
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoinduced inverse Sonogashira coupling reaction†
Lizhu
Zhang
a,
Cunbo
Wei
a,
Jiawen
Wu
a,
Dan
Liu
a,
Yinchao
Yao
a,
Zhuo
Chen
 a,
Jianxun
Liu
b,
Chang-Jiang
Yao
b,
Dinghua
Li
a,
Rongjie
Yang
a and
Zhonghua
Xia
*a
a,
Jianxun
Liu
b,
Chang-Jiang
Yao
b,
Dinghua
Li
a,
Rongjie
Yang
a and
Zhonghua
Xia
*a
aSchool of Materials Science & Engineering, Beijing Institute of Technology, Beijing 100081, China. E-mail: zhonghua.xia@bit.edu.cn
bSchool of Mechatronical Engineering, Beijing Institute of Technology, Beijing 100081, China
First published on 30th May 2022
Abstract
Alkynes are widely used in chemistry, medicine and materials science. Here we demonstrate a transition-metal and photocatalyst-free inverse Sonogashira coupling reaction between iodoalkynes and (hetero)arenes or alkenes under visible-light irradiation. Mechanistic and computational studies suggest that iodoalkynes can be directly activated by visible light irradiation, and an excited state iodoalkyne acted as an “alkynyl radical synthetic equivalent”, reacting with a series of C(sp2)–H bonds for coupling products. This work should open new windows in radical chemistry and alkynylation method.
Alkynes are among the most important class of compounds in organic chemistry. Because of their structural rigidity, special electronic properties and numerous methods available for the functionalization of the triple bond, alkynes are important tools and structural elements both in medicinal chemistry and materials sciences.1 Therefore, the development of a new methodology to introduce carbon–carbon triple bonds is of great importance in organic chemistry. The Sonogashira coupling reaction is typically used for the formation of C(sp)–C(sp2) bonds starting from hetero(aryl) halides and terminal alkynes.2 Recently, “inverse Sonogashira coupling” involving the direct alkynylation of unreactive C(sp2)–H bonds with readily available alkynyl halides has received growing interest in the development of a complementary strategy (Fig. 1a). Various main-group and transition metals have been developed to promote this transformation.3 In addition, a photomediated Sonogashira reaction without a photocatalyst was also developed by several groups (Fig. 1b).4
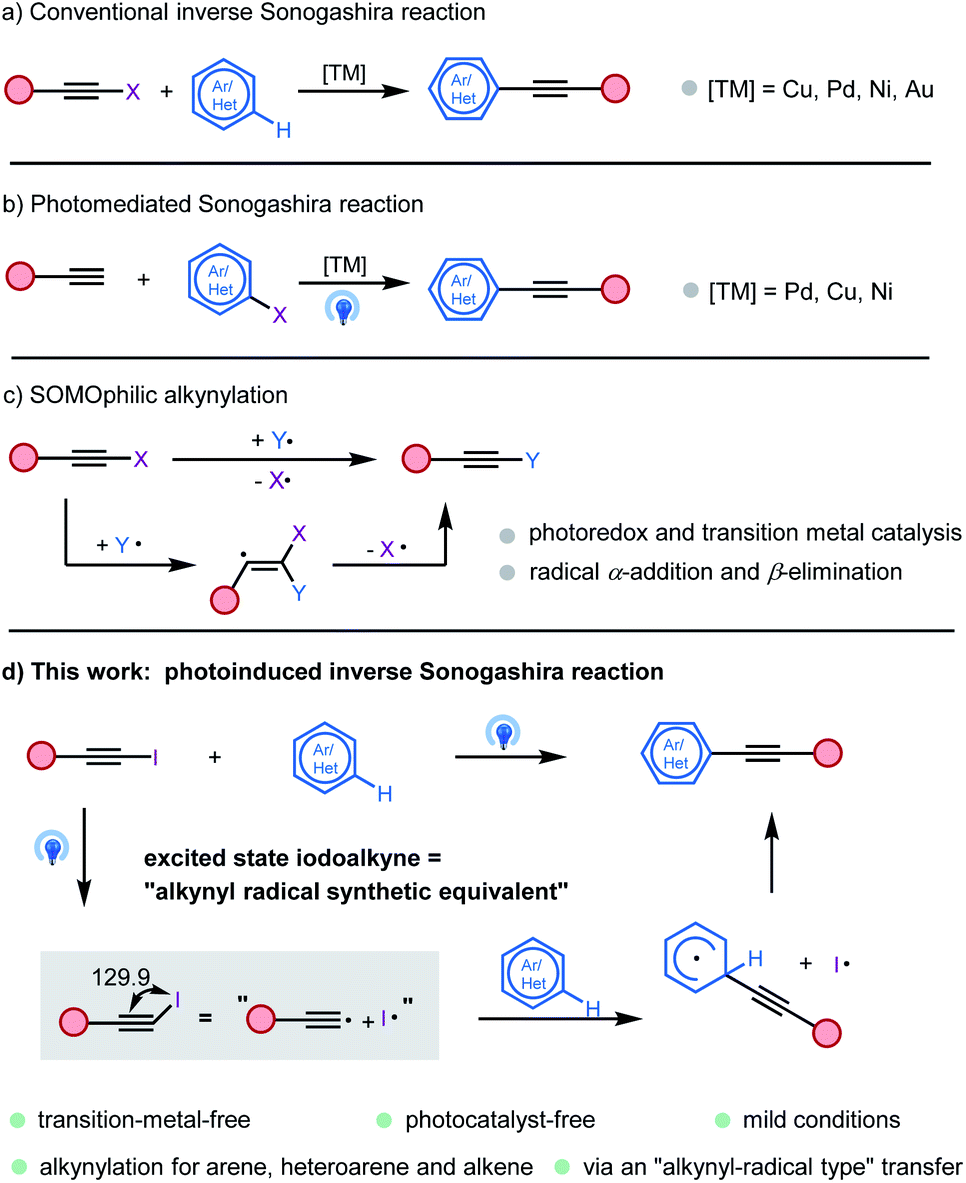 | ||
| Fig. 1 Models of alkynylation. (a) Conventional inverse Sonogashira reaction. (b) Photomediated Sonogashira reaction. (c) SOMOphilic alkynylation. (d) Photoinduced inverse Sonogashira reaction. | ||
In recent years, SOMOphilic alkylnylation (SOMO = singly occupied molecular orbital) has become an excellent method of introducing alkynyl groups (Fig. 1c).5 Based on photoredox and transition metal catalysis, numerous in situ generated radicals undergo α-addition and β-elimination to alkynyl reagents, like the broadly applicable ethynylbenziodoxolone (EBX) reagent. Various radical alkynylations were thus discovered by Li,6 Chen,7 Waser,8 and many other groups.9 However, extending the scope of radical precursors, more atom–economic reactions, and a deeper understanding of the mechanism in these transformations are still highly desirable.
After the discovering of trityl radicals by Gomberg in 1900, the “rational” era of radical chemistry has since begun.10 Now, the development of radical reactions, especially those involving C(sp3) and C(sp2) radicals, enables rapid access to drug discovery, agrochemistry, materials science, and other disciplines.11 However, the C(sp) radical remains a baffling species. Due to their very high energy, short life time, and limited and harsh preparation methods, alkynyl radicals remain an elusive species, which just exists in some extreme environments, like outer-space and the petrochemical industry.12 Even though alkynyl radicals have been proposed as intermediates for some alkynylation methods, they were regarded as mysterious species and ignored by organic chemists for a long time.13 Recently, two approaches have been developed to aid the alkynyl radical generation step. In 2015, Hashmi and collaborators reported a [Au2(μ-dppm)2]2+ catalyzed free radical–radical C(sp)–C(sp3) bond coupling reaction between iodoalkynes and aliphatic amines.14 Under irradiation of sunlight, the dimeric gold complex was proposed to reduce the iodine acetylide to an alkynyl radical. In 2017, Li developed a transition-metal-free alkynylation reaction between iodoalkyne and 2-indolinone.15 Iodoalkynes could release alkynyl radicals under high temperature conditions. In 2019, we reported an Au(I) and Ir(III) catalyzed alkynylative cyclization of o-alkylnylphenols with iodoalkynes, wherein the photosensitized energy transfer promoted the oxidative addition of a gold(I) complex with iodoalkynes.16 Based on our continuous interest in haloalkyne and photo-chemistry, we proposed that an iodoalkyne could be a potential “alkynyl radical precursor” under light irradiation. In this work, we uncovered a novel mode of transition-metal and photocatalyst-free, direct photoexcitation of iodoalkynes for the inverse Sonogashira coupling reaction with arenes, heteroarenes, and alkenes via an “alkynyl-radical type” transfer (Fig. 1d).
Optimization studies
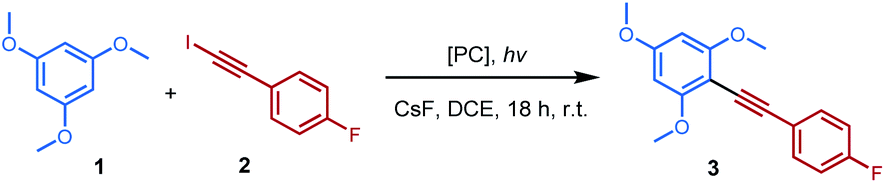
A detailed description of the reaction design for this photoinduced inverse Sonogashira reaction is provided in Table 1 by coupling 1,3,5-trimethoxylbenzene 1 with 1-fluoro-4-(iodoethynyl)benzene 2 in the presence of the Ir[dF(CF3) ppy]2(dtbbpy)PF6 ([Ir-F]) catalyst under blue LED irradiation (λmax = 460 nm). Inspiringly, the desired cross-coupling product 3 was formed in 23% yield, with two side products iodide trimethoxylbenzene 4 2%, and diyne product 5 6% (Table 1, entry 1). The screening of other photocatalysts, like Ph2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O,17 quinuclidine,18 and TiO2,19 could slightly increase the yield up to 32% (entries 2–4). Interestingly, a control experiment without a photocatalyst gave a better yield of 35% of the desired cross-coupling product 3 (entry 5). Moreover, it was found that almost all light sources could trigger this reaction, and a substantial improvement of yield was obtained as 45% under 365 nm LEDs when using 3 equivalents of 2 for 18 hours (entry 6–10). Finally, after substantial optimization it was found that the combination of 1-fluoro-4-(iodoethynyl)benzene 1, 1,3,5-trimethoxylbenzene 2 (5 equiv.), and CsF (2.5 equiv.) in DCE at room temperature under blue LED light for a prolonged time (70 hours) gave the best result, with a 76% isolated yield obtained (entry 12). Control experiments showed that other bases (KOAc and K3PO4) and solvents (MeCN, PhCF3, and CHCl3) could also promote this reaction well (see the ESI†). In contrast, no desired product could be obtained in the absence of light (entry 13) or base (CsF, entry 14). When 1-(bromoethynyl)-4-fluorobenzene was subjected to the reaction, only 3% of 3 was obtained. See the ESI† for detailed condition optimization.
O,17 quinuclidine,18 and TiO2,19 could slightly increase the yield up to 32% (entries 2–4). Interestingly, a control experiment without a photocatalyst gave a better yield of 35% of the desired cross-coupling product 3 (entry 5). Moreover, it was found that almost all light sources could trigger this reaction, and a substantial improvement of yield was obtained as 45% under 365 nm LEDs when using 3 equivalents of 2 for 18 hours (entry 6–10). Finally, after substantial optimization it was found that the combination of 1-fluoro-4-(iodoethynyl)benzene 1, 1,3,5-trimethoxylbenzene 2 (5 equiv.), and CsF (2.5 equiv.) in DCE at room temperature under blue LED light for a prolonged time (70 hours) gave the best result, with a 76% isolated yield obtained (entry 12). Control experiments showed that other bases (KOAc and K3PO4) and solvents (MeCN, PhCF3, and CHCl3) could also promote this reaction well (see the ESI†). In contrast, no desired product could be obtained in the absence of light (entry 13) or base (CsF, entry 14). When 1-(bromoethynyl)-4-fluorobenzene was subjected to the reaction, only 3% of 3 was obtained. See the ESI† for detailed condition optimization.
|
|
||||
|---|---|---|---|---|
| Entry | [PC]b | hv | 1 (eq.) | 3 yieldc |
|
a General conditions: 1 (2–5 equiv.), 2 (0.2 mmol), photocatalyst (PC) or not as shown, CsF (2.5 equiv.), DCE (0.03 M), rt, 18 h, and different light sources as shown.
b [Ir-F] = Ir[dF(CF3)ppy]2(dtbbpy)PF6 (1 mol%), Ph2CO (20 mol%), quinuclidine (20 mol%), and TiO2 (8 mg mL−1).
c Determined by H NMR using 1,4-dimethoxybenzene as an internal standard. Isolated yields in parentheses.
d Reaction for 70 h.
e Without light.
f Without CsF.
|
||||
| 1 | [Ir-F] | Blue LEDs | 2 | 23% |
| 2 | Ph2CO |
Blue LEDs | 2 | 31% |
| 3 | Quinuclidine | Blue LEDs | 2 | 29% |
| 4 | TiO2 | Blue LEDs | 2 | 32% |
| 5 | — | Blue LEDs | 2 | 35% |
| 6 | — | Blue LEDs | 3 | 37% |
| 7 | — | CFL lamp | 3 | 7% |
| 8 | — | 405 nm | 3 | 40% |
| 9 | — | 395 nm | 3 | 43% |
| 10 | — | 365 nm | 3 | 45% |
| 11d | — | 395 nm | 5 | 69% |
| 12d | — | Blue LEDs | 5 | 75% (76%) |
| 13d,e | — | Blue LEDs | 5 | 0 |
| 14d,f | — | Blue LEDs | 5 | 0 |
Scope of the reaction
This new inverse Sonogashira reaction provides an alternative method for obtaining alkynylated products in moderate to good yields, which was carried out under blue LED irradiation (Table 2). Firstly, the scope of aryl iodoalkynes was studied in reaction with 1,3,5-trimethoxylbenene 1 under optimal reaction conditions (Table 1, entry 12). Aryl iodoethynyl bearing electron-withdrawing groups in the para position, such as F, Cl, Br, and CF3, delivered the corresponding alkynylated arenes 3, 6, 7, and 8 without difficulty. Aryl iodoethynyl without substitution furnished alkynylated arenes 9 in a slightly lower yield (54%). The presence of electron-donating groups (–Me and –t-Bu) at the para position furnished alkynylated arenes 10 and 11 in good yields (60% and 79%). In addition, aryl iodoethynyl bearing methyl or chloro groups at the meta-position, or the methyl group at the ortho-position, could react smoothly with 1,3,5-trimethoxylbenene (12, 13, and 14). However, no reaction was observed with alkyl iodoalkynes bearing cyclohexyl or ethyl ester groups (15 and 16).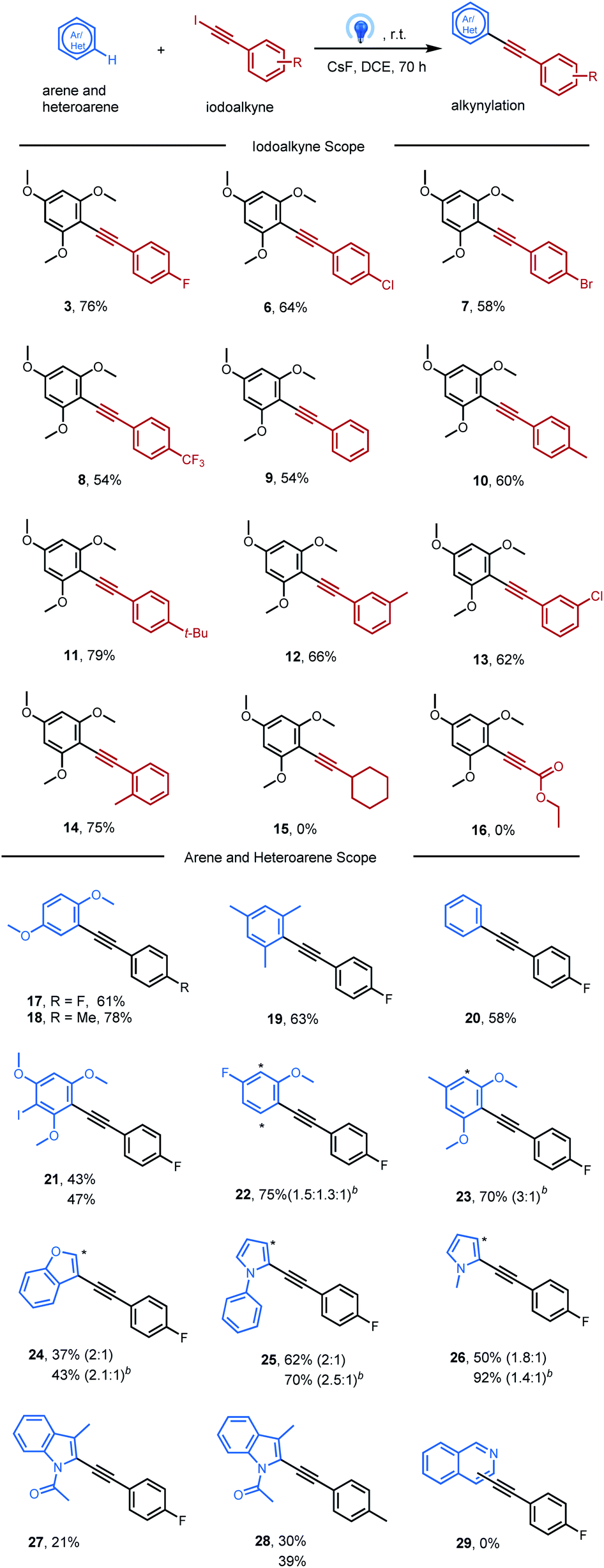
Next, this protocol allows direct alkynylation with a broad range of arenes and heteroarenes. It shows that this alkynylation approach was more effective for electron-rich arenes (3 and 17–21). For unsymmetrical aromatics with more than one reactive site, products may be formed as isomeric mixtures (22 and 23). Besides arenes, heteroarenes, including benzofuran (24), phenyl-pyrrole (25), pyrrole (26) and indole (27 and 28), could also be likewise tolerated with this photochemical procedure. And TiO2 was found to benefit the alkynylation of these heteroarenes, likely due to the surface association between TiO2 and nitrogen-containing heteroarenes.19 Isoquinoline was not tolerated in this system may be due to its intrinsic electron-deficient properties (29).
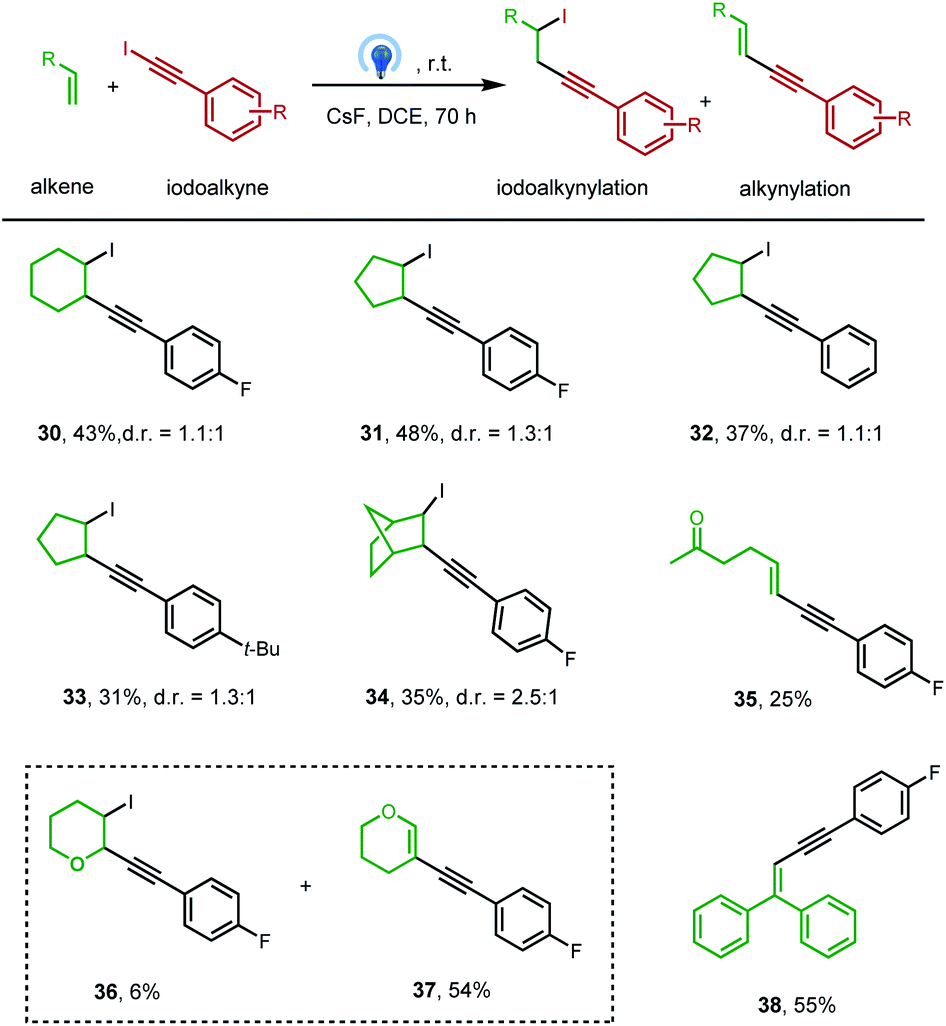
To our delight, this clean photochemical alkynylation protocol was also suitable with a variety of alkenes, and the iodoalkynylation product rather than the alkynylation product was mainly obtained under previously optimized conditions (Table 3). The success example was firstly carried out between iodoalkyne 2 and cyclohexene, affording iodoalkynylated cyclohexane in 43% yield (30), with two diastereomers (d.r. = 1.1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), which is consistent with the radical pathway. Notably, a transitional metal (like Pd, Ru, or Au) is often required to obtain this kind of haloalkynylation product.20 The extension of this transitional-metal-free method to other unactivated alkenes, like cyclopentene, norbornene, and monosubstituted alkene, hex-5-en-2-one, also proved successful (31–35). Interestingly, the activated alkene, 3,4-dihydro-2H-pyran, reacted with iodoalkyne delivering both iodoalkynylation and alkynylation products (36 and 37). And 1,1-diphenylethylene reacted with the iodoalkyne 2 delivered the corresponding alkynylation product 38 in 55% yield.
:1), which is consistent with the radical pathway. Notably, a transitional metal (like Pd, Ru, or Au) is often required to obtain this kind of haloalkynylation product.20 The extension of this transitional-metal-free method to other unactivated alkenes, like cyclopentene, norbornene, and monosubstituted alkene, hex-5-en-2-one, also proved successful (31–35). Interestingly, the activated alkene, 3,4-dihydro-2H-pyran, reacted with iodoalkyne delivering both iodoalkynylation and alkynylation products (36 and 37). And 1,1-diphenylethylene reacted with the iodoalkyne 2 delivered the corresponding alkynylation product 38 in 55% yield.
| a General conditions: alkene (10 equiv.), iodoalkyne (0.2 mmol), CsF (2.5 equiv.), DCE (0.03 M), blue LEDs (λmax = 460 nm), rt, 70 h, and isolated yields. |
|---|
|
The alkynylation and iodoalkynylation products are valuable scaffolds for further elaboration, notably through the potential reactivity of the alkyne and the iodo moiety. For instance, the alkyne moiety can be hydrogenated by formic acid under palladium (0) catalysis to Z-alkene or E-isomer,21 and the iodide group provides possibilities in the diversification of cross-coupling products.
Mechanistic investigations
Having demonstrated that inverse Sonogashira alkynylation was possible for a broad scope of iodoalkynes, arenes, heteroarenes, and alkenes, we attempted to gain a better understanding of this transformation, and three reaction pathways were envisaged. We first assumed the typical photoinduced oxidation process, under light irradiation, the ground state of iodoalkyne will transfer to its excited state, and then the excited state iodoalkyne acted as an oxidant to oxidize another substrate, and this process was recently verified by the Wase group with an EBX reagent (Scheme 1a).22 Alternatively, photoinduced homolysis, which is often taught in textbooks, is another direct pathway to generate two radicals, the alkynyl radical and the iodo radical (Scheme 1b).23 Finally, considering the extremely high energy of the alkynyl radical (the BDE(Csp-H) > 130 kcal mol−1),24 we postulated a third pathway that an activated iodoalkyne might behave as an “alkynyl radical synthetic equivalent”, rather than a “free alkynyl radical” under our blue LED irradiation conditions (Scheme 1c).8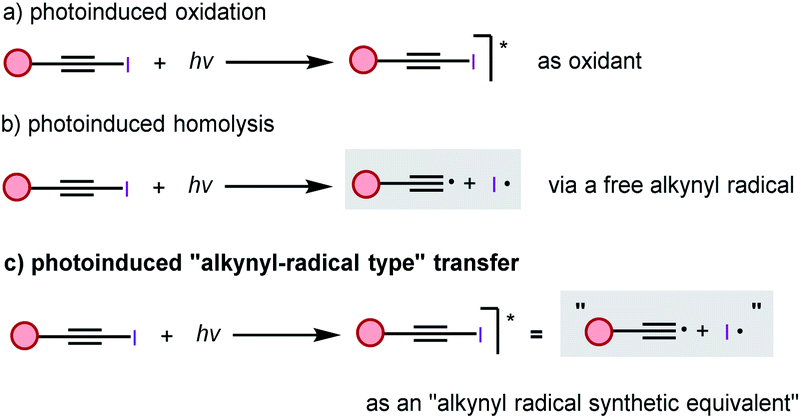 | ||
| Scheme 1 Three possible reaction pathways. | ||
Firstly, radical inhibition experiments were performed (Fig. 2a). The yield of the alkynylation product was suppressed from 76% to 55% or 43% in the presence of increasing amounts of radical scavenger, 2,2,6,6-tetramethyl-1-piperidinoxyl (TEMPO), or 5,5-dimethyl-1-pyrroline N-oxide (DMPO). Considering that the diyne side product 5 was almost always formed in our reaction, the individual iodoalkyne 2 was added into the standard conditions, the same as in Table 2 (Fig. 2b). Indeed, the diyne side product 5 was isolated in 35% yield. These control experiments revealed that the radical pathway might be involved in this reaction.
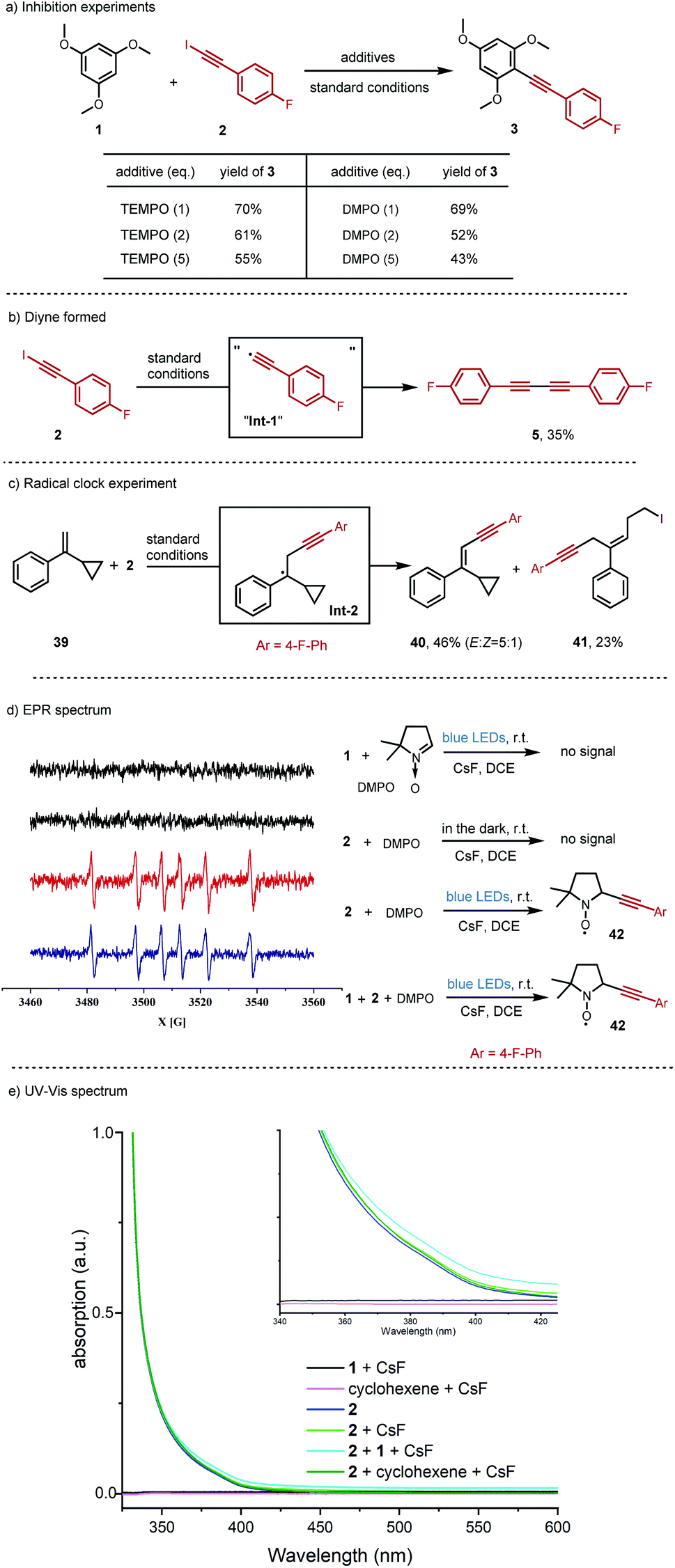 | ||
| Fig. 2 Mechanistic investigations. (a) Inhibition experiments. (b) Diyne formed. (c) Radical clock experiment. (d) Electron paramagnetic resonance (EPR) spectrum (X band, 9.8 GHz) at 298 K. (e) UV-Vis spectrum. All standard conditions are the same as in Table 2. | ||
It is well known that the ring-opening of the cyclopropylcarbinyl radical can serve as a radical clock.25 Consistent with this hypothesis, a radial clock compound, (1-cyclopropylvinyl)benzene 39, was added into the reaction conditions to react with iodoalkyne 2 (Fig. 2c). Interestingly, aside from the alkynyl alkene 40 isolated in 46% yield, the-cyclopropane ring-opening product 41 was generated in 23% yield, indicating “the alkynyl radical Int-1” and alkyl radical Int-2 in the reaction.
Moreover, the key “alkynyl radical Int-1” was observed in an electron paramagnetic resonance (EPR) spin-trapping experiment with 5,5-dimethyl-1-pyrroline N-oxide (DMPO) (Fig. 2d). There was no signal observed either with only the trimethoxylbenene 1 in the light condition, or just the iodoalkyne 2 in the dark. Delightedly, the radical signal peaks derived from the proposed radical trapping adduct 42 were detected when the iodoalkyne 2 was irradiated with light. The standard reaction conditions produced the same radical signal peaks as the individual iodoalkyne 2 did.
With these results in hand, we turned to luminescence studies to have further support for the photoactivity of iodoalkyne 2 under our reaction conditions (Fig. 2e and S6, S7†). For UV-Vis absorption, at the same concentration as the standard reaction conditions in Table 2, the iodoalkyne 2 was identified as the only absorbing species, and the absorption band did still tail off into the visible light region (Fig. 2e, plain blue line). And for the fluorescence spectroscopy, the emission peak of iodoalkyne 2 is around 360 nm, and fluorescence quenching was observed between the iodoalkyne 2 and trimethoxylbenzene 1 or cyclohexene (Fig. S6 and S7). See the ESI† for more information.
Finally, in order to further exclude the photoinduced oxidation mechanism (Scheme 1a), cyclic voltammetry studies were performed (Fig. S5†). As the reductive potential of 1-fluoro-4-(iodoethynyl)benzene 2 at the ground state is E1/2(2) = −1.40 V versus the saturated calomel electrode (SCE), the estimate of the excited state is calculated as E1/2(2*/2˙−) = +1.3 V vs. SCE,22,26 and it does not support the oxidation of the arenes, heteroarenes and alkenes in our reaction (as most oxidative potentials range from +1.7 to +2.3 V).27 More detailed mechanism studies are in the ESI.†
Computational studies
Theoretical calculations have been carried out to further investigate the mechanistic hypothesis of a photoinduced homolysis and “alkynyl-radical type” reactivity (Scheme 1b and c). It found that the activated iodoalkyne 2* pathway via an “alkynyl-radical type” transfer occurred relatively easily, with lower activation energy 2* and transition state energy TS than homolytic cleavage of iodoalkyne via a “free alkynyl radical Int-3” pathway (56.5 and 59.7 vs. 79.3 kcal mol−1) (Fig. 3). The bent structure of iodoalkyne 2* found in its triplet state (supported with phosphorescence data, see Fig. S7†) appears to be one of the key points that explain that the reaction of iodoalkyne 2 and benzene requires photoactivation to proceed. Under visible light irradiation, the modelled geometry of the excited state iodoalkyne 2* is bent (I–C–C angle of 130°) versus 180° for 2 in its ground state, and the C–I bond length increases from 2.01 Å up to 2.20 Å, and this change of geometry makes it more reactive. The reactive bent structure of the iodoalkyne 2* would provide access to the benzene, and undergoes a transition state TS of concerted nucleophile addition/release of the iodine radical to Int-4, followed by oxidation and deprotonation to 3. Moreover, based on the calculated energy of homolytic cleavage, the bonding dissociation energy of the Csp–I bond (BDE Csp–I) is estimated to be 79.3 kcal mol−1, which would correspond to a homolytic cleavage upon excitation at 361 nm.12c,28 Therefore, the pathway of photoactivated iodoalkyne via an “alkynyl-radical type” transfer, in which the calculated energy of the excited state was 56.5 kcal mol−1, is more feasible than the photoinduced homolysis under blue LED irradiation conditions (λmax = 460 nm).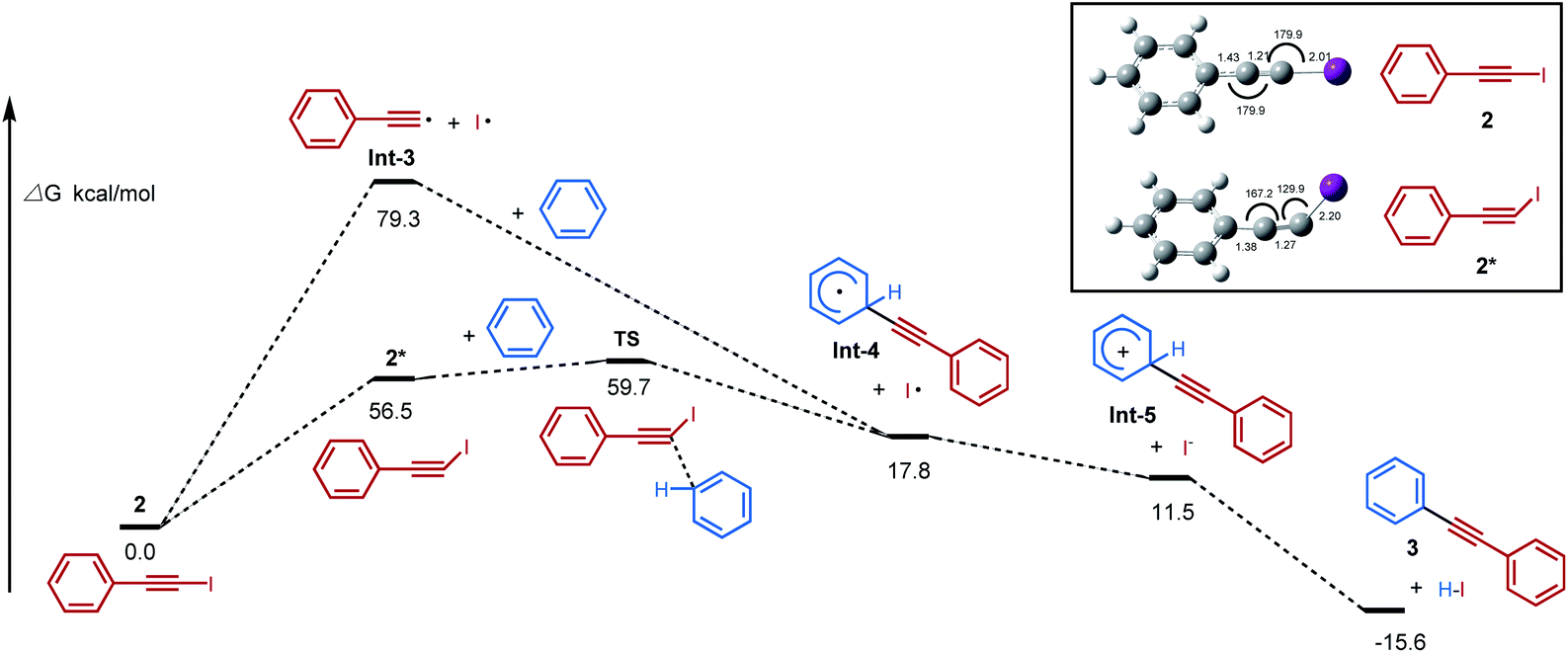 | ||
| Fig. 3 Free energy profiles for the photoinduced inverse Sonogashira coupling reaction. | ||
Based on previous literature, mechanism investigations, and computational studies, “alkynyl-radical type” transfer is believed to be involved in this photoinduced inverse Sonogashira coupling reaction, and the proposed reaction pathway is illustrated in Scheme 2. Under visible light irradiation, the geometry of the excited state iodoalkyne is bent, which becomes more reactive and acted as an “alkynyl radical synthetic equivalent”, reacting with arenes or heteroarenes via a concerted transition state to deliver a cyclohexadienyl radical intermediate. Then the cyclohexadienyl radical intermediate is oxidized by the iodo radical and the deprotonation leads to the final alkynylation product.
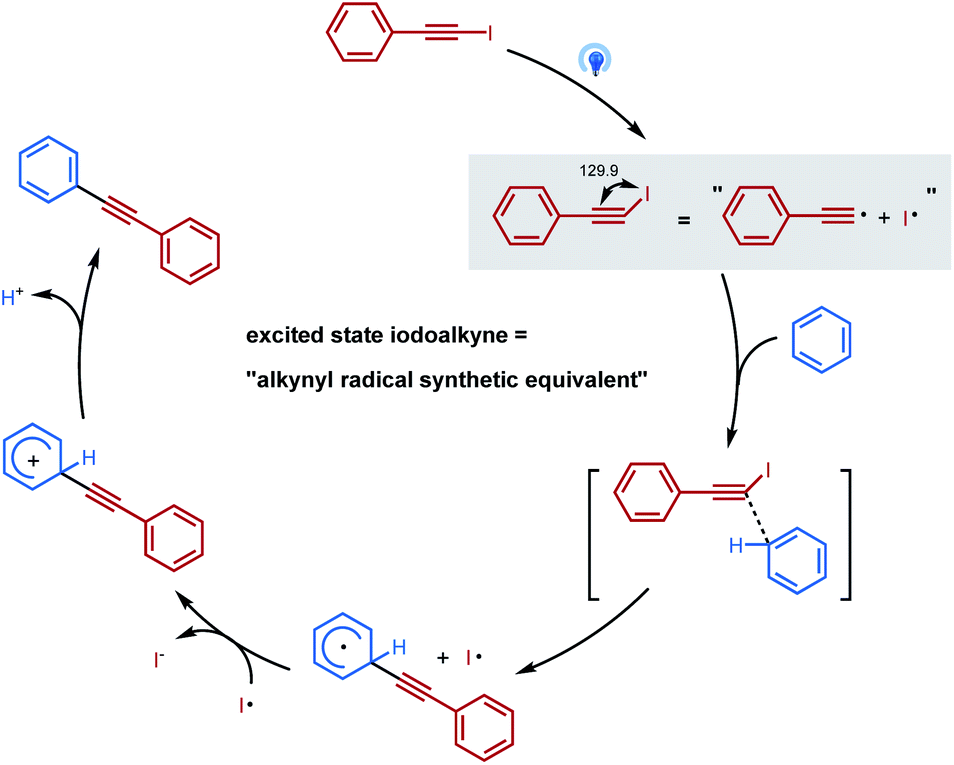 | ||
| Scheme 2 Proposed reaction mechanism. | ||
Conclusions
This work describes a facile photoinduced inverse Sonogashira coupling reaction, involving photoinduced activation of iodoalkynes to allow a C(sp2)–C(sp) and C(sp3)–C(sp) cross-coupling reaction, useful for the alkynylation of arenes, heteroarenes, and alkenes. Beyond the transition-metal catalyzed inverse Sonogashira coupling reaction, this visible light-induced alkynylation and haloalkynylation with a series of C(sp2) compounds will provide an alternative method to construct C(sp)–C bonds under very clean and mild conditions. Even though the alkynyl radical has been regarded as an elusive species for a long time, our study will help to answer some important questions and promote more research in this undeveloped and interesting field. Our work suggested that the photoactivated iodoalkyne pathway via an “alkynyl-radical type” transfer is more favorable than the “free alkynyl radical” to react with a series of C(sp2)–H bonds under visible light irradiation conditions. The excited state iodoalkyne with a bent C–C–I bond structure becomes very reactive and acted as an “alkynyl radical synthetic equivalent”, which has been successfully characterized through a radical clock experiment, EPR study, luminescence study, and theoretical calculations. Therefore, this work directly supports the transfer of an alkynyl radical, which until now has been limited to speculation and circumstantial evidence. The process described in this article enables a transition-metal-free and photocatalyst-free, direct inverse Sonogashira coupling reaction via an “alkynyl-radical type” transfer under visible light irradiation, and it opens a new door in the field of radical chemistry and alkynylation methods.Author contributions
L. Z. and C. W. performed the synthetic experiments and undertook all the physicochemical analyses. J. W. carried out computational studies. D. L., Y. Y., Z. C., J. L. and C.-J. Y. performed and analysed the luminescence and cyclic voltammetry experiments. Z. X. conceived the idea, designed the research and prepared the manuscript. All the authors commented on the final data of the manuscript and contributed to the analysis and interpretation of the data.Data availability
Experimental data including experimental procedures, characterization data, and NMR spectra for new compounds; mechanistic experiments; detailed computational studies, calculated structures, and energies is accessible in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. Qiang Zhu, Dr. Shuang Luo (Guangzhou Institutes of Biomedicine and Health, Chinese Academy of Sciences) and Prof. Honggen Wang (Sun Yat-sen University) for helpful discussions. Financial support from the Beijing Institute of Technology Research Fund Program for Young Scholars (grant number: 2020CX04260) is gratefully acknowledged.Notes and references
- For select reviews:
(a) H. C. Kolb and K. B. Sharpless, Drug Discovery Today, 2003, 8, 1128–1137 CrossRef CAS PubMed
; (b) J. Lehmann, M. H. Wright and S. A. Sieber, Chem.–Eur. J., 2016, 22, 4666–4678 CrossRef CAS PubMed
-
(a) E. Negishi and L. Anastasia, Rev., 2003, 103, 1979–2018 CAS
-
(a) I. V. Seregin, V. Ryabova and V. Gevorgyan, J. Am. Chem. Soc., 2007, 129, 7742–7743 CrossRef CAS PubMed
-
(a) A. Sagadevan and K. C. Hwang, Adv. Synth. Catal., 2012, 354, 3421–3427 CrossRef CAS
-
(a) F. Le Vaillant and J. Waser, Chem. Sci., 2019, 10, 8909–8923 RSC
- X. Liu, Z. Wang, X. Cheng and C. Li, J. Am. Chem. Soc., 2012, 134, 14330–14333 CrossRef CAS PubMed
-
(a) H. Huang, G. Zhang, L. Gong, S. Zhang and Y. Chen, J. Am. Chem. Soc., 2014, 136, 2280–2283 CrossRef CAS PubMed
-
(a) F. Le Vaillant, T. Courant and J. Waser, Angew. Chem., Int. Ed., 2015, 54, 11200–11204 CrossRef CAS PubMed
-
(a) G. A. Russel and P. Ngoviwatchai, Tetrahedron Lett., 1986, 27, 3479–3482 CrossRef
- M. Gomberg, J. Am. Chem. Soc., 1900, 22, 757–771 CrossRef
-
(a) M. Yan, J. C. Lo, J. T. Edwards and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 12692–12714 CrossRef CAS PubMed
-
(a) G. Martelli, P. Spagnolo and M. Tiecco, J. Chem. Soc. D, 1969, 282–283 RSC
-
(a) L. Wang, W. Wei, D. Yang, H. Cui, H. Yue and H. Wang, Tetrahedron Lett., 2017, 58, 4799–4802 CrossRef CAS
- J. Xie, S. Shi, T. Zhang, N. Mehrkens, M. Rudolph and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2015, 54, 6046–6050 CrossRef CAS PubMed
- H.-Y. Huang, L. Cheng, J.-J. Liu, D. Wang, L. Liu and C.-J. Li, J. Org. Chem., 2017, 82, 2656–2663 CrossRef CAS PubMed
- Z. Xia, V. Corcé, F. Zhao, C. Przybylski, A. Espagne, L. Jullien, T. Le Saux, Y. Gimbert, H. Dossmann, V. Mouriès-Mansuy, C. Ollivier and L. Fensterbank, Nat. Chem., 2019, 11, 797–805 CrossRef CAS PubMed
- A. Dewanji, P. E. Krach and M. Rueping, Angew. Chem., Int. Ed., 2019, 58, 3566–3570 CrossRef CAS PubMed
- O. Dumele, D. Wu, N. Trapp, N. Goroff and F. Diederich, Org. Lett., 2014, 16, 4722–4725 CrossRef CAS PubMed
- S. P. Pitre, T. P. Yoon and J. C. Scaiano, Chem. Commun., 2017, 53, 4335–4338 RSC
-
(a) Y. Li, X. Liu, H. Jiang, B. Liu, Z. Chen and P. Zhou, Angew. Chem., Int. Ed., 2011, 50, 6341–6345 CrossRef CAS PubMed
- R. Shen, T. Chen, Y. Zhao, R. Qiu, Y. Zhou, S. Yin, X. Wang, M. Goto and L.-B. Han, J. Am. Chem. Soc., 2011, 133, 17037–17044 CrossRef CAS PubMed
- S. G. E. Amos, D. Cavalli, F. Le Vaillant and J. Waser, Angew. Chem., Int. Ed., 2021, 60, 23827–23837 CrossRef CAS PubMed
- Photoinduced homolysis:
(a) D. E. Votkina, P. V. Petunin, M. E. Trusova, P. S. Postnikov, G. Audran and S. R. A. Marque, Phys. Chem. Chem. Phys., 2020, 22, 21881–21887 RSC
-
(a) K. M. Ervin, S. Gronert, S. E. Barlow, M. K. Gilles, A. G. Harrison, V. M. Bierbaum, C. H. DePuy, W. C. Lineberger and G. B. Ellison, J. Am. Chem. Soc., 1990, 112, 5750–5759 CrossRef CAS
-
(a) D. Griller and K. U. Ingold, Acc. Chem. Res., 1980, 13, 317–323 CrossRef CAS
- L. Buzzetti, A. Prieto, S. R. Roy and P. Melchiorre, Angew. Chem., Int. Ed., 2017, 56, 15039–15043 CrossRef CAS PubMed
-
(a) H. G. Roth, N. A. Romero and D. A. Nicewicz, Synlett, 2015, 27, 714–723 CrossRef
- Y. H. Kim, J. Ree, C. S. Lee and H. K. Shin, J. Chem. Phys., 1994, 101, 4668–4681 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc01933g |
| This journal is © The Royal Society of Chemistry 2022 |