
In situ regeneration of copper catalysts for long-term electrochemical CO2 reduction to multiple carbon products†
Cornelius A.
Obasanjo
 a,
Ali Shayesteh
Zeraati
b,
Hadi Shaker
Shiran
b,
Tu N.
Nguyen
ac,
Sharif Md.
Sadaf
d,
Md Golam
Kibria
*b and
Cao-Thang
Dinh
*a
a,
Ali Shayesteh
Zeraati
b,
Hadi Shaker
Shiran
b,
Tu N.
Nguyen
ac,
Sharif Md.
Sadaf
d,
Md Golam
Kibria
*b and
Cao-Thang
Dinh
*a
aDepartment of Chemical Engineering, Queen's University, Kingston, ON K7L 3N6, Canada. E-mail: caothang.dinh@queensu.ca
bDepartment of Chemical and Petroleum Engineering, University of Calgary, 2500 University Drive, NW Calgary, Alberta T2N 1N4, Canada. E-mail: md.kibria@ucalgary.ca
cHelen Scientific Research and Technological Development Co., Ltd, Ho Chi Minh City 700000, Vietnam
dCentre Energie, Matériaux et Télécommunications, Institut National de la Recherche Scientifique (INRS)-Université du Québec, 1650 Boulevard Lionel-Boulet, Varennes, Quebec J3X 1S2, Canada
First published on 19th July 2022
Abstract
The valorization of carbon dioxide (CO2) via electrochemical CO2 reduction (ECR) has attracted great interest as a pragmatic approach to tackle greenhouse gas emissions. Multiple carbon (C2+) products, such as ethylene (C2H4), ethanol (C2H5OH), and propanol (C3H7OH), are highly valuable chemicals and of great demand. Copper (Cu)-based catalysts are so far the only electrocatalytic materials that allow CO2 reduction to C2+ products at industrially relevant current densities (≥100 mA cm−2). However, most Cu-based catalysts are unstable in long-term reactions (>100 hours), with the main reasons being the potential-induced surface reconstruction, deposition of impurities, and catalyst aggregation and leaching, among others. Herein, we report an in situ catalyst regeneration strategy that can extend the operation time of Cu-based catalysts. By periodically adding segments of anodic currents to electrolysis, a Cu catalyst is partially oxidized to CuOx in each cycle, as confirmed by in situ Raman studies, leading to the restoration of the catalytically active sites for C2+ products. We found that the oxidation current density and time significantly affect the selectivity and stability of Cu catalysts. Applying this strategy to a Cu catalyst – which is stable for ∼5 h towards C2+ products during a continuous electroreduction under neutral-pH conditions, we were able to extend the operating time to ∼120 h in a flow cell system. The catalyst maintained a high faradaic efficiency (FE) for C2H4 of ≥50% at a fixed cathodic current density of 150 mA cm−2 for over 60 h and continued to operate with a C2H4 FE ≥ 40% for the entire length of the reaction time. This work opens up an avenue to enhance the stability of Cu electrocatalysts, via controlling the operating procedure during electrolysis.
 Cao-Thang Dinh | Dr. Cao Thang Dinh is an assistant professor of Chemical Engineering at Queen's University. He received his master's and PhD degrees in Chemical Engineering from Laval University in 2010 and 2014, respectively. From 2014 to 2019, Dr Dinh was a postdoctoral fellow in the Department of Electrical and Computer Engineering at the University of Toronto. His research focuses on developing electrochemical processes for renewable fuel and chemical production from carbon dioxide, water and renewable electricity. |
Introduction
The imminent threat of rising global temperatures has resulted in shifting weather patterns and disruption of nature's normal balance. Reducing carbon dioxide (CO2) emissions is critical and urgent; otherwise, humanity will be impacted by more and more severe weather events, together with food insecurity and global health issues. Carbon capture, utilization, and sequestration play an essential role in mitigating CO2 emissions.1,2 The development of CO2 utilization technologies such as electrochemical CO2 reduction (ECR) offers a pragmatic strategy to tackle emissions,2 as CO2 is converted into numerous valuable products without concerns of its immediate re-emission to the atmosphere.3A wide range of electrocatalytic materials for ECR have been investigated and reported in the literature. Electrocatalysts with high selectivity (FE > 80%) and long-term stability (>100 h) when performing at industrially relevant current densities (>100 mA cm−2) have been reported for single carbon (C1) products such as carbon monoxide4–7 and formic acid/formate.8–10 Cu and Cu-based catalysts have drawn great interest for ECR studies, not only because of their natural abundance and low cost, but also due to their unique feature as the only catalysts capable of converting CO2 into valuable C2+ products at high current density.11–17 Recent improvements in Cu-based catalysts and cell design have promoted ECR at elevated current densities even beyond 1 A cm−2.14 In some cases, high selectivity under alkaline and neutral-pH conditions was achieved, with FE values in the range of 40–72% and 41–52% for C2H413,14 and C2H5OH,12,15 respectively. Interestingly some high-performing ECR Cu-based electrocatalysts with C2H4 ≥ 75%18,19 have also been demonstrated with stability ≤ 100 h and current density < 100 mA cm−2. Nevertheless, the reported stability of ECR has been limited to a few operating hours13,20,21 for these C2+ products. In most cases, the Cu-based catalysts are rapidly deactivated, particularly when ECR reactions are conducted in neutral-pH or acidic media, as the competing hydrogen evolution reaction (HER) prevails after a short period.22 Notably, performing ECR under alkaline conditions has previously been shown to extend the catalyst operating time while displaying good selectivity for C2+ products. However, under these conditions, CO2 directly reacts with KOH and forms bi/carbonate salts, necessitating additional energy expenses for electrolyte and CO2 regeneration.13 Thus, there is a need to design suitable electrocatalysts and/or devise electrolysis procedures that can stably produce C2+ products in neutral or acidic environments to make the ECR technology more economically competitive.
Improving the stability of Cu electrocatalysts in low pH media still remains a great challenge.20,23–26 A wide array of reasons for Cu catalyst electrode failure have been reported, including poisoning by reaction products,27 deposition of metal impurities from the electrolyte,28 potential-induced surface reconstruction exposing inactive facets,29–37 and dissolution and redistribution of the catalyst outer surface.38,39 For gas diffusion electrodes (GDEs), which allow ECR to operate at industrially relevant current densities, catalyst deactivation is also due to the flooding of the porous gas diffusion layer and the precipitation of bi/carbonate salts that block the transport of CO2 gas to catalytic sites. To address the instability of Cu-based catalysts, strategies such as pulse electrolysis25,26,40–42 or employing electrolytically-cleaned electrolytes have been reported.43 It is also worth knowing that pulsed ECR has also been extensively studied towards enhancing C2+ product selectivity.44–47 Pulsed potential electrolysis has been exploited towards enhancing the stability of Cu-based electrocatalysts. Jännsch Y. et al.26 used 25 s at −1.38 V and 5 s at −1.0 V (vs. Ag/AgCl) to increase the stability of C2H4 production from less than 8 h to at least 16 h. Engelbrecht A. et al.25 extended a 16 h potentiostatic electrolysis to about 85 h and 95 h via variation of cathodic pulse potential (tc = 25 s, ta = 5 s, Uc = −1.6 V, Ua = −0.18 V, vs. Ag/AgCl) in combination with two different modes of electrolyte management, respectively. They both achieved C2H4 FEs below 40% which were demonstrated at current densities below 100 mA cm−2. However, to the best of our knowledge, none of these approaches have been able to demonstrate long-term stability towards C2+ products with high FE and at a high reaction rate in a flow cell configuration.
Herein, we report an in situ surface regeneration strategy to extend the operating time of a Cu electrocatalyst in neutral-pH media. The strategy is based on previous observation that oxide-derived Cu (OD-Cu) catalysts are effective for selective CO2 reduction to C2+ products.48,49 We exploited the copper oxide formed during the regeneration step of a thoroughly optimized oxidation and reduction cycle to target an extended ECR to C2+ products. With this in situ regeneration strategy, we achieved an order of magnitude improvement in operating time, with the Cu electrode operating at a cathodic current density of 150 mA cm−2 for ∼120 h. It maintained FE values for C2H4 ≥ 50% for over 60 h and remained operating with C2H4 FE ≥ 40% for the entire reaction time.
Results and discussion
Performance of Cu/PTFE catalysts at high current densities in a neutral-pH electrolyte
To fabricate Cu gas diffusion electrodes, we deposited metallic Cu on a porous polytetrafluoroethylene (PTFE) membrane with an average pore size of 0.45 microns, using the sputtering technique. The PTFE membrane was chosen because it has been demonstrated to be a durable gas diffusion layer, significantly suppressing flooding, allowing stable ECR to C2+ products in flow-cell configurations under highly alkaline conditions.13 The nominal thickness of the Cu layer was controlled, varying from 200 to 1000 nm. The morphologies of Cu/PTFE samples were characterized by scanning electron microscopy (SEM) (Fig. 1a–c). Cu particles were conformally deposited on PTFE fibers, forming a continuous Cu layer shell on the PTFE fiber core. The size of the Cu/PTFE fiber increases with increasing the Cu nominal thickness, leading to smaller pore size in the PTFE membrane (Fig. 1a–c and S1†). High magnification images show a similar Cu surface texture of small Cu particles with multiple grain boundaries for all Cu thicknesses (Fig. 1a–c and S1†).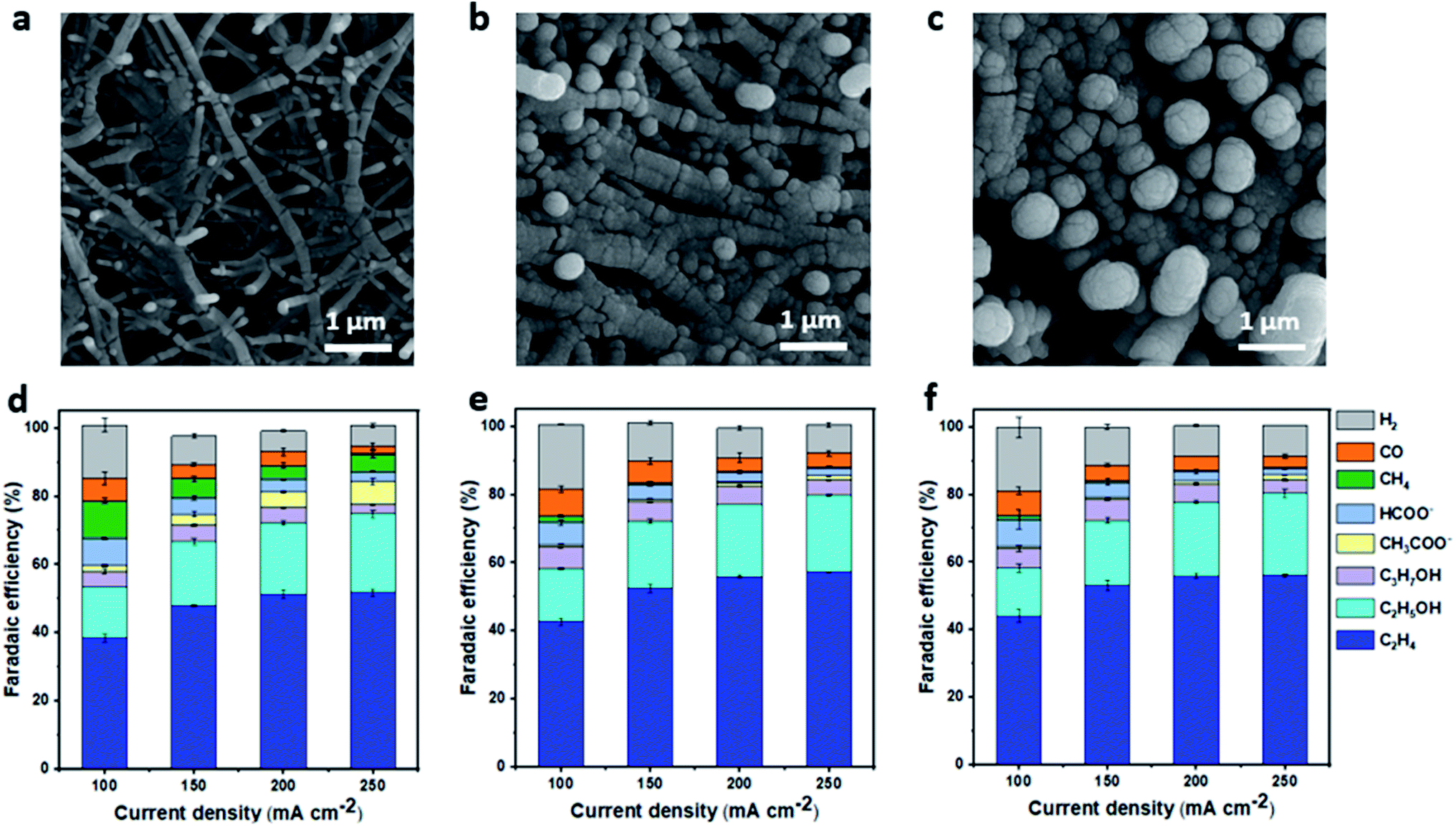 | ||
| Fig. 1 Characterization and ECR selectivity of Cu/PTFE. Scanning electron microscopy (SEM) images of Cu/PTFE with nominal Cu thicknesses of 200 nm (a), 700 nm (b) and 1000 nm (c). CO2 reduction product distribution at different cathodic current densities in 1 M KHCO3 electrolyte using Cu/PTFE with nominal thicknesses of 200 nm (d), 700 nm (e) and 1000 nm (f). The FE error bar was calculated based on data collected from different experimental trials (n = 3). Potential data for the determination of ECR selectivity of Cu/PTFE for all samples are provided in the ESI (Fig. S3).† | ||
ECR performance of Cu/PTFE GDEs was evaluated with a flow-cell reactor using 1 M KHCO3 catholyte. A bipolar membrane was used to separate the anode and cathode compartments. Nickel (Ni) foam and 1 M KOH solution were used as the anode and anolyte, respectively. The ECR of Cu/PTFE catalysts was studied in the cathodic current density range of 100–250 mA cm−2 using chronopotentiometry. Gaseous products were analyzed with in-line gas chromatography while liquid products were collected and analyzed using nuclear magnetic resonance (NMR) (Fig. S2†). All Cu/PTFE samples showed FE values for C2H4 between 38 and 55% in the studied current range, with a total C2+ (C2H4, C2H5OH, C3H7OH, and C2H3O2−) FE of >70% achieved at current densities of 150–250 mA cm−2 (Fig. 1d–f). This performance is consistent with previous reports on Cu/PTFE catalysts using similar testing conditions.21,50 We observed that Cu thickness showed little impact on the overall product selectivity within the explored current density range. While the total C2+ was lower at 100 mA cm−2 for all Cu/PTFE samples, the value increased about 15–20% at current densities between 150 and 250 mA cm−2. At these higher current densities, the Cu/PTFE GDEs maintained a good C2+ FE of >70%. However, at current densities beyond 300 mA cm−2 we observed a decrease in the total C2+ FE with a corresponding increase in H2 and CH4. The FE value of CH4 was found to be the highest on the 200 nm Cu/PTFE sample and was approximately 10 times higher at all current densities explored. These results suggest that the thickness of Cu/PTFE can be optimized to either achieve a high FE for C2+ products (thick Cu/PTFE) or a high FE for CH4 (thin Cu/PTFE).
To study the stability of the GDEs, we performed the ECR reaction at a fixed current density of 150 mA cm−2 and analyzed the gaseous products over time. The thickness of the Cu layer had a significant effect on the stability of Cu/PTFE. For the 200 nm Cu/PTFE, the C2H4 FE was stable at around 50% for about 1 h before it started decreasing rapidly, reaching 15% after 1.5 h of continuous reaction (Fig. 2a). After 1 h of reaction, the FE values of both CH4 and H2 started increasing, with H2 being the dominant product after 1.5 h (Fig. 2a–c). The stability of Cu/PTFE catalysts was significantly improved with an increase in Cu nominal thickness. The 1000 nm Cu/PTFE maintained a C2H4 FE of 50% for 5 hours (Fig. 2a). For all Cu/PTFE samples, CH4 and H2 were the two main products when the C2H4 FE started decreasing (Fig. 2b and c), while CO selectivity slowly decreased over time (Fig. S4†).
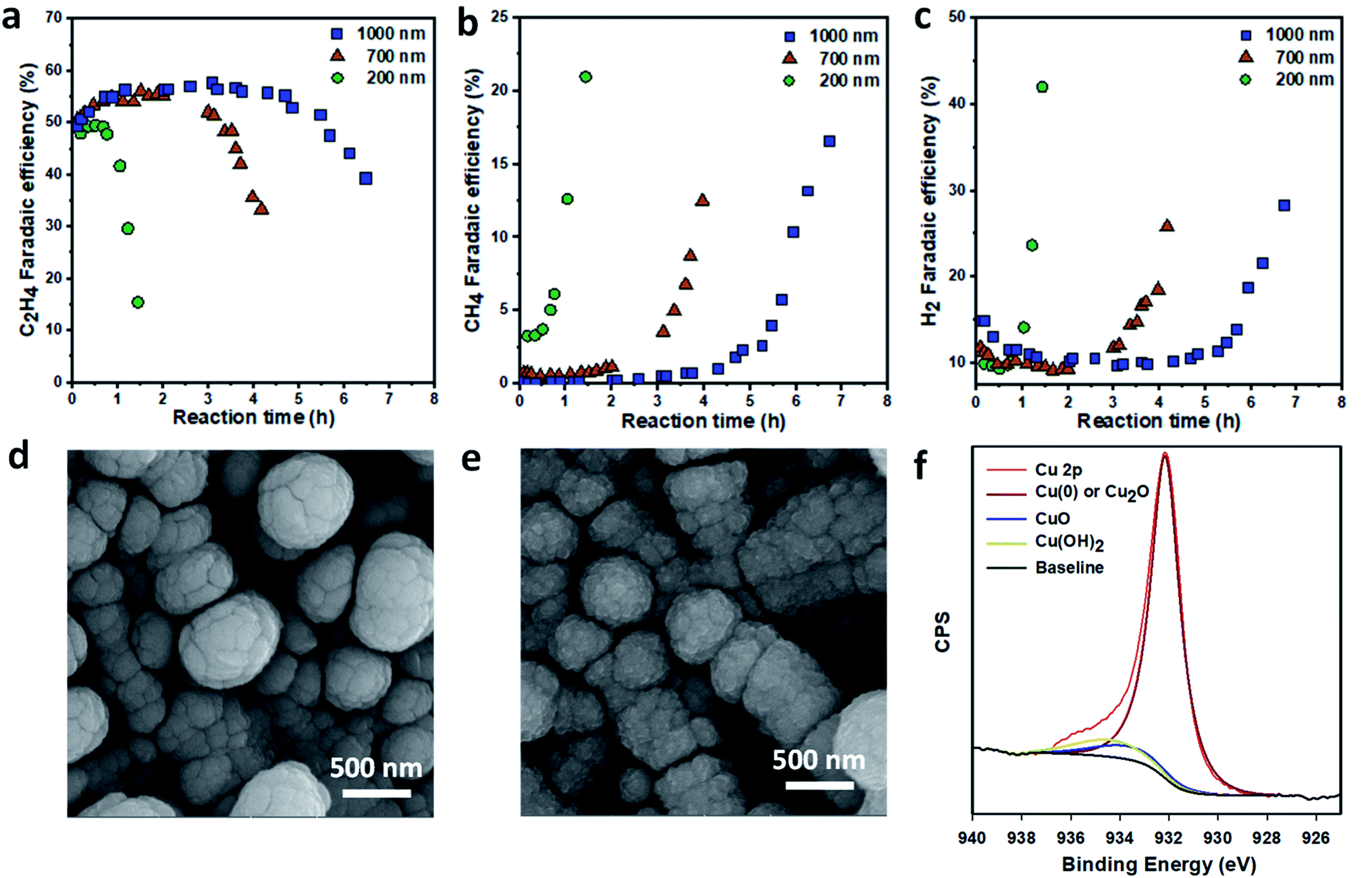 | ||
| Fig. 2 Stability of Cu/PTFE in ECR. Variation of gas products over reaction time at a constant current density of 150 mA cm−2 in 1 M KHCO3 electrolyte: (a) C2H4, (b) CH4 and (c) H2. SEM images of Cu/PTFE catalysts with a nominal thickness of 1000 nm before (d) and after continuous electrolysis for 7 hours (e). High resolution Cu 2p XPS spectrum of Cu/PTFE catalysts after continuous ECR tests showing the presence of metallic Cu, copper oxide and copper hydroxide (f). Chronopotentiometric plots for the three samples are provided in the ESI (Fig. S5–S7).† | ||
Previous studies on the stability of Cu catalysts for ECR in aqueous electrolytes have identified three main deactivation mechanisms among others. First, the Cu surface undergoes a reconstruction process, exposing the Cu sites (e.g. (111) facet) that are more selective towards CH4 formation and H2 production.51 This surface reconstruction usually occurs on the sub-10 nm scale. Second, metal impurities such as Fe and/or Ni present in the electrolyte, which can be deposited on the surface of Cu catalysts during electrolysis, form highly active sites for H2 production.52 The last mechanism involves the reduction of oxidized Cu species (Cu+, Cu2+) in Cu-based catalysts during the ECR process. Because oxidized Cu species and/or sub-surface oxygen are critical for CO2 reduction to C2+ products, the disappearance of these species makes the catalyst less selective toward C2+ products over time.53–55 Furthermore, for gas-phase ECR in flow systems using gas diffusion electrodes, the flooding of the gas diffusion layer and the precipitation of bi/carbonate salts can block the diffusion of the CO2 reactant, leading to the suppression of CO2 reduction.3
To identify the deactivation mechanism in our system, we first checked if flooding and salt precipitation were the main reasons. ECR at 150 mA cm−2 was performed continuously until the C2H4 FE decreased to around 25%. The current density was then reduced to 125, 100, 75 and 50 mA cm−2 and the gaseous products were analyzed accordingly. The FEs for H2 and CH4 remained high with lowering the current density, implying that CO2 diffusion limitation was not the reason for the drop in C2H4 FE (Fig. S8†). To further confirm that flooding was not the major mechanism for the deactivation of our Cu/PTFE sample, we investigated electrode flooding in our flow cell system using a capacitance change measurement.56 We quantified the electrochemical double-layer capacitance in an attempt to track the ingress of the electrolyte into the GDE during electrolysis. This capacitance measurement obtained through the cyclic voltammetry technique, in the non-faradaic region with variable scan rates, can offer insights into the changes that occurred at the electrode–electrolyte interface. The cyclic voltammetry was carried out between −0.05 V and −0.20 V vs. Ag/AgCl with scan rates of 10, 50, 100, 150 and 200 mV s−1. We observed no significant change in the electrochemical double-layer capacitance (Fig. S9–S11†). The capacitance was found to be around 0.5756 mF cm−2 at the open circuit potential before the start of continuous electrolysis, 0.5309 mF cm−2 after 3 h of electrolysis and 0.5143 mF cm−2 after approximately 6–7 h of continuous operation (Fig. S11†). The FE of C2H4 was about 56% after 3 h and decreased to about 42% at the end of the continuous operation.
To check for a change in the catalyst surface morphology, we compared the SEM images of the Cu/PTFE sample before and after continuous ECR tests (Fig. 2d and e). The surface texture of Cu/PTFE appeared to change after the reaction as the particle size became smaller. While this could be due to the surface construction, it may also have originated from the oxidation of the Cu surface when the sample was exposed to air after the reaction. High resolution Cu 2p X-ray photoelectron spectroscopy (XPS) of the sample (1000 nm) shows the presence of CuO and Cu(OH)2 after the reaction (Fig. 2f). The survey XPS spectra show no peaks for Fe and Ni in all samples while small peaks of C, F and Cl were detected (Fig. S12†). These data suggest that impurity could not be the main cause of lower C2H4 selectivity. We therefore reason that surface reconstruction and changing of the Cu oxidation state and sub-surface oxygen could be the main contributors to the deactivation of Cu/PTFE catalysts.
Regeneration of Cu catalysts by in situ oxidation
The development of highly selective OD-Cu catalysts for CO2 reduction to C2+ products48,49,57–60 motivated us to pursue an in situ regeneration of the deactivated Cu catalyst, using an electrochemical oxidation process to transform Cu back to Cu oxide. We reason that this oxidation step may address the main deactivation mechanisms of Cu-based catalysts since it can re-produce the selective OD-Cu catalysts. In addition, it might also enable the dissolution of metal impurities such as Fe on the Cu surface back to the electrolyte.To investigate the recovery of Cu catalyst selectivity toward C2H4via in situ electrochemical oxidation, we first performed continuous ECR tests at a current density of 150 mA cm−2 until the C2H4 FE decreased to around 10–15% (Fig. 3a). Next, we performed repeated oxidation–reduction cycles until a stable C2H4 FE was achieved (Fig. 3a). The oxidation current density was varied between 0.4 and 1 mA cm−2 while the reduction current density and time were fixed at 150 mA cm−2 and 5 min, respectively. The FE of C2H4 was analyzed at the end of each reduction cycle. The oxidation time was studied to maximize C2H4 FE. In all cases, the oxidation charges (Q = I × t) explored were either 36, 24, or 12 mC. As shown in Fig. 3b–e, the C2H4 FE increases after each oxidation–reduction cycle while the FEs for H2 and CH4 slowly decrease (Fig. S13–S16†). The FE for CO remains relatively constant with the oxidation–reduction cycle (Fig. S13–S16†). C2H4 FE values >50% were achieved at all oxidation current densities, with higher applied charges allowing the catalysts to operate with higher C2H4 FE for each oxidation current density. This suggests that a higher oxidation current density only needs a shorter time to recover the high FE for C2H4.
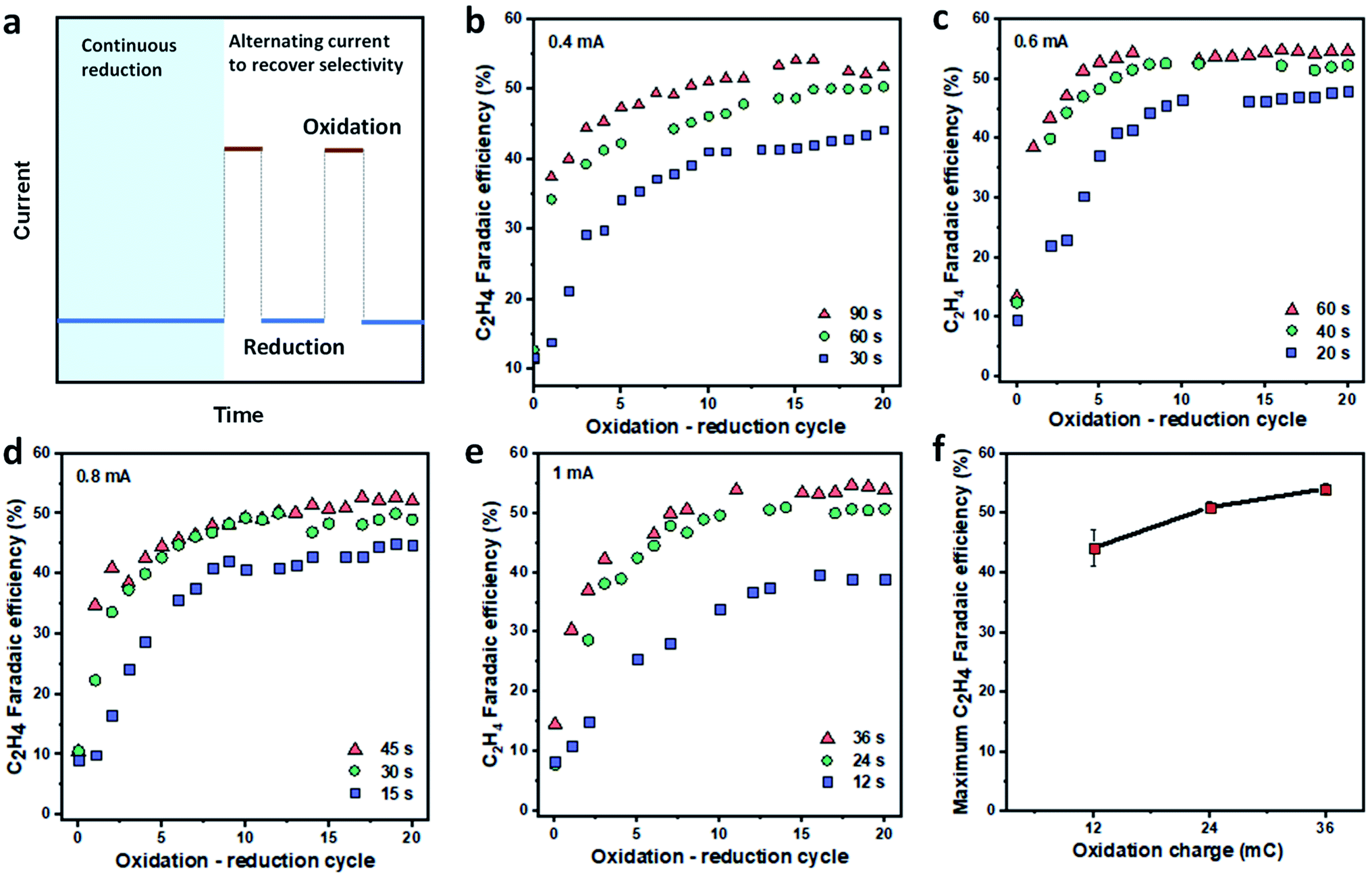 | ||
| Fig. 3 Regeneration of Cu catalysts by in situ oxidation. (a) Oxidation–reduction program for the recovery of C2H4 FE. All samples were operated continuously at a fixed reduction current density of 150 mA cm−2 until the C2H4 FE decreased to around 10–15%. The catalysts were then subjected to 20 repeated oxidation–reduction cycles. Each oxidation–reduction cycle involves an oxidation step at a designated current and time, followed by ECR at 150 mA cm−2 for 5 minutes. The gas products were analyzed at the end of each reduction cycle. The recovery of FE for C2H4 is shown with oxidation current densities of 0.4 mA cm−2 (b), 0.6 mA cm−2 (c), 0.8 mA cm−2 (d) and 1 mA cm−2 (e) at different oxidation periods. The dependence of maximum C2H4 FE on oxidation charge (f). The deviation was calculated based on data at different oxidation current densities but with similar oxidation charges (n = 4). The FE values for CO, H2 and CH4 during the oxidation–reduction cycle are provided in the ESI (Fig. S13–S16†). The morphology of Cu/PTFE after in situ electrochemical oxidation is presented in the ESI (Fig. S17†). | ||
The maximal FE for C2H4 was found to be a function of the oxidation charge regardless of the oxidation current density applied in our studied range (Fig. 3f) with a FE of 53–56% being recovered when the oxidation charge was ≥24 mC. In addition, at each given oxidation current density and time condition, we found that the C2H4 FE quickly recovered in the first 10 cycles before reaching a plateau. The presence of such a plateau suggests that (i) the 5 min ECR reaction after each oxidation step is not long enough to cause catalyst deactivation and (ii) the ratio between the catalytic sites for C2H4 and other products is dependent on the applied charge of the oxidation current density, which governs the final C2H4 FE.
In situ Raman studies on oxidized Cu species
To shed light on how the oxidation current density and time affect product selectivity, we performed operando Raman analysis. This technique allowed us to investigate the catalyst structure and surface adsorbates during reduction and oxidation. We first studied the surface of Cu/PTFE catalysts during a continuous ECR process. The CO intermediate is usually observed by surface enhanced Raman scattering (SERS) at three Raman shift regions: 280–290 cm−1 for the restricted rotation of adsorbed CO, 360–370 cm−1 for Cu–CO stretching, and 1800–2100 cm−1 for C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O stretching.61,62 All in situ Raman measurements in this study were performed using a 785 nm laser which exhibits suitable SERS activity. Signal enhancements were performed and control experiments with two different laser wavelengths were carried out to study the effect of laser excitement wavelength (Fig. S18†). The Raman shift in the range of 520–530 cm−1 can be assigned to the Cu–OH band. It has been shown that higher intensity of the Cu–CO stretching band can be assigned to a higher coverage of the CO intermediate and higher C2+ selectivity.63 A significant decrease in the intensity of restricted rotation of adsorbed CO and Cu–CO stretching compared to the Cu–OH can be seen by increasing the reduction time (Fig. 4a). This is a sign of the deactivation of copper by less CO coverage resulting in a reduced amount of C2H4. These observations are in agreement with the drop in C2H4 FE over the reaction time.
O stretching.61,62 All in situ Raman measurements in this study were performed using a 785 nm laser which exhibits suitable SERS activity. Signal enhancements were performed and control experiments with two different laser wavelengths were carried out to study the effect of laser excitement wavelength (Fig. S18†). The Raman shift in the range of 520–530 cm−1 can be assigned to the Cu–OH band. It has been shown that higher intensity of the Cu–CO stretching band can be assigned to a higher coverage of the CO intermediate and higher C2+ selectivity.63 A significant decrease in the intensity of restricted rotation of adsorbed CO and Cu–CO stretching compared to the Cu–OH can be seen by increasing the reduction time (Fig. 4a). This is a sign of the deactivation of copper by less CO coverage resulting in a reduced amount of C2H4. These observations are in agreement with the drop in C2H4 FE over the reaction time.
 | ||
| Fig. 4 In situ Raman studies on oxidized Cu species of Cu/PTFE. (a) In situ SERS of Cu/PTFE conducted at different times of reaction in KHCO3 electrolyte at a current density of 30 mA cm−2. (b) The structure and surface adsorbates during reduction and oxidation cycles at 0.6 mA cm−2 oxidation current density for 20 s. | ||
Given the observed removal of adsorbed CO species and a significant drop in C2H4 formation, we performed in situ Raman studies using the oxidation–reduction strategy as a remedy against this deactivation. As the oxidation current density was applied, the Cu–CO and Cu–OH species disappeared, and the CuOx bands emerged in the range of 450–700 cm−1. These oxide species were visible after 20 s of oxidation current (Fig. 4b). The CuOx bands disappeared entirely while the Cu–CO and Cu–OH species reappeared after the reduction cycle. It has been shown that these CuOx crystals reduced into small Cu grains developing OD catalysts which facilitated the C–C coupling and accounted for enhanced C2H4 selectivity.63–65 Interestingly, by applying cathodic current the restricted rotation of adsorbed CO and Cu–CO stretching is recovered, confirming the re-coverage of CO. By repeating the oxidation–reduction cycles, more intense Cu–CO bonds were formed, which indicates enhanced CO coverage. Roldán Cuenya et al.63 showed that the intensity of ratio of the Cu–CO stretching band to the CO rotation band can be correlated to the CO coverage and a higher ratio leads to more C–C coupling and C2+ formation. Our SERS analysis illustrates an increasing trend of this ratio with increasing the number of oxidation–reduction cycles. This confirms a better CO coverage on the surface of the catalyst leading to the sustained C2H4 FE (Fig. S19†).
Extending catalyst lifetime with the alternating current strategy
Having identified the oxidation conditions for recovering C2H4 FE, we set out to prolong the catalyst lifetime by performing an alternating oxidation–reduction current procedure (Fig. 5a). The reduction current density was fixed at 150 mA cm−2 while the oxidation current density and time, as well as the reduction time, were varied to optimize the operation conditions. We first fixed the oxidation charge of 36 mC and varied the oxidation current density (from 0.4 to 1 mA cm−2) and oxidation time (36 to 90 s). The reduction current density and time were 150 mA cm−2 and 5 min, respectively. We found that all operational procedures showed similar stability, maintaining C2H4 FE ≥ 50% for at least 10 hours (Fig. 5b), doubling the time of the continuous reduction procedure. The FE for CO slowly decreased while H2 and CH4 FEs increased over time (Fig. S20†). Therefore, varying the oxidation current density and time plays a negligible role if the oxidation charge is fixed.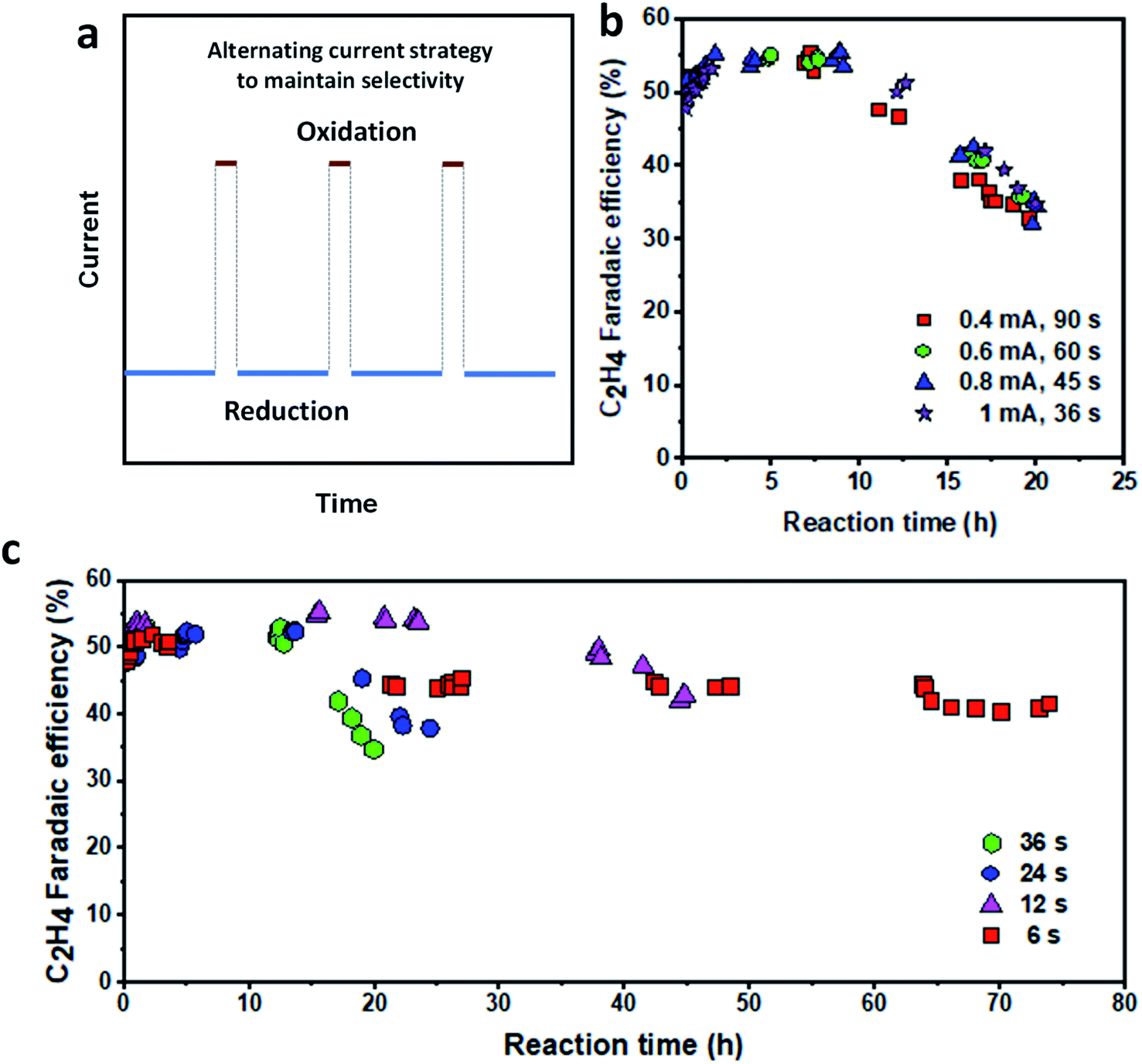 | ||
| Fig. 5 Stability of the Cu/PTFE catalysts with the alternating current strategy. (a) Schematic illustration of the alternating current strategy. (b) Oxidation charge optimization at a fixed reduction current density of 150 mA cm−2 for 5 min reduction time. The cathodic potential versus time plot is provided in Fig. S21.† (c) Oxidation time optimization at a fixed current density of 150 mA cm−2 for 5 min reduction time and an oxidation current density of 1 mA cm−2 (the oxidation time was varied between 6 s and 36 s). The cathodic potential versus time plot is provided in Fig. S23.† | ||
To further explore the effect of the oxidation charge, we then fixed the oxidation current density at 1 mA cm−2 and varied the oxidation time from 6 to 36 s. The reduction current density and time are 150 mA cm−2 and 5 min, respectively. While larger oxidation charge leads to higher C2H4 FE, the catalyst becomes less stable when the oxidation time (oxidation charge) is increased (Fig. 5c). With a 36 s oxidation time, the catalyst showed the highest C2H4 FE of 54% but was stable at above 50% for 14–16 h before dropping quickly to about 30% in 20 h of operation. In contrast, when the oxidation time was reduced to 6 s, the catalyst operated stably for up to 75 h, although with a lower C2H4 FE of 40% and above (Fig. 5c). Under all operating conditions, the FE of CO slowly decreased while those of H2 and CH4 increased steadily with the reaction time (Fig. S22†). These results suggest that the oxidation time could not be tuned to achieve both high C2H4 FE and long stability simultaneously. While the catalyst can operate with an initial C2H4 FE of about 50% at all oxidation times, each operating condition tends to temporarily arrive at a constant FE as mentioned earlier, beyond which the cumulative effect of the total oxidation time over the catalyst operating lifetime begins to significantly impact the catalyst performance. Therefore, it can be hypothesized that the total amount of charge (the product of a given oxidation charge and the number of times it was cycled) throughout the lifetime of the operating catalyst determines the overall stability of the Cu/PTFE catalyst electrode.
To extend the lifetime of the catalyst, we turned our attention to the reduction time. We reason that if the stability of the catalyst depends only on the oxidation charge, the overall operating time can be extended by increasing the reduction time. We fixed the reduction current density (150 mA cm−2), oxidation current density (1 mA cm−2) and time (24 s) and varied the reduction time. As shown in Fig. 6a, increasing the reduction time extended the overall catalyst lifetime (time to maintain a C2H4 FE ≥ 40%). Based upon our optimized operating conditions, the longest catalyst lifetime of ∼120 h was achieved at a reduction time of 45 min. At this condition, the C2H4 FE was maintained at over 50% for more than 60 h, which is around ten times longer than the continuous operation (5 h). The CO FE decreased rapidly in the first 20 h and is then relatively stable (Fig. S24†). The CH4 FE slowly increased over time while the H2 FE varied in the range of 10–15% (Fig. S25 and S26†). Furthermore, under these operating conditions, we found that a further increase of the reduction time and/or oxidation charge does not improve the catalyst lifetime, at least for when we aim to maintain a C2H4 FE ≥ 40%. An operation of the catalyst under the same conditions, but with a higher oxidation charge (60 mC), was carried out for ∼58 h (Fig. S28†).
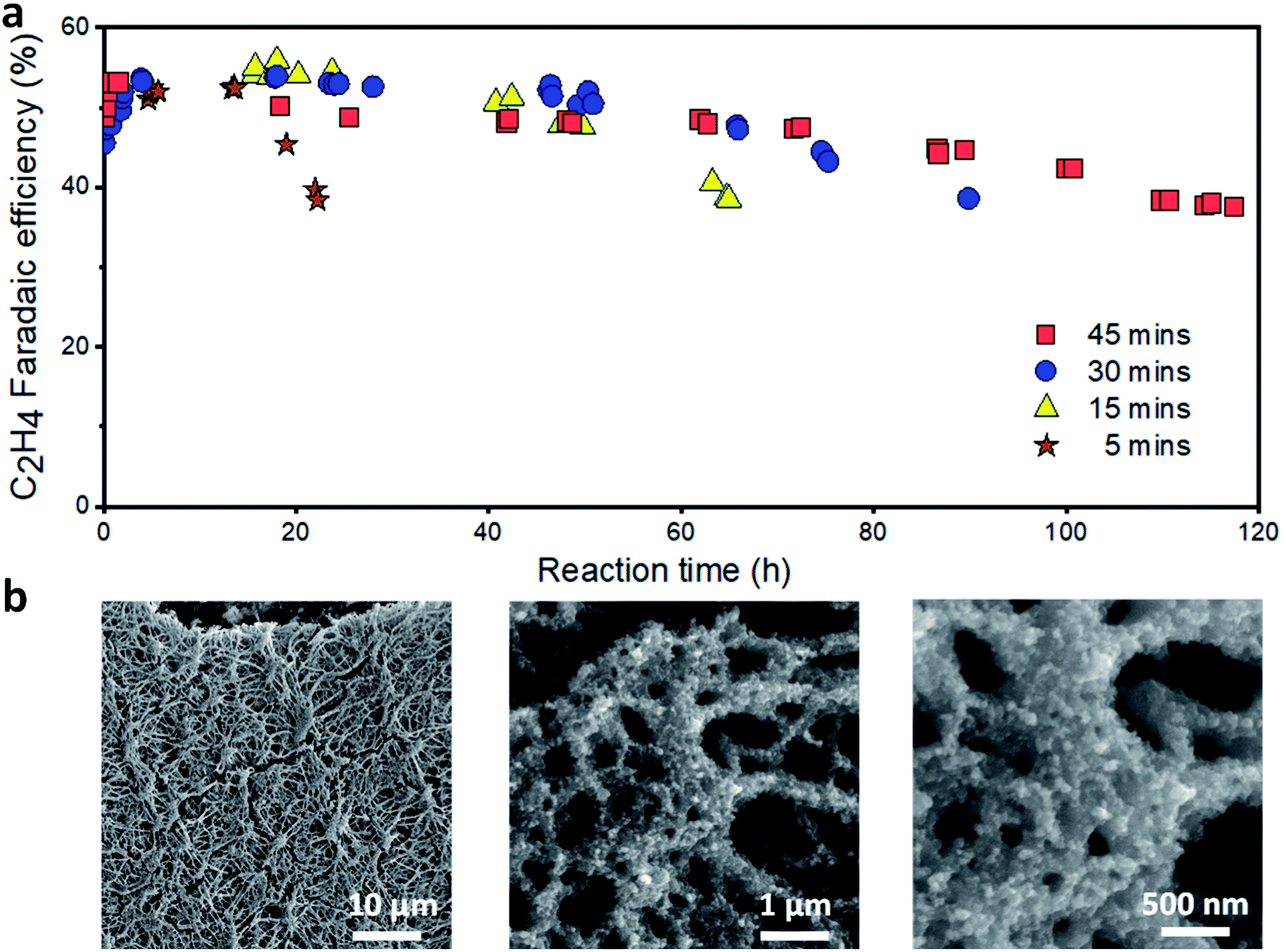 | ||
| Fig. 6 Stability of the Cu/PTFE catalysts with the optimized alternating current strategy in 1 M KHCO3. (a) Reduction time optimization (varying from 5 to 45 min) at 150 mA cm−2 and a fixed oxidation current density and time of 1 mA cm−2 and 24 s, respectively. (b) SEM of Cu/PTFE after the oxidation–reduction strategy using 45 min reduction at a current density of 150 mA cm−2 and a fixed oxidation current density and time of 1 mA cm−2 and 24 s, respectively. The cathodic potential versus time plot is provided in Fig. S27.† | ||
A limited Cu catalyst lifetime with the optimized oxidation and reduction conditions implies a major deactivation mechanism different from the potential induced surface reconstruction and surface impurities discussed above. To identify the major cause of the gradual decrease in C2H4 FE over the operating time, we characterized the Cu catalyst after the reaction using SEM. The catalyst layer (Fig. 6b) was found to be much thinner than that for the catalysts before (Fig. 2d) or after continuous ECR tests (Fig. 2e). We reason that the Cu at the Cu/PTFE interface and at the top of the catalyst layer can migrate during the oxidation cycle i.e., when Cu is oxidized to Cu oxide. This gradual migration of Cu species over the reaction time could lead to the redistribution of Cu on the PTFE substrate, forming Cu/PTFE locations with different Cu thicknesses. In a flow cell configuration, phenomena such as catalyst fragmentation66 and agglomeration,37,67 Ostwald ripening,36,68 particle dissolution and (electro-) redeposition39 of dissolved copper species in the form of new smaller Cu fragments, as we observed in Fig. 6b, cannot be neglected. Over time, these effects could result in the formation of some areas with very thick Cu while the Cu layer may disappear in other areas, leading to the degradation of the Cu/PTFE catalyst layer when using the alternating current approach. In addition, the impact of other associated failure mechanisms resulting from the type of electrolyzer system architecture being used can lead to a systemic failure of the overall cell system. For a flow cell architecture, as we employed in this study, the additional impact of flooding and salt formation within the PTFE pores cannot be ignored for an extended testing time. These failure mechanisms, through the interdependent nature of the cell system architecture, directly impact and combine with the major failure resulting from our strategy to suppress the stability performance of the electrolyzer system and thereby limit its overall operating time.
Conclusions
In this work, we have introduced an in situ regeneration of Cu catalysts for stable CO2 reduction to multiple carbon products. Our strategy involves an alternating current operating procedure in which the catalysts are periodically regenerated by the oxidizing Cu surface. By optimizing the oxidation and reduction current density and time, a ten-fold increase in Cu catalyst lifetime has been achieved. Our optimal oxidation–reduction strategy enables the Cu catalyst to operate for an extended time (∼120 h) and maintain a significant FE of ≥50% at a fixed current density of 150 mA cm−2 for the first 62 h, while the reaction continued with a selectivity of ≥40% for the entire length of the operation. In situ Raman characterization confirmed the presence of copper oxide species during the oxidation cycle. This approach could potentially open up an opportunity for the long-term use of Cu and Cu-based catalysts for effective ECR to C2+ products and the realization of the electrochemical CO2 conversion technology.Experimental
Electrode preparation
The copper cathode gas diffusion electrodes were prepared by magnetron sputtering using a Kurt J. Lesker PVD 75 vacuum deposition system. Pure Cu sputtering (99.99%) metal targets (3′′ diameter) were used with a DC source in pure argon plasma in a sputter-down configuration. The samples were affixed to a 150 mm diameter rotating platen with Kapton tape, with a target-to-platen distance of ∼165 mm. Cu was sputtered onto a porous hydrophobic polytetrafluoroethylene (PTFE) gas diffusion layer with a 450 nm mean pore size using 120 W DC power and 4 mTorr pressure to achieve a deposition rate in the range of 0.76–1.06 angstroms per second. Approximately 200 nm, 700 nm, and 1000 nm nominally thick Cu films were sputtered onto the PTFE substrate.Electrochemical CO2 reduction
Electrochemical CO2 reduction experiments were performed in a customized gas-fed flow cell configuration. The flow cell is made up of three compartments which are for CO2 gas, catholyte, and anolyte. Aqueous solutions of 1 M KHCO3 (Sigma-Aldrich) and 1 M KOH (Sigma-Aldrich) were used as the catholyte and anolyte, respectively. A bipolar membrane (Fumasep) was used for the experiment. The sputtered Cu/PTFE gas diffusion electrode (GDE), which is the working electrode, was carefully positioned between the catholyte chamber and a continuous flow of CO2 gas stream through the back of the cathode compartment. The catalyst side of the cathode compartment of the GDE was made to face the electrolyte. The cathode chamber was equipped with a leak-free Ag/AgCl in 3 M KCl reference electrode. The orientation of the bipolar membrane used was reverse biased with respect to the electrodes, where the cation-exchange layer (CEL) was made to face the cathode and the anion-exchange layer (AEL) was made to face the anode, thus maintaining separate pH in both sides of the electrolyzer compartment. Nickel foam (MTI Corp.) was used as the counter electrode in the anode compartment. Gaseous CO2 was allowed to flow past the back of the GDL at a constant flow of 30 sccm by means of a mass flow controller. Since only a fraction of the CO2 gas is used in the reaction process towards CO2RR products, the electrolyzer outlet flow rate from the CO2 gas chamber was measured using a digital volumetric flow meter and was used in the calculation of the faradaic efficiency of the reaction products. The electrolytes were circulated in both compartments using peristaltic pumps. The electrochemical measurement experiments were performed using a potentiostat (Metrohm). The electrochemical potentials were compared against Ag/AgCl (3 M KCl) reference.The gaseous products were quantified using online gas chromatography (GC, PerkinElmer Clarus 590). The GC was equipped with a thermal conductivity detector (TCD) and a flame ionization detector (FID). The chromatography instrument was directly connected to the outlet of the CO2 gas chamber of the customized gas-fed flow cell for continuous online analysis. The liquid products were analyzed using nuclear magnetic resonance spectroscopy (NMR). The 1H NMR spectra of freshly collected liquid products were acquired on an Auto-400 ultrashield Bruker instrument operating at a denoted spectrometer frequency given in megahertz (MHz) at 25 °C in D2O using water suppression mode, with dimethyl sulfoxide (DMSO) as a reference.
All potential data were presented versus Ag/AgCl. However, iR-compensation was performed, and the details are presented in the supplementary section (Fig. S29 and S30†).
Capacitance change measurement
The double layer capacitance was evaluated by regressing the measured working electrode charging current as a function of potential scan rate. The scan rates of 10, 50, 100, 150 and 200 mV s−1 were applied. The potential range of −0.20 V to −0.05 V vs. Ag/AgCl was chosen only within limits where the HER and OER would be avoided and where capacitive current could be adequately evaluated. Charging current was determined as an average of the absolute values of the anodic and cathodic currents. The capacitance was determined as the value of the slope of the charging current versus the potential scan rate.Sample characterization
The surface morphology and composition of the catalysts were investigated using scanning electron microscopy (TESCAN-SEM system) operated at 5–10 kV acceleration voltage. X-ray photoelectron spectroscopy (XPS) measurements were carried out with a VG Escalab 220i-XL spectrometer using polychromatic Al Kα radiation (1486.6 eV).In situ Raman
Raman spectroscopy was executed with a Renishaw Raman spectrometer equipped with a λ = 785 nm laser as the excitation source and a 63× immersion objective lens. All in situ experiments were carried out with a laser power of 0.1 mW over 10 acquisitions with an exposure time of 10 seconds. The custom-designed flow cell had an electrolyte reservoir and a gas channel separated by a gas diffusion electrode. The immersion objective was dipped in the electrolyte reservoir to collect the spectra. A Pt wire as the anode, sputtered copper on PTFE as the cathode, and Ag/AgCl as the reference electrode were dipped in the electrolyte reservoir filled with 1.0 M KHCO3 electrolyte. The area of the cathode in this configuration was 1 cm2. During the experiments, a continuous 30 sccm CO2 gas flow was delivered to the cathode through the gas channel. The sputtered copper samples were reduced at 30 mA cm−2 for at least 2 h before the oxidation/reduction cycles.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. A. O. acknowledges the financial support from Queen’s University. T. N. N. acknowledges the support from Helen Co., Ltd. M. G. K. acknowledges the support from Canada First Research Excellence Fund at the University of Calgary. C. T. D. acknowledges the financial support from the Natural Sciences and Engineering Research Council of Canada (NSERC) and Queen's University. The authors thank Dr Graham Gibson at Nanofabrication Kingston for his assistance in sample preparation.Notes and references
- S. Overa, B. H. Ko, Y. Zhao and F. Jiao, Electrochemical approaches for CO2 conversion to chemicals: a journey toward practical applications, Acc. Chem. Res., 2022, 55(5), 638–648 CrossRef CAS PubMed.
- S. Nitopi, et al., Progress and perspectives of electrochemical CO2 reduction on copper in aqueous electrolyte, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- D. Wakerley, et al., Gas diffusion electrodes, reactor designs and key metrics of low-temperature CO2 electrolysers, Nat. Energy, 2022, 7, 130–143 CrossRef CAS.
- B. Endrodi, et al., High carbonate ion conductance of a robust PiperION membrane allows industrial current density and conversion in a zero-gap carbon dioxide electrolyzer cell, Energy Environ. Sci., 2020, 13, 4098–4105 RSC.
- C. T. Dinh, F. P. García De Arquer, D. Sinton and E. H. Sargent, High rate, selective, and stable electroreduction of CO2 to CO in basic and neutral media, ACS Energy Lett., 2018, 3, 2835–2840 CrossRef CAS.
- H. Yang, et al., Carbon dioxide electroreduction on single-atom nickel decorated carbon membranes with industry compatible current densities, Nat. Commun., 2020, 11, 1–8 CrossRef PubMed.
- R. B. Kutz, et al., Sustainion imidazolium-functionalized polymers for carbon dioxide electrolysis, Energy Technol., 2017, 5, 929–936 CrossRef CAS.
- L. Fan, C. Xia, P. Zhu, Y. Lu and H. Wang, Electrochemical CO2 reduction to high-concentration pure formic acid solutions in an all-solid-state reactor, Nat. Commun., 2020, 11, 3633 CrossRef CAS PubMed.
- I. Grigioni, et al., CO2 electroreduction to formate at a partial current density of 930 mA cm−2 with InP colloidal quantum dot derived catalysts, ACS Energy Lett., 2021, 6, 79–84 CrossRef CAS.
- Y. Chen, et al., A robust, scalable platform for the electrochemical conversion of CO2 to formate: identifying pathways to higher energy efficiencies, ACS Energy Lett., 2020, 5, 1825–1833 CrossRef CAS.
- C. Choi, et al., Highly active and stable stepped Cu surface for enhanced electrochemical CO2 reduction to C2H4, Nat. Catal., 2020, 3, 804–812 CrossRef CAS.
- Y. C. Li, et al., Binding site diversity promotes CO2 electroreduction to ethanol, J. Am. Chem. Soc., 2019, 141, 8584–8591 CrossRef CAS PubMed.
- C. T. Dinh, et al., CO2 electroreduction to ethylene via hydroxide-mediated copper catalysis at an abrupt interface, Sci. 80, 2018, 360, 783–787 CrossRef CAS PubMed.
- F. P. García de Arquer, et al., CO2 electrolysis to multicarbon products at activities greater than 1 A cm−2, Sci. 80, 2020, 367, 661–666 CrossRef PubMed.
- X. Wang, et al., Efficient electrically powered CO2-to-ethanol via suppression of deoxygenation, Nat. Energy, 2020, 5, 478–486 CrossRef CAS.
- T. T. H. Hoang, et al., Nanoporous copper-silver alloys by additive-controlled electrodeposition for the selective electroreduction of CO2 to ethylene and ethanol, J. Am. Chem. Soc., 2018, 140, 5791–5797 CrossRef CAS PubMed.
- J. Kim, et al., Branched copper oxide nanoparticles induce highly selective ethylene production by electrochemical carbon dioxide reduction, J. Am. Chem. Soc., 2019, 141, 6986–6994 CrossRef CAS PubMed.
- B. Zhang, et al., Highly electrocatalytic ethylene production from CO2 on nanodefective Cu nanosheets, J. Am. Chem. Soc., 2020, 142, 13606–13613 CrossRef CAS PubMed.
- S. Sultan, et al., Interface rich CuO/Al2CuO4 surface for selective ethylene production from electrochemical CO2 conversion, Energy Environ. Sci., 2022, 15, 2397–2409 RSC.
- Y. Xu, et al., Self-cleaning CO2 reduction systems: unsteady electrochemical forcing enables stability, ACS Energy Lett., 2021, 6, 809–815 CrossRef CAS.
- F. Li, et al., Molecular tuning of CO2-to-ethylene conversion, Nature, 2020, 577, 509–513 CrossRef CAS PubMed.
- Y. Wang, et al., Catalyst synthesis under CO2 electroreduction favours faceting and promotes renewable fuels electrosynthesis, Nat. Catal., 2020, 3, 98–106 CrossRef CAS.
- Z. Weng, et al., Self-cleaning catalyst electrodes for stabilized CO2 reduction to hydrocarbons, Angew. Chem., Int. Ed., 2017, 56, 13135–13139 CrossRef CAS PubMed.
- C. S. Chen, et al., Stable and selective electrochemical reduction of carbon dioxide to ethylene on copper mesocrystals, Catal. Sci. Technol., 2015, 5, 161–168 RSC.
- A. Engelbrecht, et al., On the electrochemical CO2 reduction at copper sheet electrodes with enhanced long-term stability by pulsed electrolysis, J. Electrochem. Soc., 2018, 165, J3059–J3068 CrossRef CAS.
- Y. Jännsch, et al., Pulsed potential electrochemical CO2 reduction for enhanced stability and catalyst reactivation of copper electrodes, Electrochem. Commun., 2020, 121, 106861 CrossRef.
- D. W. DeWulf, T. Jin and A. J. Bard, Electrochemical and surface studies of carbon dioxide reduction to methane and ethylene at copper electrodes in aqueous solutions, J. Electrochem. Soc., 1989, 136, 1686–1691 CrossRef CAS.
- Y. Hori, et al., ‘Deactivation of copper electrode’ in electrochemical reduction of CO2, Electrochim. Acta, 2005, 50, 5354–5369 CrossRef CAS.
- G. H. Simon, C. S. Kley and B. Roldan Cuenya, Potential-dependent morphology of copper catalysts during CO2 electroreduction revealed by in situ atomic force microscopy, Angew. Chem., Int. Ed., 2021, 60, 2561–2568 CrossRef CAS PubMed.
- T. H. Phan, et al., Emergence of potential-controlled Cu-nanocuboids and graphene-covered Cu-nanocuboids under operando CO2 electroreduction, Nano Lett., 2021, 21(5), 2059–2065 CrossRef CAS PubMed.
- H. Matsushima, A. Taranovskyy, C. Haak, Y. Gründer and O. M. Magnussen, Reconstruction of Cu(100) electrode surfaces during hydrogen evolution, J. Am. Chem. Soc., 2009, 131, 10362–10363 CrossRef CAS PubMed.
- Y. G. Kim, J. H. Baricuatro, A. Javier, J. M. Gregoire and M. P. Soriaga, The evolution of the polycrystalline copper surface, first to Cu(111) and then to Cu(100), at a fixed CO2RR potential: a study by operando EC-STM, Langmuir, 2014, 30, 15053–15056 CrossRef CAS PubMed.
- F. Scholten, K. L. C. Nguyen, J. P. Bruce, M. Heyde and B. Roldan Cuenya, Identifying structure–selectivity correlations in the electrochemical reduction of CO2: a comparison of well-ordered atomically clean and chemically etched copper single-crystal surfaces, Angew. Chem., Int. Ed., 2021, 60, 19169–19175 CrossRef CAS PubMed.
- Y. G. Kim, et al., Surface reconstruction of pure-Cu single-crystal electrodes under CO-reduction potentials in alkaline solutions: a study by seriatim ECSTM-DEMS, J. Electroanal. Chem., 2016, 780, 290–295 CrossRef CAS.
- Y. G. Kim, J. H. Baricuatro and M. P. Soriaga, Surface reconstruction of polycrystalline Cu electrodes in aqueous KHCO3 electrolyte at potentials in the early stages of CO2 reduction, Electrocatalysis, 2018, 9, 526–530 CrossRef CAS.
- S. Popović, et al., Stability and degradation mechanisms of copper-based catalysts for electrochemical CO2 reduction, Angew. Chem., Int. Ed., 2020, 59, 14736–14746 CrossRef PubMed.
- J. Huang, et al., Potential-induced nanoclustering of metallic catalysts during electrochemical CO2 reduction, Nat. Commun., 2018, 9, 1–9 CrossRef PubMed.
- F. D. Speck and S. Cherevko, Electrochemical copper dissolution: A benchmark for stable CO2 reduction on copper electrocatalysts, Electrochem. Commun., 2020, 115, 0–3 CrossRef CAS.
- S. Popovic, M. Bele and N. Hodnik, Reconstruction of copper nanoparticles at electrochemical CO2 reduction reaction conditions occurs via two-step dissolution/redeposition mechanism, ChemElectroChem, 2021, 8, 2634–2639 CrossRef CAS.
- J. Yano, T. Morita, K. Shimano, Y. Nagami and S. Yamasaki, Selective ethylene formation by pulse-mode electrochemical reduction of carbon dioxide using copper and copper-oxide electrodes, J. Solid State Electrochem., 2007, 11, 554–557 CrossRef CAS.
- R. Shiratsuchi, Y. Aikoh and G. Nogami, Pulsed electroreduction of CO2 on copper electrodes, J. Electrochem. Soc., 1993, 140, 3479–3482 CrossRef CAS.
- R. Shiratsuchi, Y. Aikoh and G. Nogami, Pulsed electroreduction of CO2 on copper electrodes - II, J. Electrochem. Soc., 1994, 140, 3479–3482 CrossRef.
- Y. Hori, Electrochemical CO2 reduction on metal electrodes, Mod. Asp. Electrochem., 2008, 42, 89–189 CAS.
- H. S. Jeon, et al., Selectivity control of Cu nanocrystals in a gas-fed flow cell through CO2 pulsed electroreduction, J. Am. Chem. Soc., 2021, 143(19), 7578–7587 CrossRef CAS PubMed.
- R. C. DiDomenico and T. Hanrath, Pulse symmetry impacts the C2 product selectivity in pulsed electrochemical CO2 reduction, ACS Energy Lett., 2021, 292–299 Search PubMed.
- C. Kim, L.-C. Weng and A. T. Bell, Impact of pulsed electrochemical reduction of CO2 on the formation of C2+ products over Cu, ACS Catal., 2020, 10, 12403–12413 CrossRef CAS.
- K. W. Kimura, et al., Controlled selectivity of CO2 reduction on copper by pulsing the electrochemical potential, ChemSusChem, 2018, 11, 1781–1786 CrossRef CAS PubMed.
- Q. Lei, et al., Investigating the origin of enhanced C2+ selectivity in oxide-/hydroxide-derived copper electrodes during CO2 electroreduction, J. Am. Chem. Soc., 2020, 142, 4213–4222 CrossRef CAS PubMed.
- H. Mistry, et al., Highly selective plasma-activated copper catalysts for carbon dioxide reduction to ethylene, Nat. Commun., 2016, 7, 12123 CrossRef PubMed.
- F. Li, et al., Cooperative CO2-to-ethanol conversion via enriched intermediates at molecule–metal catalyst interfaces, Nat. Catal., 2020, 3, 75–82 CrossRef CAS.
- W. Luo, X. Nie, M. J. Janik and A. Asthagiri, Facet dependence of CO2 reduction paths on Cu electrodes, ACS Catal., 2016, 6, 219–229 CrossRef CAS.
- S. J. Raaijman, N. Arulmozhi and M. T. M. Koper, Morphological stability of copper surfaces under reducing conditions, ACS Appl. Mater. Interfaces, 2021, 13(41), 48730–48744 CrossRef CAS PubMed.
- G. Liu, et al., CO2 reduction on pure Cu produces only H2 after subsurface O is depleted: theory and experiment, Proc. Natl. Acad. Sci. U. S. A., 2021, 118(23), e2012649118 CrossRef CAS PubMed.
- H. Y. Wang, et al., Direct evidence of subsurface oxygen formation in oxide-derived Cu by X-ray photoelectron spectroscopy, Angew. Chem., Int. Ed., 2022, 61, e202111021 CAS.
- W. Zhang, et al., Atypical oxygen-bearing copper boosts ethylene selectivity toward electrocatalytic CO2 reduction, J. Am. Chem. Soc., 2020, 142, 11417–11427 CrossRef CAS PubMed.
- M. E. Leonard, L. E. Clarke, A. Forner-Cuenca, S. M. Brown and F. R. Brushett, Investigating electrode flooding in a flowing electrolyte, gas-fed carbon dioxide electrolyzer, ChemSusChem, 2020, 13, 400–411 CrossRef CAS PubMed.
- Y. Lum, B. Yue, P. Lobaccaro, A. T. Bell and J. W. Ager, Optimizing C-C coupling on oxide-derived copper catalysts for electrochemical CO2 reduction, J. Phys. Chem. C, 2017, 121, 14191–14203 CrossRef CAS.
- D. Cheng, et al., The nature of active sites for carbon dioxide electroreduction over oxide-derived copper catalysts, Nat. Commun., 2021, 12, 1–8 CrossRef PubMed.
- C. Chen, et al., The in situ study of surface species and structures of oxide-derived copper catalysts for electrochemical CO2 reduction, Chem. Sci., 2021, 12, 5938–5943 RSC.
- A. Verdaguer-Casadevall, et al., Probing the active surface sites for CO reduction on oxide-derived copper electrocatalysts, J. Am. Chem. Soc., 2015, 137, 9808–9811 CrossRef CAS PubMed.
- M. Moradzaman and G. Mul, In situ Raman study of potential-dependent surface adsorbed carbonate, CO, OH, and C species on Cu electrodes during electrochemical reduction of CO2, ChemElectroChem, 2021, 8, 1478–1485 CrossRef CAS.
- J. Wang, H. Y. Tan, Y. Zhu, H. Chu and H. M. Chen, Linking the dynamic chemical state of catalysts with the product profile of electrocatalytic CO2 reduction, Angew. Chem., Int. Ed., 2021, 60, 17254 CrossRef CAS PubMed.
- C. Zhan, et al., Revealing the CO coverage-driven C-C coupling mechanism for electrochemical CO2 reduction on Cu2O nanocubes via Operando Raman spectroscopy, ACS Catal., 2021, 11, 7694–7701 CrossRef CAS PubMed.
- S. C. Lin, et al., Operando time-resolved X-ray absorption spectroscopy reveals the chemical nature enabling highly selective CO2 reduction, Nat. Commun., 2020, 11, 1–12 CrossRef PubMed.
- R. M. Arán-Ais, F. Scholten, S. Kunze, R. Rizo and B. Roldan Cuenya, The role of in situ generated morphological motifs and Cu(i) species in C2+ product selectivity during CO2 pulsed electroreduction, Nat. Energy, 2020, 5, 317–325 CrossRef.
- H. Jung, et al., Electrochemical fragmentation of Cu2O nanoparticles enhancing selective C-C coupling from CO2 reduction reaction, J. Am. Chem. Soc., 2019, 141, 4624–4633 CrossRef CAS PubMed.
- J. C. Meier, et al., Stability investigations of electrocatalysts on the nanoscale, Energy Environ. Sci., 2012, 5, 9319–9330 RSC.
- N. Hodnik, G. Dehm and K. J. J. Mayrhofer, Importance and challenges of electrochemical in situ liquid cell electron microscopy for energy conversion research, Acc. Chem. Res., 2016, 49, 2015–2022 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ta02709g |
| This journal is © The Royal Society of Chemistry 2022 |