe 2p3/2 and Fe 2p1/2, respectively (Fig. 2c).61 ITO/tpy-[Co(bis-tpy)]5 shows a similar XPS signal at 781.4 eV which is the characteristics peak of Co 2p3/2, while the peak at 797 eV represents Co 2p1/2, indicating the in situ formation of an on-surface Co(II) terpyridine coordination compound (Fig. 2d and f). Overall, the XPS result confirms the formation of on-surface octahedral compounds and that both the metal ions are in 2+ oxidation states.62,63
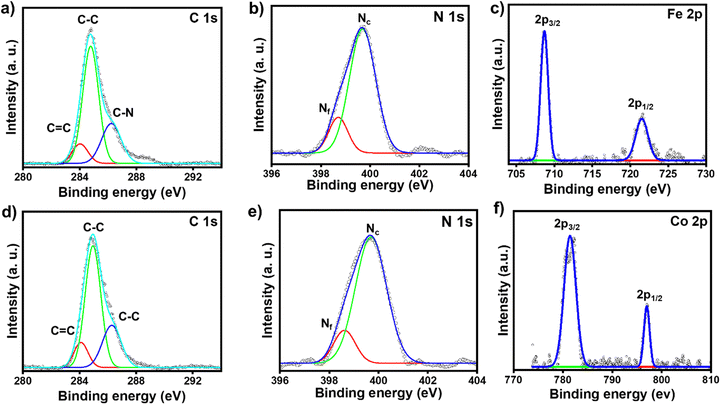 |
| | Fig. 2 (a–c) C 1s, N 1s, and Fe 2p XPS spectra of ITO/tpy-[Fe(bis-tpy)]5 and (d–f) C 1s, N 1s, and Co 2p XPS spectra of ITO/tpy-[Co(bis-tpy)]5. The solid lines represent fitted data, while the open circles indicate experimental data. Nf and Nc stand for free nitrogen and coordinated nitrogen that binds metal ions (Fe2+ or Co2+). | |
Field-emission scanning electron microscopy for thickness analysis
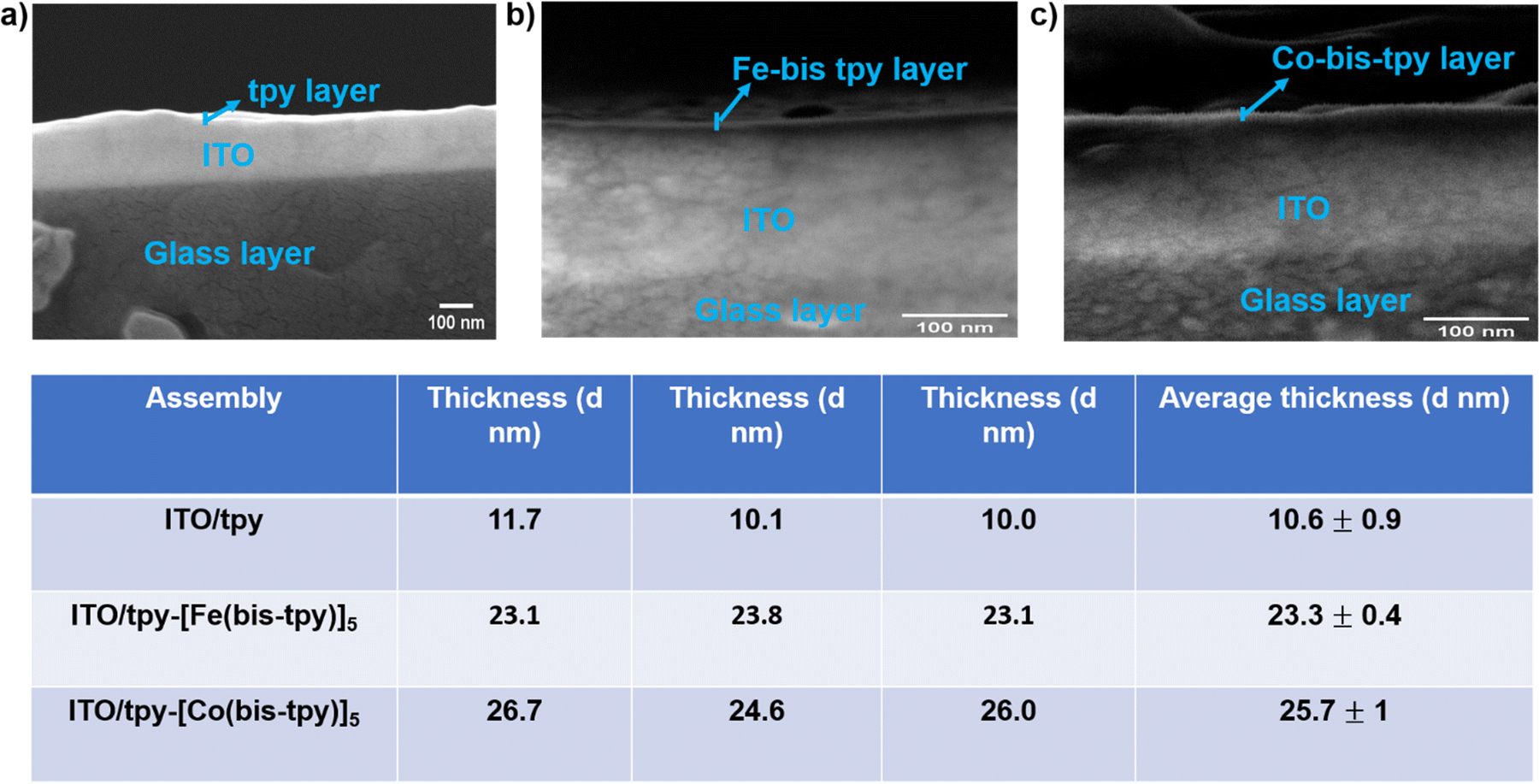
To determine the thicknesses of the molecular layers, cross-section field-emission scanning electron microscopy (FE-SEM) was performed on both the template layer and metal-ion-containing surface-confined molecular assemblies. To record the FE-SEM images, an ultrathin layer (∼5 nm) of Au was deposited on each sample via a sputter-coater. Such a thin film of Au is desirable to prevent electrical charging on the surface.64 The cross-section FE-SEM analysis determined the thickness of ITO/tpy (template layer), ITO/tpy-[Fe(bis-tpy)]5, and ITO/tpy-[Co(bis-tpy)]5, estimated as 10.6 ± 0.9, 23.3 ± 0.4, and 25.7 ± 1 nm, respectively. To ensure robust thickness measurements, each measurement was carried out on three different positions and the average thicknesses are provided in tabular form (Fig. 3). Since the layer thickness varies from sample to sample, non-uniformity in thickness cannot be neglected and this is one of the major drawbacks of electrochemically grafted molecular layers.
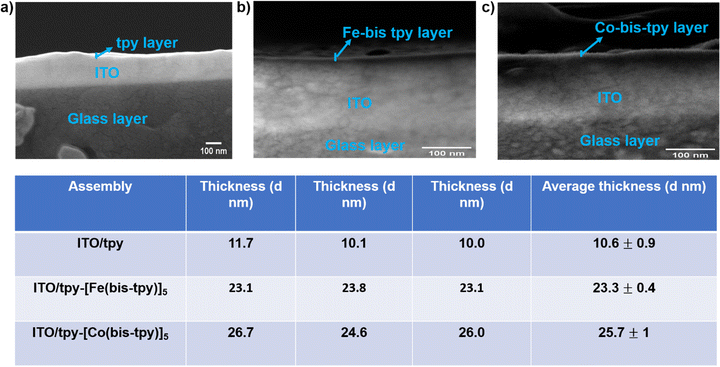 |
| | Fig. 3 (a–c) Cross-sectional FE-SEM images to deduce the thickness of electrochemically grafted ITO/tpy ITO/tpy-[Fe(bis-tpy)]5 and ITO/tpy-[Co(bis-tpy)]5. The thicknesses from three different measurements and the average thicknesses are provided in the table for the respective layers. | |
UV-vis spectra
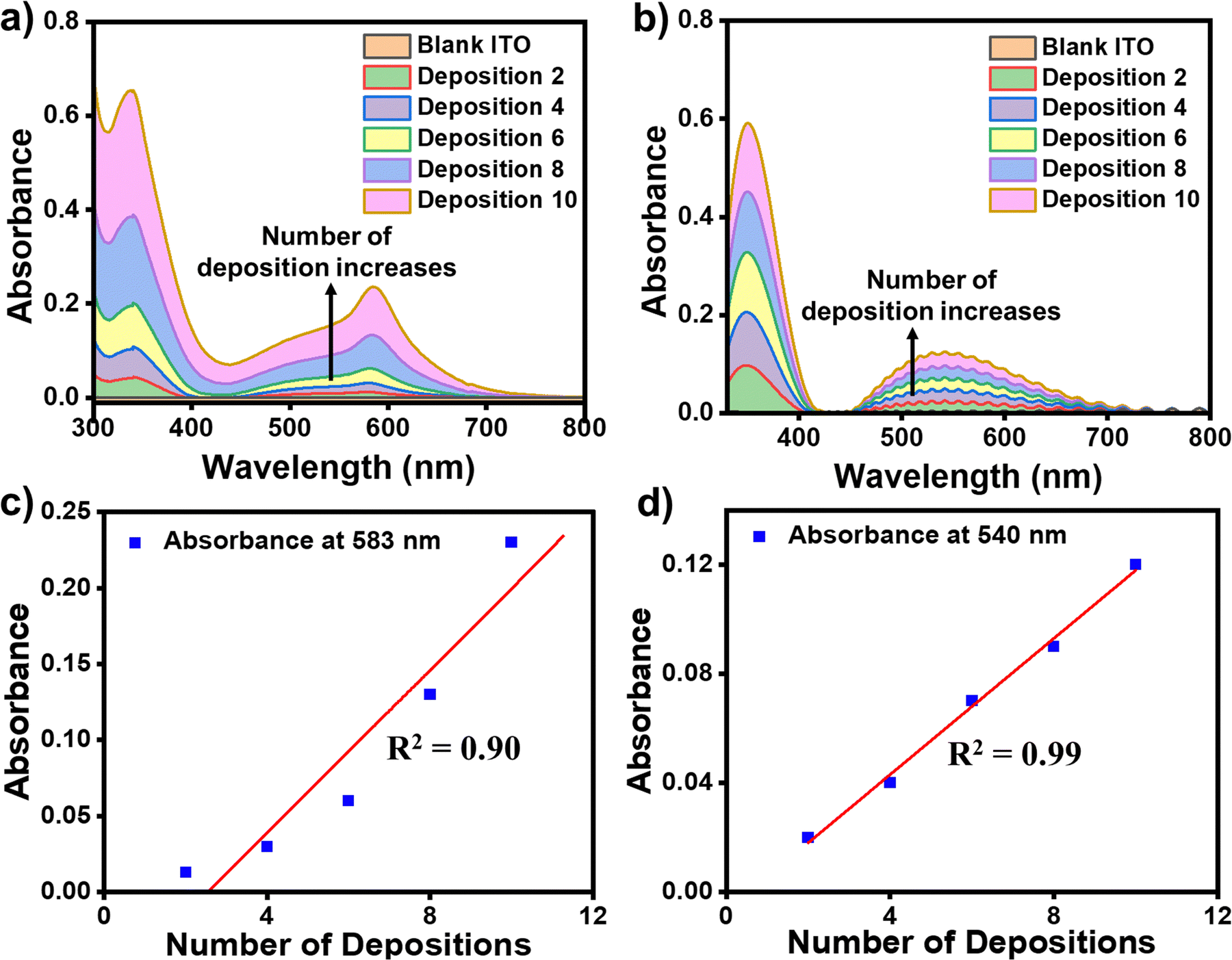
The formation of the successive metal ions/bis-tpy layers on top of the template layer (ITO/tpy) was monitored by UV-vis spectroscopy in the wavelength range of 300–800 nm. The ITO/tpy system shows absorbance at 344 nm due to π–π* electronic transitions. The UV-vis spectra were recorded after the sequential deposition of either Fe(ClO4)2 or Co(ClO4)2 followed by bis-tpy ligand coordination to form nanometric assemblies. UV-vis absorption spectra show an increase in absorbance for ITO/tpy-[Fe(bis-tpy)]10 and ITO/tpy-[Fe(bis-tpy)]10 demonstrating the formation of metal ions-chelating ligand binding (Fig. 4a and b). In addition, a linear increase (R2 = 0.9–0.99) in absorbance with metal ions/ligand coordination was obtained at λmax = 583 and 540 nm, due to metal-to-ligand charge-transfer (MLCT) transitions occurring at Fe2+ to bis-tpy ligands, and Co2+ to bis-tpy ligands, respectively (Fig. 4c and d). In another study, we combined ITO/tpy-[Fe(bis-tpy)]5 and ITO/tpy-[Co(bis-tpy)]5 keeping gel electrolyte between them and recorded a UV-vis spectrum that shows two MLCT bands, at λmax = 579 nm 529 nm for Fe2+ to bis-tpy and Co2+ to bis-tpy, respectively (Fig. S6, ESI†).
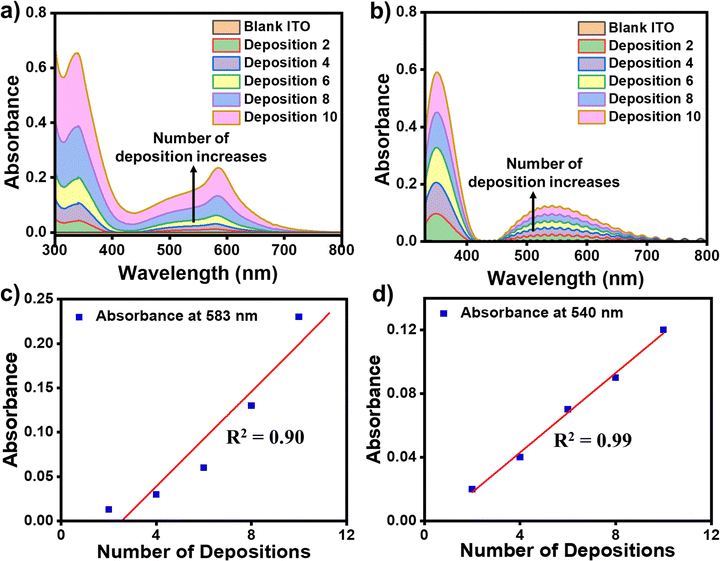 |
| | Fig. 4 (a) Absorption spectra recorded on ITO/tpy-[Fe(bis-tpy)10] after different numbers of Fe2+/bis-tpy deposition steps. (b) Absorption spectra of ITO/tpy-[Co(bis-tpy)10] after different numbers of Co2+/bis-tpy deposition steps. (c and d) Plots of Abs vs. no. of deposition steps for Fe2+- and Co2+-based molecular assemblies showing linear relationships (R2 = 0.90 and 0.99, respectively). | |
Cyclic voltammetry (CV) studies of molecular thin films
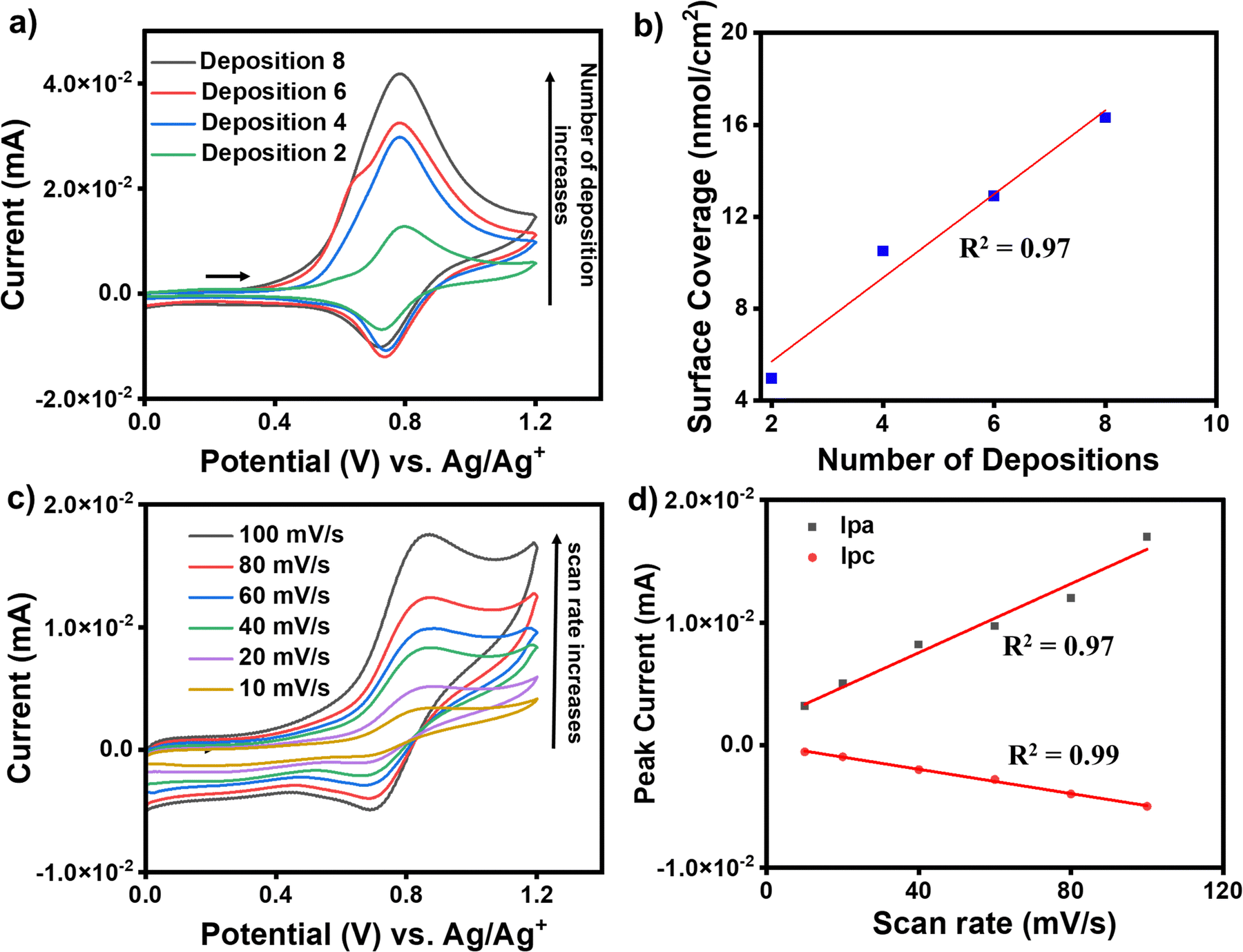
For the growth of an individual redox-active molecular layer after metal ion and ligand deposition, the modified electrodes were used as the working electrode to record cyclic voltammograms (CVs). ITO/tpy-[M(bis-tpy)n (where M = Co or Fe, n = 2, 4, 6, 8th deposition steps)] were employed for electrochemical studies to understand their redox behavior. The CVs were recorded in ambient conditions using a conventional three-electrode electrochemical setup, where a Pt wire and Ag/Ag+ were used as counter electrode and reference electrode, respectively. The CVs were recorded in deoxygenated (purged with argon for 15 min) 100 mM solution of tetrabutylammonium perchlorate in acetonitrile on a freshly prepared modified ITO electrode and their current compared due to charge transfer between ITO and redox probes, Fe2+ or Co2+ that are present in the molecular assemblies. An increase in faradaic current (cathodic and anodic) was observed after every metal ion and bis-tpy ligand deposition, confirming the formation of an Fe2+-bis-tpy assembly on ITO (Fig. 5a). The ITO/tpy-[Fe(bis-tpy)]n assembly shows anodic (Epa) and cathodic (Epc) peak potentials at +0.92, and +0.8 V (vs. Ag/AgCl), respectively, for an Fe2+/3+ redox couple. The conducting nature of the film grown on ITO was tested in the presence of a redox probe such as ferrocene, allowing charge transfer to occur with a reversible Fc/Fc+ faradaic process (Fig. S7, ESI†). Similarly, for a Co2+/Co3+ redox couple in ITO/tpy-[Co(bis-tpy)]n, anodic (Epa) and cathodic (Epc) peak potentials were observed at −0.02 and −0.54 V (vs. Ag/Ag+), respectively, (Fig. S8, ESI†). The half-wave potentials of Fe (EFe1/2) and Co (ECo1/2) appeared at +0.86 and −0.37 V, respectively. In the case of both metal complexes on the same ITO electrode, the half wave potentials of Fe (EFe1/2) and Co (ECo1/2) were 0.77 and −0.11 V, respectively (vs. Ag/Ag+) (Fig. S9, ESI†). The surface coverage and linear relationship as a function of deposition steps indicate enhancement of faradaic current (Fig. 5b). In addition, CVs were recorded on Fe2+ and Co2+ containing molecular assemblies at varying scan rates (10, 20, 40, 60, 80, 100 mV s−1) (Fig. 5c). A linear relationship (R2 = 0.97 for anodic and 0.99 for cathodic) between the faradaic currents and scan rates confirms the on-surface redox phenomena, i.e., a diffusion-less charge transfer process between the attached redox-probes and the ITO working electrode, which is in good agreement with our system of studies (Fig. 5d). Room-temperature (298 K) electrochemical parameters that are obtained at different scan rates were further employed to deduce electron transfer rate constants, KET, for the oxidation and reduction process involved in Fe2+-containing molecular assemblies. To obtain the KET values, cathodic and anodic peak potentials were plotted as a function of scan rates and the linear regime chosen to deduce the charge transfer coefficients, α. Using the well-established Laviron method, the KET values were estimated as 16.1 and 13. 6 s−1, which is an indication of fast-electron transfer kinetics for such a redox-active ‘on-surface’ coordination compound. Detailed discussion of the KET determinations is discussed in the (Fig. S10, ESI†).
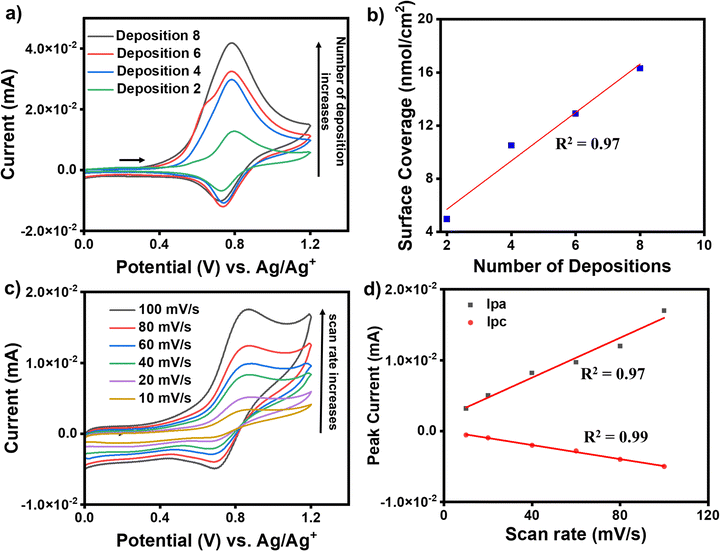 |
| | Fig. 5 Room-temperature cyclic voltammograms of (a) ITO/tpy-[Fe(bis-tpy)]n with an increasing number of Fe2+/bis-tpy ligand deposition steps (n = 2 to 8). (b) Plot of surface coverage as a function of the number of deposition steps, showing a linear relationship (R2 = 0.97). (c) Cyclic voltammograms of ITO/tpy-[Fe(bis-tpy)]8 recorded at different scan rates. (d) Plots of anodic (Ipa) and cathodic (Ipc) peak current as a function of the scan rate (mV s−1), revealing linear relationships (R2 = 0.97 and 0.99, respectively). | |
DC-based current–voltage (I–V) measurements
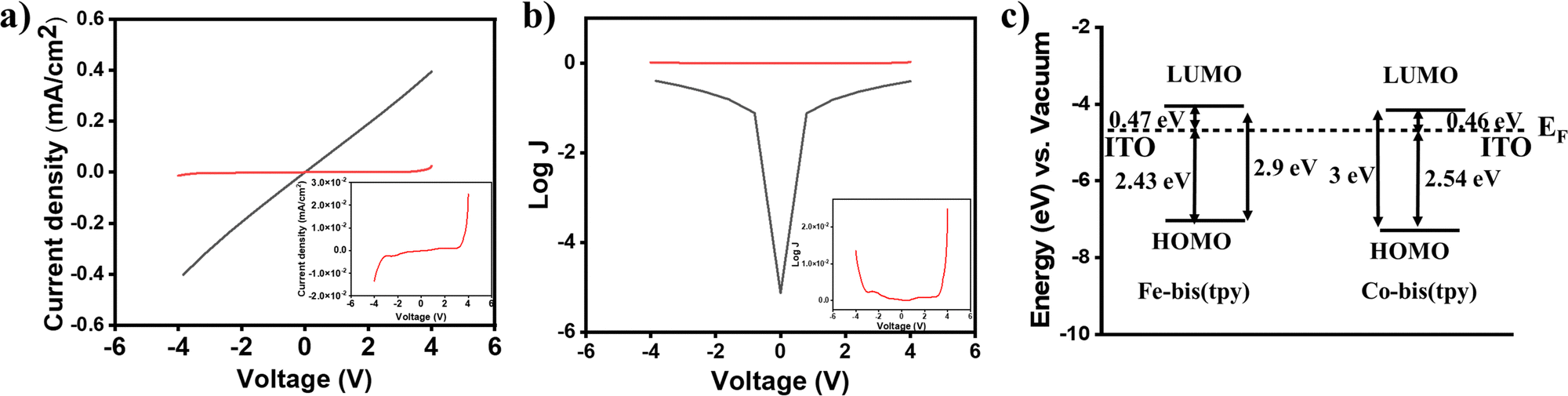
Redox-active molecular thin films have been employed for understanding the current–voltage (I–V) response. In this regard, heterostructure devices were fabricated by combining two ITO/tpy-[Fe(bis-tpy)]5/gel electrolyte/[Co(bis-tpy)]5-tpy/ITO, where the gel electrolyte and redox films were freshly prepared to ensure that they did not get oxidized. Using a Keithley source meter unit and a four-probe measurement setup, current was monitored in response to an applied voltage. The I–V plots were recorded by applying a potential of ±4 V and were converted to the corresponding current density (in J, milliamp cm−2) by considering the area of the devices (Fig. 6a). The J–V response is shown up to nearly ±2 V, but the junctions do not show any conduction. However, above ±2 V, the junctions show conduction to occur in both directions, which is a bit asymmetrical. The origin of the asymmetric conduction is the difference in energy of EF–LUMO/EF–HOMO for Fe2+ and Co2+ molecular assemblies. In a controlled experiment, similar devices were prepared but with two bare ITOs and such reference junctions (ITO/gel electrolyte/ITO) show a linear I–V relationship following Ohm's law. Fig. 6b shows a semilog plot of current density as a function of applied voltage, and molecular junctions and reference junctions are compared. The energy profile diagram (calculated from combined UV-vis spectra and electrochemical measurements) suggests that electron transport occurs from the Fermi level of the ITO to the LUMO of the Fe2+/Co2+-bis-tpy compounds, as this energy barrier is much lower than that of EF–HOMO, which are nearly 2.43 and 2.54 eV for Fe2+ and Co2+-tpy thin films (Fig. 6c).
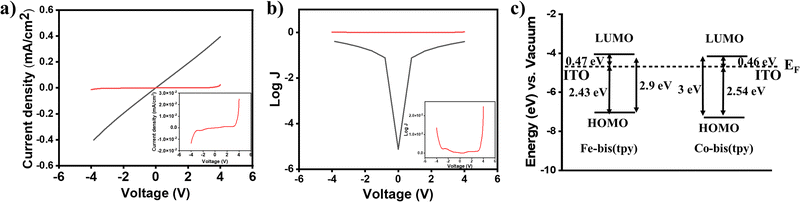 |
| | Fig. 6 (a) A comparison of the J–V plot of a reference junction (solid black line) and that of a molecular junction (solid red line). (b) Log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) J vs. voltage plots and (c) energy level diagrams of ITO (EF 4.7 eV) and the HOMO and LUMO energies of Fe2+-bis(tpy) and Co2+-bis(tpy). J vs. voltage plots and (c) energy level diagrams of ITO (EF 4.7 eV) and the HOMO and LUMO energies of Fe2+-bis(tpy) and Co2+-bis(tpy). | |
Electrical impedance measurements of a molecular junction
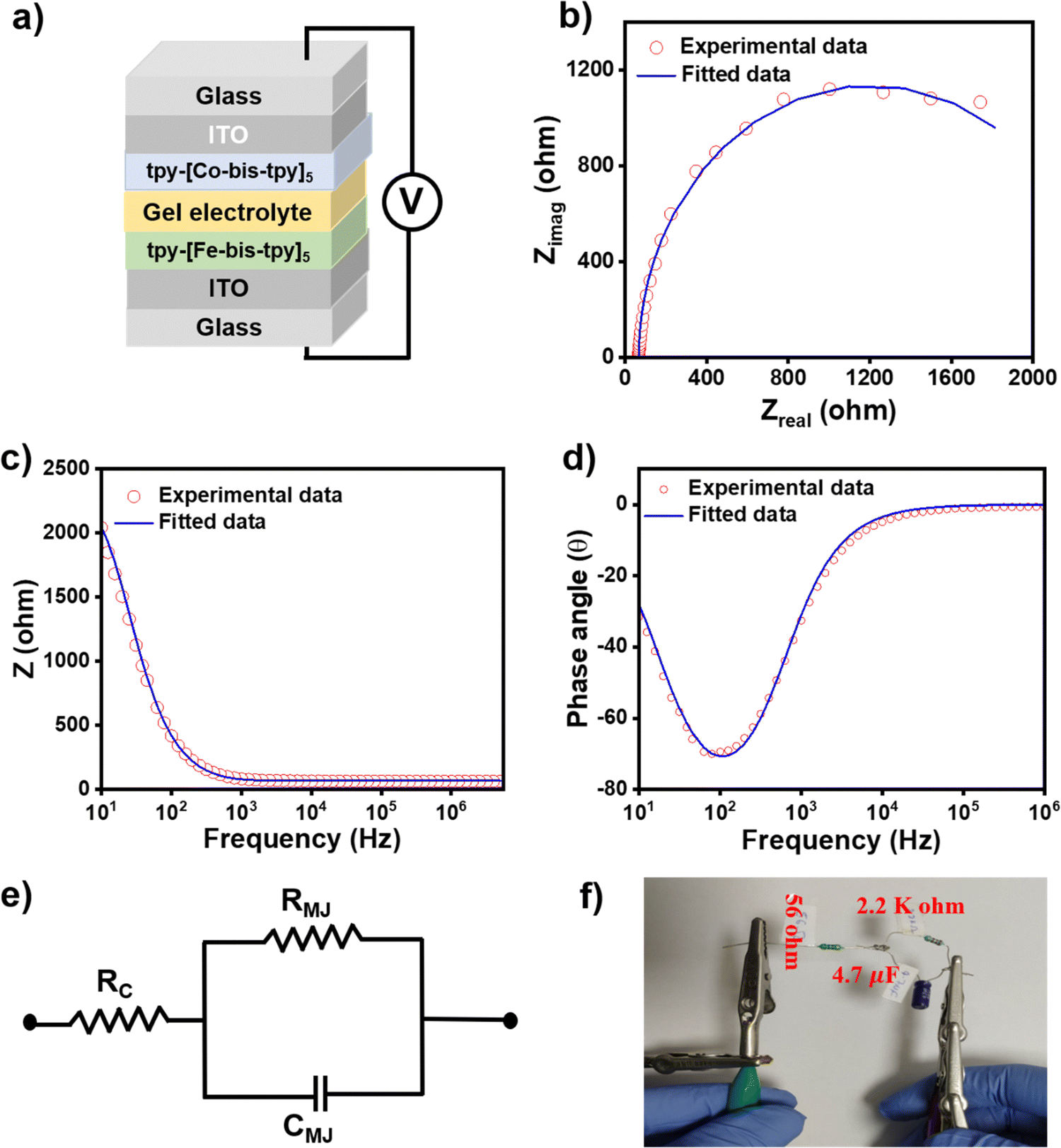
It is desirable to understand the frequency-dependent electrical response in electronic devices via AC-based electrical measurements, as DC-based electrical measurement does not provide such information. Similarly, for real applications of molecular junctions, it is important to understand the frequency response of such molecular junctions, and electrical impedance measurement (EIS) studies help us understand individual electrical components of the devices, such as resistor, capacitance, uncompensated resistance, and inductance.65 To proceed with the AC-based measurements, we made sure that the molecular junctions were not short-circuited before the EIS measurements, so cyclic voltammograms were recorded at a scan rate of 20 mV s−1 that showed reversible oxidation and reduction signals, ensuring the films are intact on the ITO. Using a Gamry potentiostat, EIS was performed on ITO/tpy-[Fe(bis-tpy)]5/gel electrolyte/[Co(bis-tpy)]5-tpy/ITO (Fig. 7a). The applied frequency range went from 5 MHz to 10 Hz in 10 mV AC bias while the DC voltage was fixed at 0 V. Both the experimental and fitted Nyquist plot (zrealvs. zimag.) and Bode plot (zrealvs. frequency and phase angle vs. frequency) of the junction are shown in Fig. 7b–d. From the plot it is clear that the Nyquist plot does not reveal a direct frequency response or phase angle (θ°) dependency, whereas the Bode plot is effective. The Bode plot permits us to comprehend both the real impedance and the phase angle as a function of frequency. For instance, it shows that the junction behaves like a capacitor in a lower frequency regime (101 to 103 Hz) and the real impedance decreases exponentially with an increase in frequency (Fig. 7c). On the other hand, at higher frequency, the real impedance reaches nearly zero Ohm and is almost flat with increasing frequency, which corresponds to the capacitive behavior of the junctions. The similar frequency response of the capacitor and resistor components can be explained by the changes in the phase angle (θ°) with respect to frequency. A phase angle at θ = −75° shows the maximum response of capacitance, which decrease further in a higher frequency regime (Fig. 7d). The phase angle reaching nearly θ = 0° indicates resistance behavior of the molecular junctions. The EIS data were further employed in the Randles circuit model using Gamry electrochemical analyst software, where RMJ, RCT, Cdl components were used.66 This circuit model consists of uncompensated resistance (67.5 Ohm), which is in a series and parallel combination of the capacitor (3.8 × 10−6 F) and molecular junction resistance (2.2 × 103 Ohm) (Fig. 7e). A physical equivalent circuit was fabricated by combining a similar resistor and capacitor following the rule of an equivalent circuit, which performed with very similar I–V responses. The circuit is shown in Fig. 7f. These circuit-validated data show that our electrical measurements originate from the ‘on-surface’ coordination compounds but not from any other artifacts.
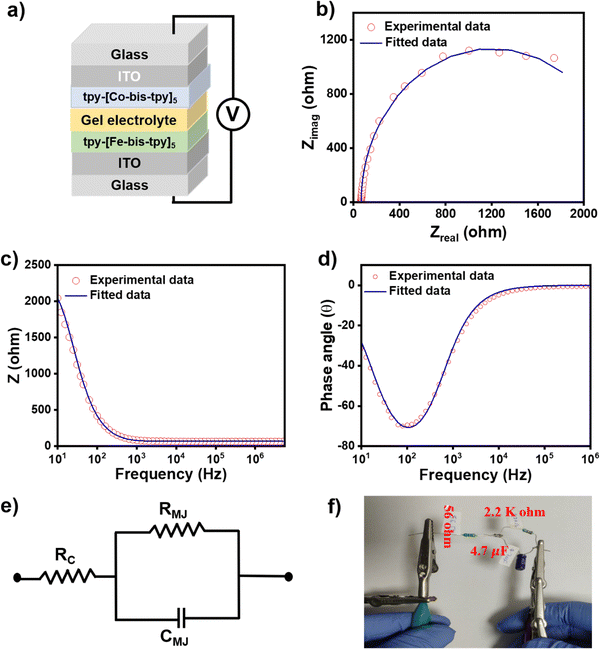 |
| | Fig. 7 (a) A schematic description of a molecular junction composed of ITO/tpy-[Fe(bis-tpy)]5/gel electrolyte/[Co(bis-tpy)]5-tpy/ITO. (b) The Nyquist plot of the junction. (c and d) Bodes plot at varying frequencies. (e) The Randles equivalent circuit and f) a real circuit for the experimental validation of the fitted model. | |
Conclusions
We utilize a versatile, elegant electrochemical technique to fabricate template layers for covalent interfaces between a transparent conducting oxide and terpyridine. The chelating ligand with three pyridines binds transition metal ions followed by interaction with the bis-terpyridine ligand. We explore a layer-by-layer technique, a classical approach for controlling the ‘on-surface’ chemical composition, the number of deposition units, and the thickness. The molecular thin film growth process can be monitored via optical and electrochemical methods, elemental analysis, and thickness measurements after each metal ion/ligand deposition step. The ‘on-surface’ opto-electro-active molecular assemblies not only show a reversible, one-electron transfer and fast redox process, but they also allow Fc/Fc+ electron transfer, showing the permeability of the films. We further combine two modified ITO electrodes incorporating a conductive gel between them for current–voltage measurements, a clean-room-free technique. The devices show semiconducting behavior below ±2 V, but they do not show conductivity; above that they conduct due to the lower energy barrier between the Fermi energy level and the LUMO. The frequency-dependent capacitance and resistance behaviors of the molecular junctions are explained based on solid-state electrical impedance spectroscopy, and the fitted circuit model was further validated experimentally. Such individual electrical components are vital for designing molecular electronic devices suitable for practical applications. Though the electrochemical reduction method is a versatile platform for growing molecular thin films, the technique is limited to diazonium salts and needs a potentiostat, three electrodes, and an electrochemical cell. If an aryl diazonium does not contain a bulky group, it produces non-crystalline films. The opto-electroactive coordination compounds covalently grown on an ITO-modified electrode can be further utilized for use in electrochromic devices and for photovoltaic applications.
Experimental section
Reagents and materials
2-Acetyl pyridine, 4-nitrobenzaldehyde, ammonium acetate, terephthaldehyde, and tetrabutylammonium perchlorate were purchased from Sigma-Aldrich and used without further purification. Iodine, sodium hydroxide, and 5% palladium on carbon and hydrazine hydrate were purchased from Finar. ITO-coated glass substrates (100 nm coating) were purchased from Nanoshel UK Ltd (sheet resistance of 14–16 Ohm sq−1, thickness 1.1 mm and transmittance ∼83% in the visible regime).
Activation of the ITO substrate
ITO-coated glass slides were cleaned by ultra-sonication, 15 min each in hexane, acetone, and isopropyl alcohol, followed by drying with a nitrogen gun. Then the substrates were put in an oven at 100 °C for 30 min to remove water content. The activated ITO-coated glass substrates were used for further experiments.
Preparation of diazonium salts and template layers (ITO/tpy)
To prepare diazonium salts of terpyridine amine, 7 mg of 4′-NH2tpy and 342 mg of tetrabutylammonium perchlorate were dissolved in 10 mL of acetonitrile and the mixture was cooled to reach a temperature of 0–5 °C, following a well-established protocol.67 20 mM of tButyl nitrite was added to a cold solution of 4′-aminophenyl terpyridine (1 mg) and a colour change from yellow to slightly off-white was observed. The solution was used for electrochemical grafting for the formation of template layers (ITO/tpy) taken in an electrochemical cell equipped with freshly prepared ITO, Pt, and Ag/AgCl as working, counter, and reference electrodes, respectively. The aryl diazonium salt solution in anhydrous acetonitrile was deoxygenated in the presence of argon flow for 15 min before electrochemical grafting. Cyclic voltammograms were recorded (unstirred solution) at room temperature at a scan rate of 100 mV s−1 and a cathodic peak was observed at −0.38 V (vs. Ag/AgCl) in the first cycle, revealing the diazonium reduction that forms radicals which graft on the ITO working electrode. The CVs were recorded up to the 10th scan but after the first CV scan, the reduction peak was diminished, because the ITO surface blocked further formation of aryl radicals. The template layers (ITO/tpy) were further used for layer-by-layer metal ions/bis-tpy ligand deposition followed by characterization.
Preparation of gel electrolyte
The gel electrolyte was synthesized using a reported procedure.61 In brief, LiClO4 was dried in an oven at 100 °C for 24 h. Then, 210 mg of poly(methyl methacrylate) (PMMA) and 90 mg of LiClO4 were added to a 50 mL round bottom flask, followed by the addition of 0.6 mL of propylene carbonate and 1 mL of acetonitrile added dropwise and stirred for 24 h under an N2 environment. The obtained transparent gel electrolyte was used to prepare the molecular electronic and reference devices for electrical measurements.
Characterization techniques
Synthesized terpyridine (tpy) and bis-terpyridine were characterized with 1H NMR spectra recorded on a JEOLECX 500 FT (500 MHz) at 298 K and the NMR peak positions are listed with reference to TMS. The molecular mass of the compounds was obtained on a WATERS Q-TOF Premier electrospray ionization mass spectrometer. Elemental analysis was characterized using X-ray photoelectron spectroscopy (XPS) with an Auger electron spectroscopy (AES) module PHI 5000 Versa Prob II. The XPS spectrometer was equipped with a monochromatic Al Kα (hν = 1486.6 eV) radiation source. Nanometric gold coating was performed using a Quorum vacuum chamber. Cross-section FE-SEM was recorded on Carl Zeiss EVO 50 with an applied 30 KV acceleration voltage. UV-vis spectra were recorded on a JASCO UV-Visible-NIR spectrophotometer (V-770). Electrochemical measurements were performed using a Metrohm Autolab Potentiostat Electrochemical workstation (Model: 204, software nova-2.14). Current–voltage (I–V) measurements and EIS were recorded using a Keithley 2604B Source meter unit, and Gamry Instrument (Model 600+) equipped with a frequency response analyzer (FRA), respectively.
Author contributions
P. S.: experiments; writing – original draft; P. C. M.: conceptualization; funding acquisition; investigation; supervision; writing – original draft.
Conflicts of interest
The authors declare no conflicts of interest related to the present work.
Acknowledgements
P. S. would like to thank the Council of Scientific and Industrial Research (CSIR) for providing a Junior Research Fellowship. P. C. M acknowledges the Department of Science and Technology for a start-up research grant (SRG/2019/000391) and IIT Kanpur (IITK/CHM/2019044) for initiation and special grant support to establish the laboratory. Financial support from the Council of Scientific & Industrial Research (CSIR), New Delhi, Sanctioned No. 01(3049)/21/EMR-II awarded to P. C. M is greatly acknowledged. The authors acknowledge IIT Kanpur for infrastructure and equipment facilities.
References
- H. Kroemer, Nat. Mater., 2012, 11, 91 CrossRef PubMed.
- J. T. Yates and C. T. Campbell, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 911–916 CrossRef CAS.
- K. Michaeli, N. Kantor-Uriel, R. Naaman and D. H. Waldeck, Chem. Soc. Rev., 2016, 45, 6478–6487 RSC.
- Y. Hamo, M. Lahav and M. E. Boom, Angew. Chem., Int. Ed., 2020, 59, 2612–2617 CrossRef CAS PubMed.
- C. Van Dyck, T. J. Marks and M. A. Ratner, ACS Nano, 2017, 11, 5970–5981 CrossRef CAS PubMed.
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103–1170 CrossRef CAS PubMed.
- R. K. Smith, P. A. Lewis and P. S. Weiss, Prog. Surf. Sci., 2004, 75, 1–68 CrossRef CAS.
- A. Schoedel, C. Scherb and T. Bein, Angew. Chem., 2010, 122, 7383–7386 CrossRef.
- R. L. McCreery and A. J. Bergren, Adv. Mater., 2009, 21, 4303–4322 CrossRef CAS PubMed.
- H. Yang, G. Chen, A. A. C. Cotta, A. T. N’Diaye, S. A. Nikolaev, E. A. Soares, W. A. A. Macedo, K. Liu, A. K. Schmid, A. Fert and M. Chshiev, Nat. Mater., 2018, 17, 605–609 CrossRef CAS PubMed.
- S. Sanvito, Chem. Soc. Rev., 2011, 40, 3336 RSC.
- A. Moneo, A. González-Orive, S. Bock, M. Fenero, I. L. Herrer, D. C. Milan, M. Lorenzoni, R. J. Nichols, P. Cea, F. Perez-Murano, P. J. Low and S. Martin, Nanoscale, 2018, 10, 14128–14138 RSC.
- E. Coronado, Nat. Rev. Mater., 2020, 5, 87–104 CrossRef.
- D. Zacher, O. Shekhah, C. Wöll and R. A. Fischer, Chem. Soc. Rev., 2009, 38, 1418–1429 RSC.
- X. Chen, M. Roemer, L. Yuan, W. Du, D. Thompson, E. D. Barco and C. A. Nijhuis, Nat. Nanotechnol., 2017, 12, 797–803 CrossRef CAS.
- T. Schmaltz, G. Sforazzini, T. Reichert and H. Frauenrath, Adv. Mater., 2017, 29, 1605286 CrossRef PubMed.
- J. E. Greenwald, J. Cameron, N. J. Findlay, T. Fu, S. Gunasekaran, P. J. Skabara and L. Venkataraman, Nat. Nanotechnol., 2021, 16, 313–317 CrossRef CAS.
- X. Qiu, V. Ivasyshyn, L. Qiu, M. Enache, J. Dong, S. Rousseva, G. Portale, M. Stöhr, J. C. Hummelen and R. C. Chiechi, Nat. Mater., 2020, 19, 330–337 CrossRef CAS.
- B. J. Cafferty, L. Yuan, M. Baghbanzadeh, D. Rappoport, M. H. Beyzavi and G. M. Whitesides, Angew. Chem., Int. Ed., 2019, 58, 8097–8102 CrossRef CAS PubMed.
- A. Vilan, D. Aswal and D. Cahen, Chem. Rev., 2017, 117, 4248–4286 CrossRef CAS PubMed.
- T. Neumann, J. Liu, T. Wächter, P. Friederich, F. Symalla, A. Welle, V. Mugnaini, V. Meded, M. Zharnikov, C. Wöll and W. Wenzel, ACS Nano, 2016, 10, 7085–7093 CrossRef CAS PubMed.
- E. Aslanidis, E. Skotadis and D. Tsoukalas, Nanoscale, 2021, 13, 3263–3274 RSC.
- R. V. Martínez, F. García, R. García, E. Coronado, A. Forment-Aliaga, F. M. Romero and S. Tatay, Adv. Mater., 2007, 19, 291–295 CrossRef.
- A. P. Bonifas and R. L. McCreery, Nat. Nanotechnol., 2010, 5, 612–617 CrossRef CAS PubMed.
- W. Y. Lo, N. Zhang, Z. Cai, L. Li and L. Yu, Acc. Chem. Res., 2016, 49, 1852–1863 CrossRef CAS PubMed.
- R. Gupta, P. Jash and P. C. Mondal, J. Mater. Chem. C, 2021, 9, 11497–11516 RSC.
- H. Maeda, R. Sakamoto and H. Nishihara, Coord. Chem. Rev., 2017, 346, 139–149 CrossRef CAS.
- H. Atesci, V. Kaliginedi, J. A. Celis Gil, H. Ozawa, J. M. Thijssen, P. Broekmann, M. A. Haga and S. J. Van Der Molen, Nat. Nanotechnol., 2018, 13, 117–121 CrossRef CAS.
- P. C. Mondal, V. Singh and M. Zharnikov, Acc. Chem. Res., 2017, 50, 2128–2138 CrossRef CAS PubMed.
- M. Lahav and M. E. van der Boom, Adv. Mater., 2018, 30, 1–7 Search PubMed.
- R. Gupta, P. Jash, P. Sachan, A. Bayat, V. Singh and P. C. Mondal, Angew. Chem., Int. Ed., 2021, 60, 26904–26921 CrossRef CAS PubMed.
- P. Sachan and P. C. Mondal, Analyst, 2020, 145, 1563–1582 RSC.
- X. Yao, X. Sun, F. Lafolet and J.-C. Lacroix, Nano Lett., 2020, 20, 6899–6907 CrossRef CAS PubMed.
- Q. Nguyen, U. Tefashe, P. Martin, M. L. Della Rocca, F. Lafolet, P. Lafarge, R. L. McCreery and J. Lacroix, Adv. Electron. Mater., 2020, 6, 1901416 CrossRef.
- S. K. Saxena, U. M. Tefashe and R. L. McCreery, J. Am. Chem. Soc., 2020, 142, 15420–15430 CrossRef CAS PubMed.
- Q. Nguyen, U. Tefashe, P. Martin, M. L. Della Rocca, F. Lafolet, P. Lafarge, R. L. McCreery and J. Lacroix, Adv. Electron. Mater., 2020, 6, 1901416 CrossRef.
- A. K. Farquhar, M. Supur, S. R. Smith, C. Dyck and R. L. McCreery, Adv. Energy Mater., 2018, 8, 1802439 CrossRef.
- A. L. Gui, H. M. Yau, D. S. Thomas, M. Chockalingam, J. B. Harper and J. J. Gooding, Langmuir, 2013, 29, 4772–4781 CrossRef CAS.
- S. M. Khor, G. Liu, C. Fairman, S. G. Iyengar and J. J. Gooding, Biosens. Bioelectron., 2011, 26, 2038–2044 CrossRef CAS PubMed.
- T. Wu, C. M. Fitchett, P. A. Brooksby and A. J. Downard, ACS Appl. Mater. Interfaces, 2021, 13, 11545–11570 CrossRef CAS PubMed.
- D. Bélanger and J. Pinson, Chem. Soc. Rev., 2011, 40, 3995–4048 RSC.
- E. M. Dief, Y. B. Vogel, C. R. Peiris, A. P. Le Brun, V. R. Gonçales, S. Ciampi, J. R. Reimers and N. Darwish, Langmuir, 2020, 36, 14999–15009 CrossRef CAS PubMed.
- A. Mattiuzzi, Q. Lenne, J. Carvalho Padilha, L. Troian-Gautier, Y. R. Leroux, I. Jabin and C. Lagrost, Front. Chem., 2020, 8, 1–6 CrossRef PubMed.
- J. M. Buriak, Chem. Rev., 2002, 102, 1271–1308 CrossRef CAS PubMed.
- C. R. Peiris, Y. B. Vogel, A. P. Le Brun, A. C. Aragonès, M. L. Coote, I. Díez-Pérez, S. Ciampi and N. Darwish, J. Am. Chem. Soc., 2019, 141, 14788–14797 CrossRef CAS PubMed.
- M. James, S. Ciampi, T. A. Darwish, T. L. Hanley, S. O. Sylvester and J. J. Gooding, Langmuir, 2011, 27, 10753–10762 CrossRef CAS PubMed.
- Y. B. Vogel, N. Darwish, M. B. Kashi, J. J. Gooding and S. Ciampi, Electrochim. Acta, 2017, 247, 200–206 CrossRef CAS.
- L. Zhang, Y. B. Vogel, B. B. Noble, V. R. Gonçales, N. Darwish, A. Le Brun, J. J. Gooding, G. G. Wallace, M. L. Coote and S. Ciampi, J. Am. Chem. Soc., 2016, 138, 9611–9619 CrossRef CAS.
- S. Zhang, S. Ferrie, C. R. Peiris, X. Lyu, Y. B. Vogel, N. Darwish and S. Ciampi, J. Am. Chem. Soc., 2021, 143, 1267–1272 CrossRef CAS.
- X. Chen, E. Luais, N. Darwish, S. Ciampi, P. Thordarson and J. J. Gooding, Langmuir, 2012, 28, 9487–9495 CrossRef CAS.
- M. Lahav and M. E. van der Boom, Adv. Mater., 2018, 30, 1706641 CrossRef.
- T. Gupta, P. C. Mondal, A. Kumar, Y. L. Jeyachandran and M. Zharnikov, Adv. Funct. Mater., 2013, 23, 4227–4235 CrossRef CAS.
- R. Sakamoto, K.-H. Wu, R. Matsuoka, H. Maeda and H. Nishihara, Chem. Soc. Rev., 2015, 44, 7698–7714 RSC.
- K. Terada, K. Kanaizuka, V. M. Iyer, M. Sannodo, S. Saito, K. Kobayashi and M. Haga, Angew. Chem., Int. Ed., 2011, 50, 6287–6291 CrossRef CAS PubMed.
- M. R. Gill and J. A. Thomas, Chem. Soc. Rev., 2012, 41, 3179 RSC.
- Y. Hamo, M. Lahav and M. E. Boom, Angew. Chem., Int. Ed., 2020, 59, 2612–2617 CrossRef CAS PubMed.
- V. Marin, E. Holder, R. Hoogenboom and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 618–635 RSC.
- R. D. Hancock, Chem. Soc. Rev., 2013, 42, 1500–1524 RSC.
- V. Singh, P. C. Mondal, A. K. Singh and M. Zharnikov, Coord. Chem. Rev., 2017, 330, 144–163 CrossRef CAS.
- S. Amunugama, E. Asempa, R. C. Tripathi, D. Wanniarachchi, H. Baydoun, P. Hoffmann, E. Jakubikova and C. N. Verani, Dalton Trans., 2022, 51, 8425–8436 RSC.
- S. R. Jena and J. Choudhury, Chem. Commun., 2020, 56, 559–562 RSC.
- C. Zanardi, B. Zanfrognini, S. Morandi, F. Terzi, L. Pigani, L. Pasquali and R. Seeber, Electrochim. Acta, 2018, 260, 314–323 CrossRef CAS.
- F. M. F. de Groot, J. C. Fuggle, B. T. Thole and G. A. Sawatzky, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 42, 5459–5468 CrossRef CAS.
- E. Maleki, O. Unal and A. Amanov, Surf. Interfaces, 2018, 13, 233–254 CrossRef.
- P. Jash, R. K. Parashar, C. Fontanesi and P. C. Mondal, Adv. Funct. Mater., 2021, 2109956 Search PubMed.
- P. Chandra Mondal, U. M. Tefashe and R. L. McCreery, J. Am. Chem. Soc., 2018, 140, 7239–7247 CrossRef CAS PubMed.
- R. Gupta, P. Jash, A. Pritam and P. C. Mondal, Can. J. Chem., 2022, 1–8 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*