
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGramicidin A accumulates in mitochondria, reduces ATP levels, induces mitophagy, and inhibits cancer cell growth†
Yun-Wei
Xue
a,
Hiroaki
Itoh
 a,
Shingo
Dan
b and
Masayuki
Inoue
*a
a,
Shingo
Dan
b and
Masayuki
Inoue
*a
aGraduate School of Pharmaceutical Sciences, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: inoue@mol.f.u-tokyo.ac.jp
bDivision of Molecular Pharmacology, Cancer Chemotherapy Center, Japanese Foundation for Cancer Research, 3-8-31 Ariake, Koto-ku, Tokyo 135-8550, Japan
First published on 3rd June 2022
Abstract
Gramicidin A (1) is a linear 15-mer peptidic natural product. Because of its sequence of alternating D- and L-chirality, 1 folds into a β6.3-helix in a lipid bilayer and forms a head-to-head dimer to function as a transmembrane channel for monovalent cations (H+, Na+, and K+). The potent anticancer activity of 1 was believed to be mainly attributed to the free ion diffusion across the plasma membrane. In this study, we investigated the cytostatic action of 1 in nanomolar concentrations using the human breast cancer cell line MCF-7, and revealed the unprecedented spatiotemporal behavior of 1 for the first time. Compound 1 not only disrupted the ion concentration gradients of the plasma membrane, but also localized in the mitochondria and depolarized the inner mitochondrial membrane. The diminished H+ gradient in the mitochondria inhibited ATP synthesis. The resultant mitochondrial malfunction led to mitophagy, while the cellular energy depletion induced G1 phase accumulation. The multiple events occurred in a time-dependent fashion and ultimately caused potent inhibition of cell growth. The present study provides valuable information for the design and development of new cytostatic agents exploiting channel-forming natural products.
Introduction
The life of eukaryotic cells depends on membranes that define multiple boundaries in cells. The plasma membrane separates cytosol from the extracellular environment, while organelle membranes maintain the characteristic differences between the contents of each organelle and the cytosol. Ion gradients across the plasma and organelle membranes are precisely controlled in living cells to regulate all aspects of cellular biology. Thus, perturbation of such ion concentration differences is often detrimental to cellular homeostasis.Ionophore natural products are a family of hydrophobic molecules that disrupt ion gradients to exert various biological effects.1 They are classified as mobile carriers or channels based on the mechanism of ion transport. Mobile carriers bind with ions and the resultant complex diffuses across membranes, whereas channels constitute transmembrane nanopores that permit ion flow through the membrane. Because of the continuous pore, ion transport by channels is generally 103 to 104-fold faster than that by carriers. Over the past few decades, mobile carriers have attracted intense attention as lead compounds for developing new anticancer therapeutics. For example, salinomycin selectively targets cancer stem cells to reduce in vivo cancer growth and metastasis, and recently entered phase I/II trials.2,3 Compared with mobile carriers, the anticancer properties of channels have remained largely unexplored despite their greater ion transport efficiency.4–6
Gramicidin A (1, molecular weight = 1882 Da) and polytheonamide B (5, 5030 Da) are ion-channel forming natural products (Fig. 1a). Compound 1 is a 15-residue peptide produced by the soil bacterium Bacillus brevis,7,8 and 5 is a 48-residue peptide isolated from the marine sponge Theonella swinhoei.9 Although their side chains and molecular sizes are disparate and their natural sources are unrelated, they share a prominent structural feature that only appears in a few natural products. The linear sequences of 1 and 5 comprise alternating D- and L-amino acids except for the achiral glycines. Due to the switching Cα-stereochemistry, all the side chains are on one side of the strand, which forces the sequence into a helix by interresidue hydrogen bonding. Namely, both peptides fold into a β6.3-helix with a 4 Å diameter pore in a hydrophobic environment (Fig. 1b).10–16 As a result, 1 and 5 form transmembrane ion channels as a head-to-head dimer and as a monomer, respectively, and allow for the facile diffusion of monovalent cations. The cation selectivities of 1 and 5 are consistent (H+ > K+ > Na+), indicating their similar channel properties.17,18 The channel formation of 1 and 5 is attributed to their potent activities against cancer cells: the 50% inhibitory concentrations (IC50) against P388 mouse leukemia cells are 5.8 nM for 1 and 0.086 nM for 5.
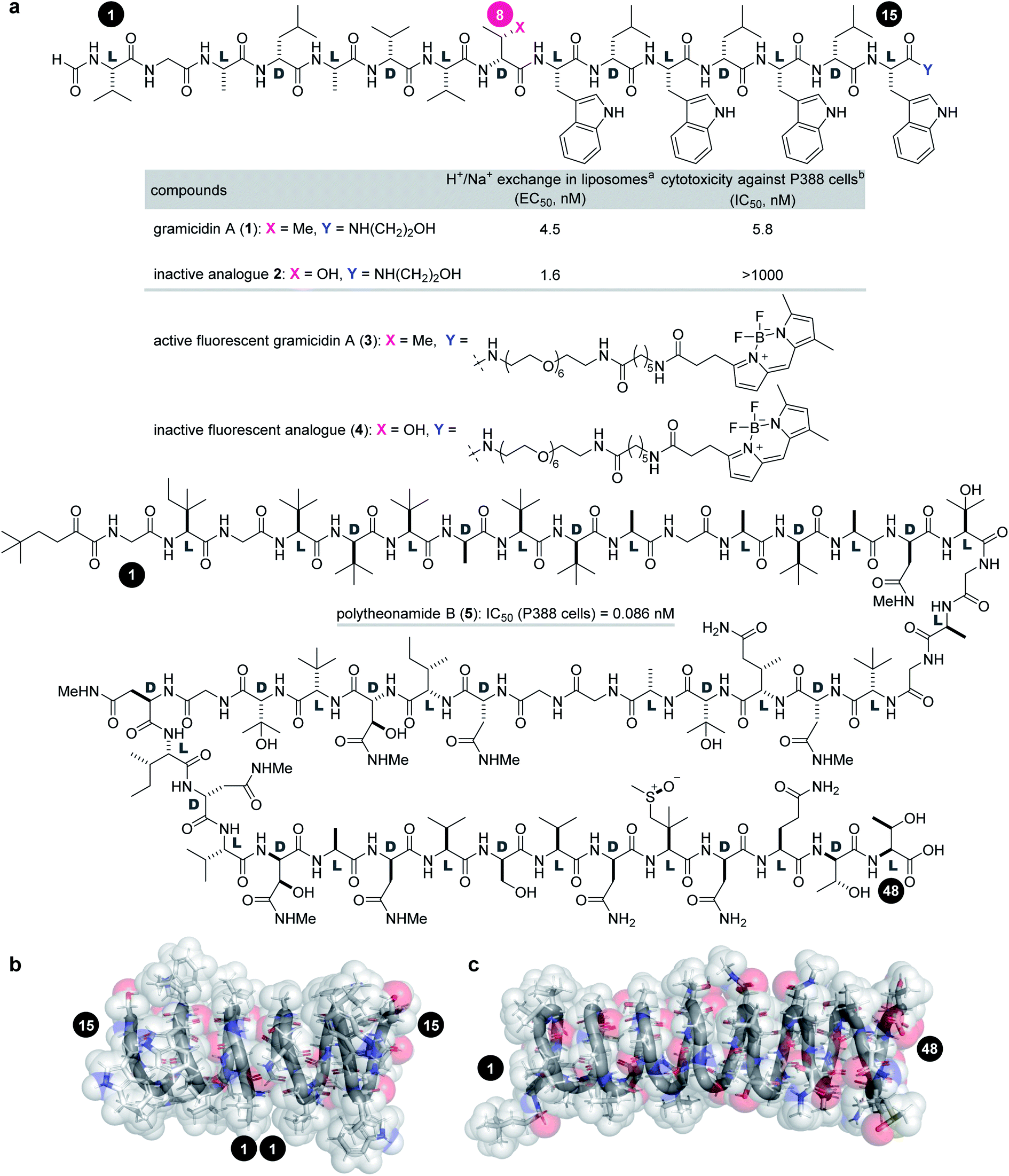 | ||
Fig. 1 Structures and functions of channel-forming peptidic natural products. (a) Structures of gramicidin A (1), inactive analogue 2, active fluorescent probe 3, inactive fluorescent probe 4, and polytheonamide B (5). (b) Head-to-head β6.3-helical dimer structure of 1 (PDB ID 1MAG). (c) β6.3-Helical structure of 5 (PDB ID 2RQO). Numbers highlighted in filled circles are residue numbers. aEC50 value of H+/Na+ transport activity toward pH-gradient liposomes comprising egg yolk phosphatidylcholine and egg yolk phosphatidylglycerol (19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). bIC50 value against the P388 mouse leukemia cell line. :1). bIC50 value against the P388 mouse leukemia cell line. | ||
We have engaged in the synthesis, biological evaluation, and functional modulation of channel-forming natural products over the past decade. In 2018, we reported the full solid-phase total synthesis of polytheonamide B (5).19–21 The efficient synthesis allowed us to uncover a unique dual mode of action of 5 in MCF-7 human breast cancer cells. Compound 5 elicits free cation transport in the plasma membrane, and then localizes in the lysosomes to diminish the H+ gradient across the lysosomal membranes. Depolarization across the plasma membrane and neutralization of the lysosomes are considered to trigger the apoptotic cell death pathway.
We became interested in clarifying whether gramicidin A (1) would emulate the dual mode of action of structurally- and functionally-related 5. Although potent antitumor effects of 1 have been demonstrated by in vitro assays using cancer cells and by in vivo experiments using a xenograft mouse model,22–27 the precise time-dependent mechanisms of the activity of 1 remained unclear. Herein, we report our investigation of the spatiotemporal cellular behavior of 1 using MCF-7 cells. Compound 1 depolarized not only the plasma membrane, but also the inner mitochondrial membrane after accumulating in the mitochondria. The resultant loss of the mitochondrial transmembrane potential attenuated the production of adenosine triphosphate (ATP), which led to eventual cell cycle arrest, ultimately resulting in the inhibition of cancer cell growth. These results revealed a new intricate mode of action of 1 and the functional differences between 1 and 5 for the first time.
Results and discussion
Preparation of 1 and chemical probes 2–4
Investigation of the biological behavior of 1 required suitable chemical probes. We first selected an inactive analogue of 1 to differentiate 1-specific and 1-nonspecific functions in the assays. During our previous high-throughput screening of thousands of synthetic gramicidin A analogues, we discovered various compounds with altered activities.28,29 Among them, 2 was found to be inactive against P388 cells (>1000 nM for 2, Fig. 1a). Despite its negligible toxicity, 2 differs from 1 only in one substituent at residue-8 (Cβ-Me for 1, Cβ-OH for 2) and retains the ion-channel activity against liposomes (EC50 = 4.5 nM for 1, 1.6 nM for 2). Because of its similar physicochemical properties with 1, analogue 2 was expected to serve as a suitable probe for negative control experiments. Hence, pure 1 and 2 were prepared by Fmoc-based solid-phase peptide synthesis (SPPS).28,30To analyze the cellular uptake and distribution by live cell imaging, fluorescent probes 3 and 4 were designed based on active 1 and inactive 2, respectively (Fig. 1a). The C-terminus was chosen as the modification site rather than the N-terminus so as not to disturb the head-to-head dimer formation. Moreover, a hydrophobic fluorophore and linker were selected to retain the intrinsic lipophilic properties of 1 and 2. Therefore, 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene (BODIPY)31 was envisioned to be conjugated with the C-termini of 1 and 2 through a tether consisting of polyethylene glycol (PEG)-6 and C6-alkyl chains. Compounds 3 and 4 were synthesized by SPPS, followed by C-terminal modification.
Scheme 1 illustrates the routes to 3 and 4. The 15-mer peptides were elongated from Fmoc-L-Trp(Boc)-trityl-ChemMatrix resin (6)32via cycles of piperidine-mediated Fmoc removal and microwave-assisted amidation33–35 at 60 °C or 40 °C using Nα-Fmoc-protected amino acids in the presence of O-(7-aza-1H-benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HATU) and 1-hydroxy-7-azabenzotriazole (HOAt).36 The N-terminus was then deprotected and formylated with p-nitrophenyl formate to afford 7a and 7b. A mixture of (CF3)2CHOH and CH2Cl2 cleaved 7a and 7b from the resin, thereby releasing 8a and 8b.
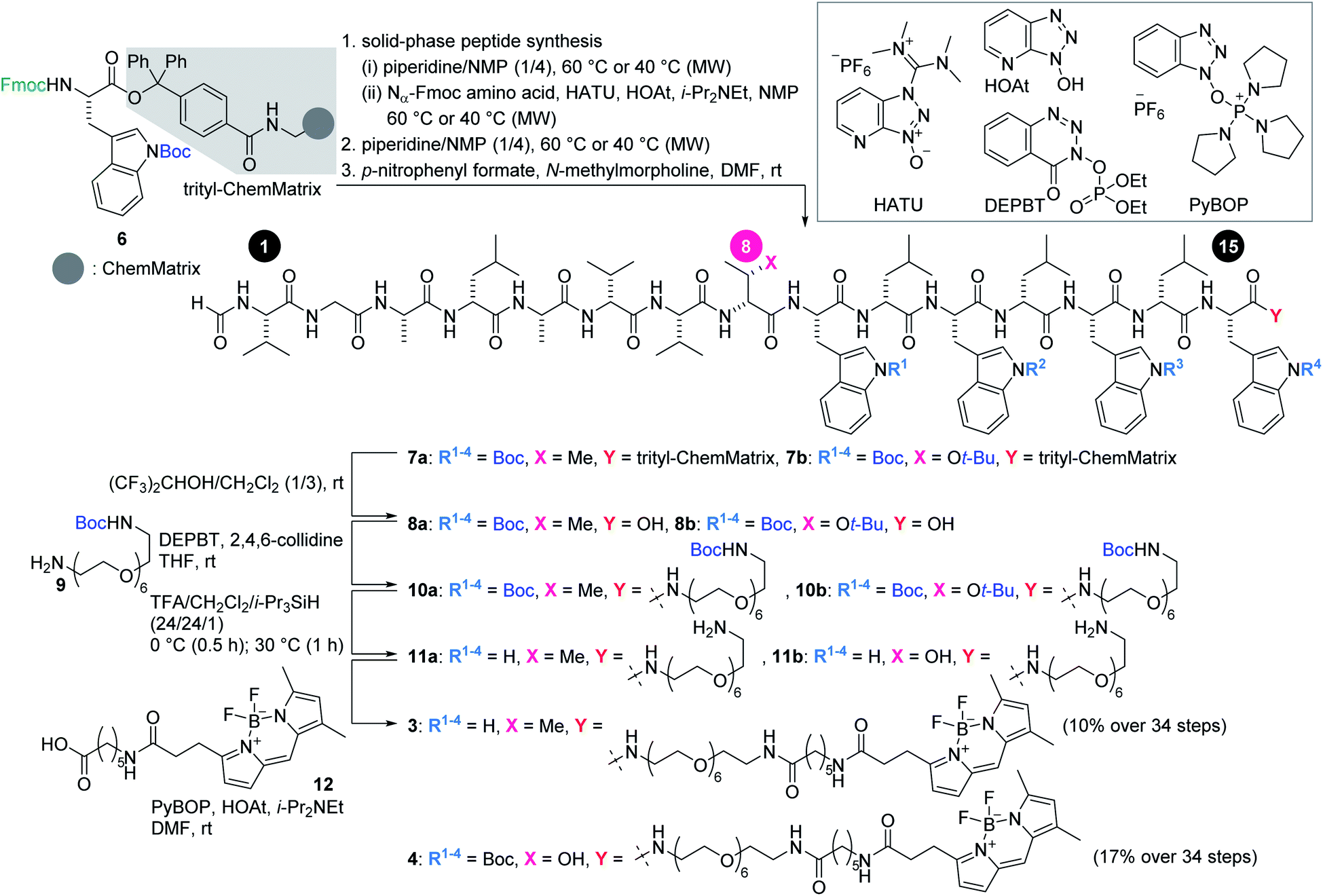 | ||
| Scheme 1 Solid-phase synthesis of fluorescent probes 3 and 4. Boc = tert-butoxycarbonyl, DEPBT = 3-(diethoxyphosphoryloxy)-1,2,3-benzotriazin-4(3H)-one, HATU = O-(7-aza-1H-benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate, HOAt = 1-hydroxy-7-azabenzotriazole, MW = microwave, PyBOP = (benzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate, t-Bu = tert-butyl, TFA = trifluoroacetic acid. | ||
Next, the linker and fluorophore were introduced in a stepwise fashion. Carboxylic acids 8a and 8b were condensed with mono-Boc-protected diamine 9 by the action of 3-(diethoxyphosphoryloxy)-1,2,3-benzotriazin-4(3H)-one (DEPBT)37 and 2,4,6-collidine to generate 10a and 10b, respectively. The five Boc groups of 10a, and the five Boc groups and one t-Bu group of 10b were simultaneously removed by treating with trifluoroacetic acid (TFA), i-Pr3SiH,38 and H2O to furnish 11a and 11b. Finally, BODIPY moiety 12 was condensed with the amine of 11a and 11b using (benzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate (PyBOP)39 and HOAt, thereby giving rise to fluorescent probes 3 and 4 in 10% and 17% yields, respectively, over 34 steps from 6.
Growth inhibition activities of 1 and chemical probes 2–4 against cancer cells
We applied 1 to JFCR39 cancer panel screening to gain preliminary insight into the mode of action (Fig. S1†).40,41 Among the 39 cancer cell lines of the panel, BSY-1 and MCF-7 (breast), U-251 (central nervous system), and DMS114 (lung) were the most susceptible strains. To our surprise, a COMPARE analysis of the growth inhibition fingerprints of 1 and 5 (Fig. S2†) revealed no correlation between them (R = −0.175).42 These results indicated that the mechanism of action of 1 completely differs from that of 5.Because of its high sensitivity to 1, MCF-7 cells were chosen for further study. As an initial step, the activities of 1–4 against MCF-7 cells were assessed by quantifying the 50% growth inhibition concentration (GI50) and 50% lethal concentration (LC50) values (Table 1).43 Interestingly, parent 1 exhibited potent growth inhibition against MCF-7 cells (GI50 = 42 nM), but was not lethal up to 5000 nM (LC50 > 5000 nM). Thus, 1 is strong as a cytostatic agent, yet weak as a cytotoxin.44 In sharp contrast, the GI50 and LD50 values of 5 were different only by 19-fold (GI50 = 0.69 nM vs. LC50 = 13 nM), demonstrating the high cytotoxic activity of 5 and the distinct effects between 1 and 5. The growth inhibition activity (GI50 = 990 nM) of 2 was attenuated 24-fold compared with 1, and no cytotoxicity (LC50 > 5000 nM) was observed. These data validated that 2 functioned as a structurally similar inactive analogue. Fluorescent probes 3 (GI50 = 170 nM, LC50 > 3000 nM) and 4 (GI50 = 2800 nM, LC50 > 5000 nM) maintained the activity profiles of 1 and 2, respectively, suggesting that the cellular behavior of 3/4 reflect those of 1/2.
| Compounds | GI50 (nM)a | LC50 (nM)a |
|---|---|---|
| a The 50% growth inhibitory concentration (GI50, nM) and 50% lethal concentration (LC50, nM) were determined by sulforhodamine B assay. Values are displayed as mean ± SD of three independent experiments. | ||
| 1 | 42 ± 2 | >5000 |
| 2 | 990 ± 130 | >5000 |
| 3 | 170 ± 20 | >3000 |
| 4 | 2800 ± 600 | >5000 |
| 5 | 0.69 ± 0.09 | 13 ± 2 |
Depolarization of the plasma membrane by gramicidin A (1) and inactive analogue 2
To uncover the origin of the strong cytostatic activity of gramicidin A (1), we first analyzed the depolarizing effect of active 1 and inactive 2 on the plasma membrane of MCF-7 cells.45 DiBAC4(3)46 was adopted as a fluorescent reporter in this assay to evaluate the time-dependent depolarization (Fig. 2a). As expected, natural product 1 rapidly enhanced fluorescence over 1 h at concentrations between 12.3 nM to 333 nM (Fig. 2b). The 50% effective concentration (EC50) was calculated to be 82 nM (Table 2). The comparable EC50 and GI50 (42 nM) values supported the well-accepted mode of action of the channel forming 1: disruption of the ion concentration gradient across the plasma membrane impaired the cellular functions. Alternatively, when inactive probe 2 was added to MCF-7 cells, no significant fluorescence change was observed up to 3000 nM (EC50 > 3000 nM) (Fig. 2b and Table 2). Considering the similar ion transport activities of 1 and 2 in the liposome assay (see Fig. 1a), inhibition of the ion-conducting channels of 2 in the MCF-7 cell assay is attributable to the different membrane environment between liposomes and MCF-7 cells. The data also disclosed that the subtle structural switch of the side chain of 1 and 2 resulted in the drastic functional change.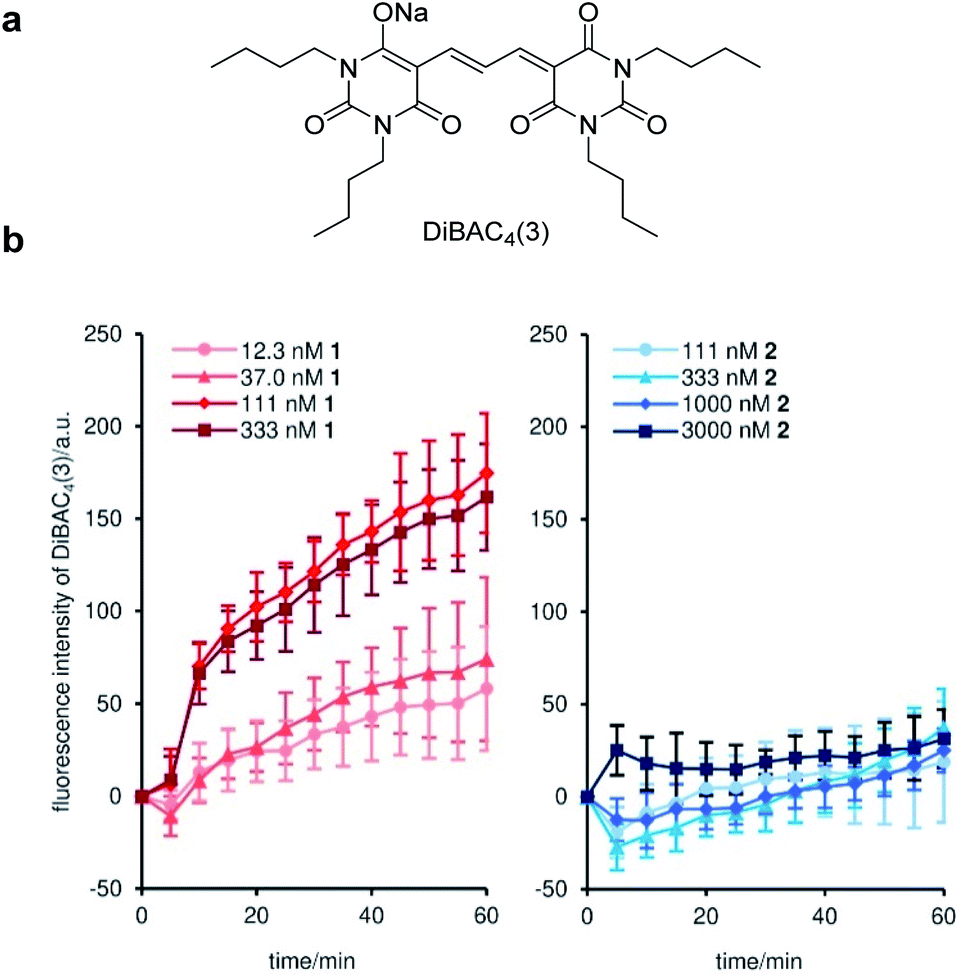 | ||
| Fig. 2 Time-dependent depolarization of the plasma membrane of MCF-7 cells by 1 and 2. (a) Structure of DiBAC3(4). (b) Time courses of the changes in the fluorescence intensity of DiBAC3(4) upon the addition of 1 (left) and 2 (right). Each plot is displayed as mean ± SD of three replicates. a.u. = arbitrary unit. | ||
Mitochondrial and lysosomal localization of fluorescent probes 3 and 4 and mitophagy induction by 1 and 3
The cellular distribution was visualized by imaging studies of fluorescent probes 3 and 4 by confocal fluorescence microscopy (Fig. 3). When 50 nM of active probe 3 and inactive probe 4 were individually incubated with MCF-7 cells, 3 and 4 differentially localized in the intracellular structures.47 To determine the selective subcellular localization of 3 and 4, time-dependent localization analyses were conducted for 3 and 4 using two fluorescent organelle markers; MitoTracker Red CMXRos (Fig. 3a), which covalently labels mitochondria,48 and LysoTracker Red DND-99, which accumulates in acidic lysosomes (Fig. 3b). Pearson's correlation coefficient (R) was calculated to evaluate the degree of colocalization of 3 and 4 with these markers.49,50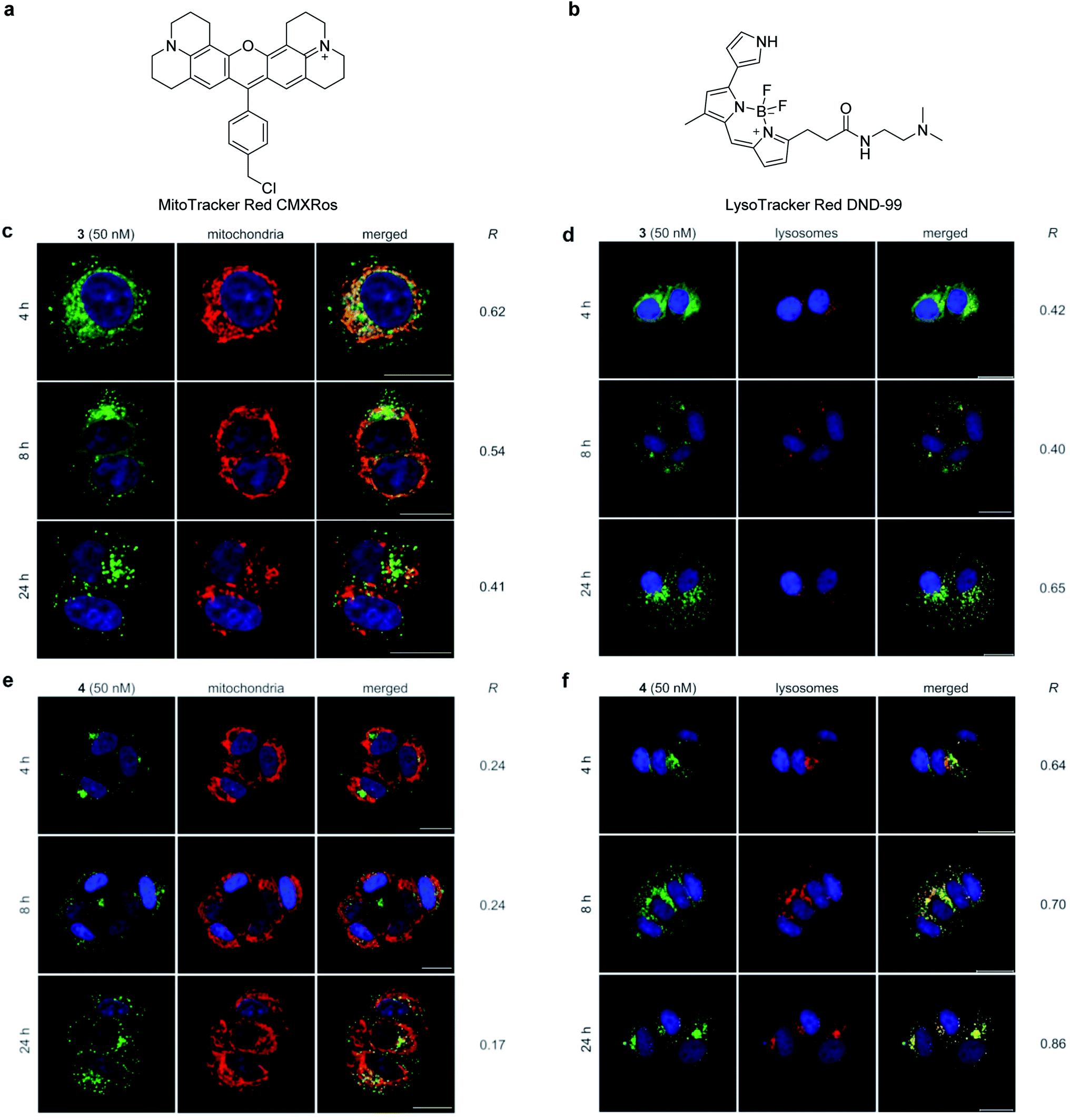 | ||
| Fig. 3 Time-dependent intracellular localization of fluorescent probes 3 and 4 in the presence of organelle markers in MCF-7 cells. (a) Structure of a mitochondrial marker, MitoTracker Red CMXRos. (b) Structure of a lysosomal marker, LysoTracker Red DND-99. (c) 3 with MitoTracker. (d) 3 with LysoTracker. (e) 4 with MitoTracker. (f) 4 with LysoTracker. The signals from the probes and organelle markers are green and red, respectively. Colocalization of the probes and organelle markers is shown as the merged images. The nuclei were stained with Hoechst 33342. The probes (50 nM) were incubated for 4 h, 8 h, and 24 h. Scale bar represents 20 μm. R value represents Pearson's correlation coefficients calculated from the green and red fluorescence in each merged image. | ||
The images of active 3 and inactive 4 in MCF-7 cells were obtained at 4 h, 8 h, and 24 h after incubation in the presence of MitoTracker and LysoTracker. After 4 h, the fluorescence signal of 3 (shown in green) mainly overlapped with that of MitoTracker (Fig. 3c, R = 0.62) (shown in red) and partly with that of LysoTracker (Fig. 3d, R = 0.42) (shown in red), establishing mitochondria as the major localization site of 3.51 Interestingly, the relative distribution of 3 between the mitochondria and lysosomes changed over time. During the 24 h incubation, fluorescence from the mitochondria time-dependently decreased (R = 0.54 for 8 h, R = 0.41 for 24 h), while fluorescence from the lysosomes increased (R = 0.40 for 8 h, R = 0.65 for 24 h). On the other hand, the negative control probe 4 selectively localized to the lysosomes (Fig. 3f) rather than the mitochondria (Fig. 3e) in the same experimental time frame (R = 0.17–0.24 with MitoTracker vs. R = 0.64–0.86 with LysoTracker). These imaging data together suggest the importance of mitochondria as an intracellular target site for gramicidin A (1) to exert its potent growth inhibition activity. Thus, the intracellular behavior of 1 should be distinct from that of lysosome-accumulating polytheonamide B (5).
Next, we tested if the internalization of 3 and 4 involves an active pathway (Fig. S3 and S4†). Along with glycolysis (2-deoxy-D-glucose) and oxidative phosphorylation (NaN3) inhibitors, 5-(N-ethyl-N-isopropyl)-amiloride markedly reduced the uptake of 3 and 4 within the cells.52 The results confirmed that 3 and 4 were not passively transported but were internalized by energy-dependent macropinocytosis.53
During the imaging experiments (Fig. 3c and d), the green fluorescence from 3 appeared to move from the mitochondria to the lysosomes. We hypothesized that the phenomenon corresponded to the selective autophagy of mitochondria, known as mitophagy.54 Mitophagy is an important mitochondrial quality control mechanism that eliminates damaged mitochondria by lysosome-mediated degradation. To examine this hypothesis, we utilized a mitochondria-selective pH-sensitive dye (Mtphagy dye, Fig. 4a).55 Mtphagy dye conjugates with mitochondria and shows weak fluorescence under neutral conditions. Upon mitophagy, it becomes highly fluorescent by the acidification of mitochondrial fragments in the lysosomes. Indeed, the red signals from Mtphagy dye were markedly enhanced after 24 h incubation of 3 (50 nM) and well-merged with the green signal from 3 (Fig. 4b, R = 0.62). When inactive 4 (50 nM) was applied, the fluorescence from the Mtphagy dye was not changed, and did not overlap with the fluorescence from 4 (R = 0.34). The imaging data analyses for active 3 and inactive 4 in Fig. 3 and 4 show the selective time-dependent behavior of 3: 3 first accumulated in the mitochondria and then localized in acidic lysosomes via mitophagy.
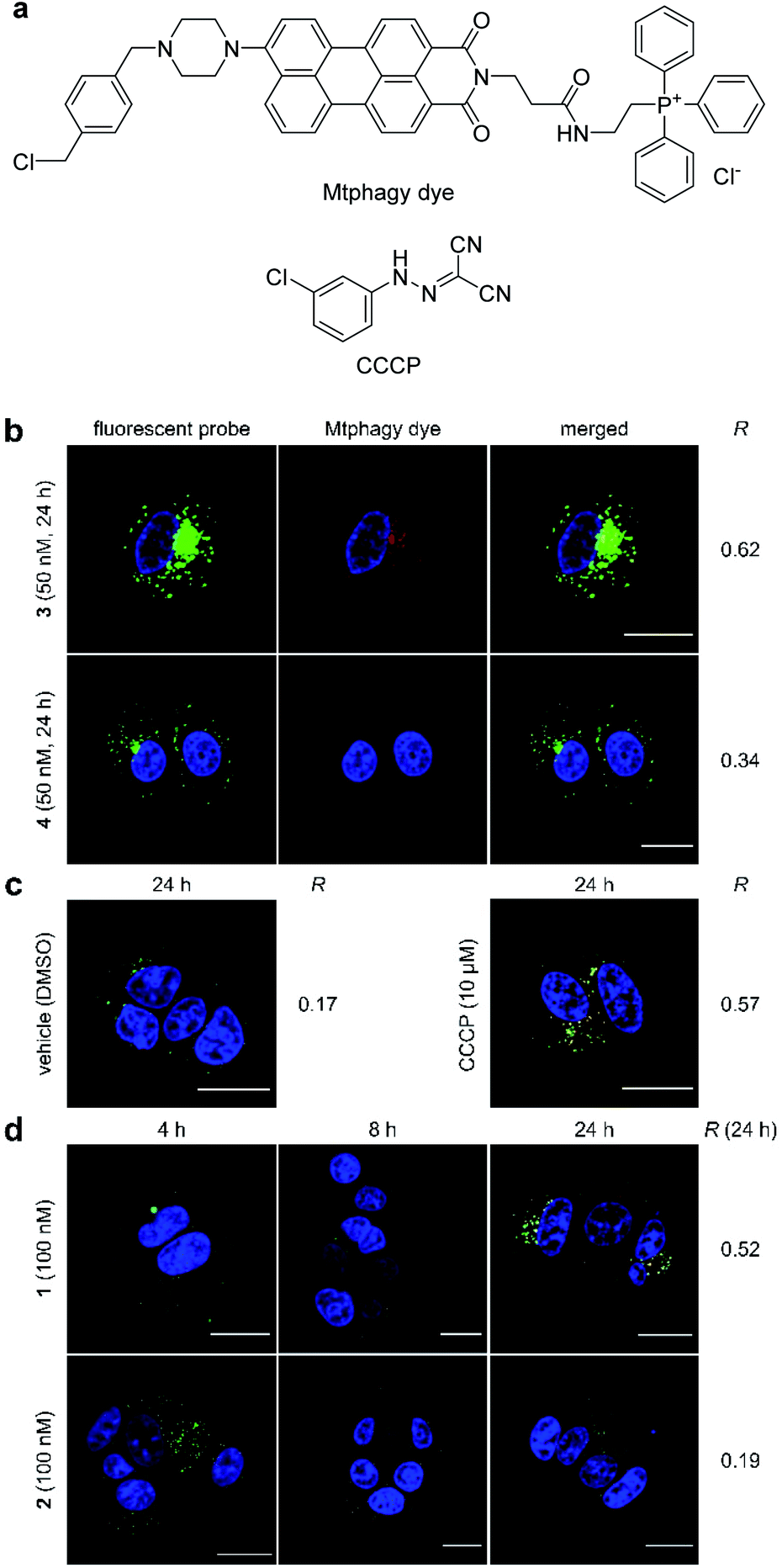 | ||
| Fig. 4 Mitophagy induced by 1–4. (a) Structures of Mtphagy dye and carbonyl cyanide m-chlorophenylhydrazone (CCCP). (b) Confocal fluorescent images obtained using fluorescent probe 3 or 4 (50 nM) in the presence of Mtphagy dye (red) after 24 h incubation with MCF-7 cells. (c) Confocal fluorescent images obtained using Mtphagy dye (red) and lysosome-selective dye (Lyso dye, green). Vehicle (DMSO) and CCCP (10 μM) were added and incubated for 24 h. (d) Confocal fluorescent images obtained using Mtphagy dye and Lyso dye after incubation with 1 or 2 (100 nM) for 4, 8, and 24 h. Subcellular regions colored in yellow in the images indicate colocalization of the Mtphagy dye and synthetic probes (b) or Lyso dye (c, d). The nuclei were stained with Hoechst 33342. Scale bar represents 20 μm. R value represents Pearson's correlation coefficients calculated from the green and red fluorescence in each merged image. | ||
The observed mitophagy was attributable to the H+ transport activity of 3, as disruption of the electrochemical H+ gradient of the inner mitochondrial membrane (IMM) causes mitochondrial malfunction and subsequent mitophagy. To further investigate the correlation between mitochondrial depolarization and mitophagy, the function of the parent natural product 1 was compared with that of carbonyl cyanide m-chlorophenylhydrazone (CCCP, Fig. 4a), which is known to disrupt the H+ gradient of the IMM.56 In this imaging study, green Lyso dye and red Mtphagy dye were co-incubated with MCF-7 cells for 24 h in the absence and presence of CCCP or 1. Whereas the fluorescence signals from Lyso dye and Mtphagy dye barely overlapped (R = 0.17) in the negative control experiment (DMSO), treatment with CCCP (10 μM) caused a significant overlap (R = 0.57) after 24 h (Fig. 4c). When gramicidin A (1, 100 nM) was used (Fig. 4d), the green and red colors were merged in a time-dependent fashion. Upon 24 h incubation, a high correlation value was obtained (R = 0.52), indicating that 1 had a potent mitophagy-inducing effect similar to CCCP. Applying the same concentration of inactive 2 (100 nM, R = 0.19, Fig. 4d) did not promote mitophagy after 24 h, further demonstrating that the mitophagy was selectively elicited by active 1. The results in Fig. 4 strongly indicated that the localization of ion-channel forming 1 to mitochondria and disruption of the H+ gradient across the IMM prompted the cellular stress response, resulting in fusion of mitochondrial fragments with lysosomes via the mitophagy process.
Mitochondrial depolarization effects of gramicidin A
Disruption of the H+ gradient across IMM of MCF-7 by 1 was quantified in separate experiments using tetramethylrhodamine methyl ester (TMRM, Fig. 5a). Cationic TMRM accumulates in the negatively polarized IMM and disperses from the mitochondria upon depolarization, thereby acting as a fluorescent reporter of the H+ gradient. Specifically, the TMRM-stained area per cell, which is calculated by image analysis, is proportional to the degree of the retained H+ gradient. When active 1 (100 nM) was employed, the H+ gradient gradually decreased during the 24 h incubation (Fig. 5b, red), strongly suggesting the H+ channel activity of 1 in the IMM. On the other hand, inactive 2 did not depolarize the IMM (Fig. 5b, cyan), even though the same concentration was added. Since 1 decreased the H+ gradient around its GI50 value (82 nM), the mitochondrial depolarization effect of 1 must associate with its potent cytostatic activity. Here, we revealed that ion-channel forming 1 depolarized not only the plasma membrane, but also the mitochondrial membrane.57–61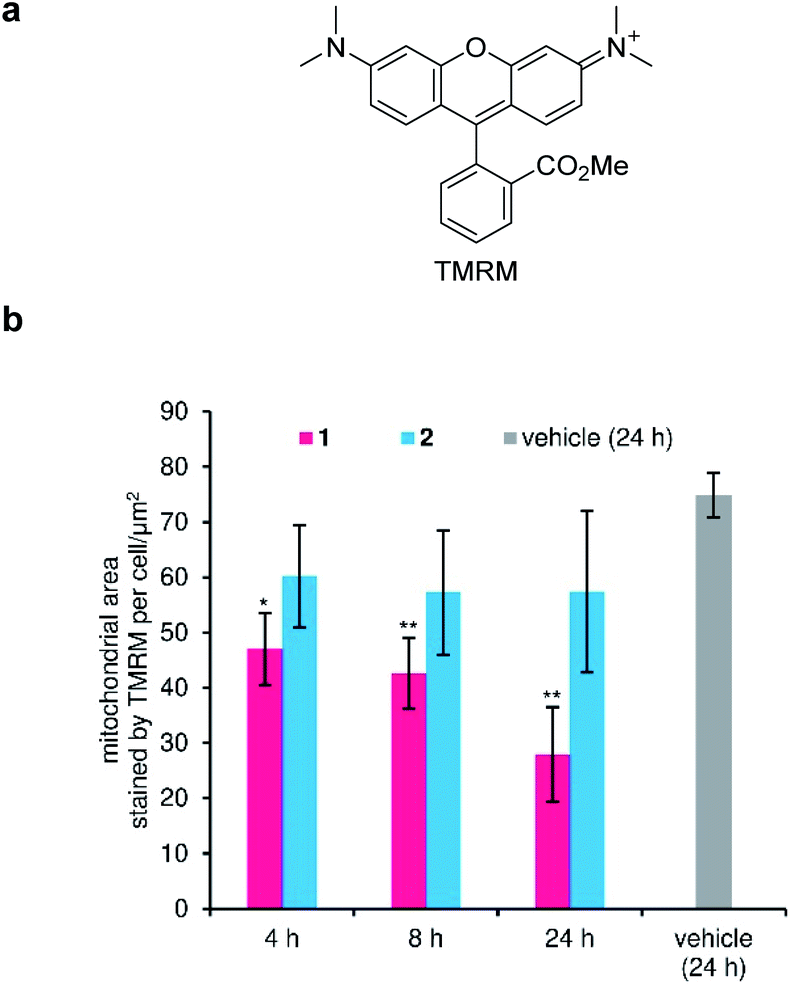 | ||
| Fig. 5 Depolarization activity of the inner mitochondrial membrane (IMM) by 1. (a) Structure of tetramethylrhodamine methyl ester (TMRM). (b) Time-dependent change in the fluorescence from TMRM. Compounds (100 nM) were added and incubated for 4, 8, and 24 h. Each plot was made from three images as mean ± SD. Asterisks indicate significant differences compared with vehicle control (DMSO, 24 h) by Dunnett's test. *P < 0.05, **P < 0.01. | ||
Inhibition of ATP production in mitochondria by gramicidin A
Mitochondria fuel cellular function through the synthesis of ATP by oxidative phosphorylation. The H+ gradient across the IMM creates potential energy, which is utilized by FoF1-ATP synthase to produce ATP.62 As depolarization of the IMM by 1 was expected to decrease ATP synthesis, we next analyzed perturbation of the mitochondrial ATP production rate by 1. In this assay, MCF-7 cells were permeabilized with digitonin to give a solution of intact mitochondria, which was applied to an enzymatic ATP-detection system.63,64 As a positive control, we adopted oligomycin A, an inhibitor of FoF1-ATP synthase (Fig. 6a).65 Under these conditions, oligomycin potently decreased the ATP concentration with an EC50 value of 1.3 ± 0.5 nM (black line, Fig. 6b). Alternatively, compound 1 inhibited ATP production in a dose-dependent manner (pink line), and the EC50 was calculated to be 67 ± 5 nM.66 This value was comparable to the required concentrations for the IMM depolarization (100 nM), mitophagy-induction (100 nM), and cytostatic activity (42 nM), corroborating the interconnection of these series of events. | ||
| Fig. 6 Inhibition of ATP production by 1 using membrane-permeabilized MCF-7 cells. (a) Structure of oligomycin A. (b) Concentration-dependent inhibition of ATP production. The ATP production rate was normalized against the rates obtained by adding vehicle (DMSO) as 100% and 5 μM oligomycin A as 0%. Each plot is displayed as mean ± SD of three replicates. The EC50 values of 1 and oligomycin A were calculated as 67 ± 5 nM (pink) and 1.3 ± 0.5 nM, respectively, by sigmoidal curve fittings. | ||
Cell cycle arrest by gramicidin A
Since ATP production in MCF-7 cells consists of 80% oxidative phosphorylation,67 mitochondrial malfunction would suppress cell growth due to the cellular energy depletion.68 To decipher the details of the cytostatic activity of 1, we examined the arrest of cell cycle progression by 1 (Fig. 7).69,70 Flow cytometric analysis of the cell population was performed after 48 h treatment of MCF-7 cells with varied concentrations of 1 (20–500 nM). The proportion of cells in G1 was dose-dependently increased in proportion to the decrease in S. On the other hand, the change in the G2/M phase was smaller than that in G1 and S phases. Because it is known that an energy-sensitive check exists at the G1/S border, these data indicated that the depleted ATP levels induced by 1 did not satisfy the energy requirement for passage through G1 into S phase. The cell cycle arrest at the energetic checkpoint again supported the significance of the depolarization of mitochondria and the ensuing ATP reduction as the mode of action of 1.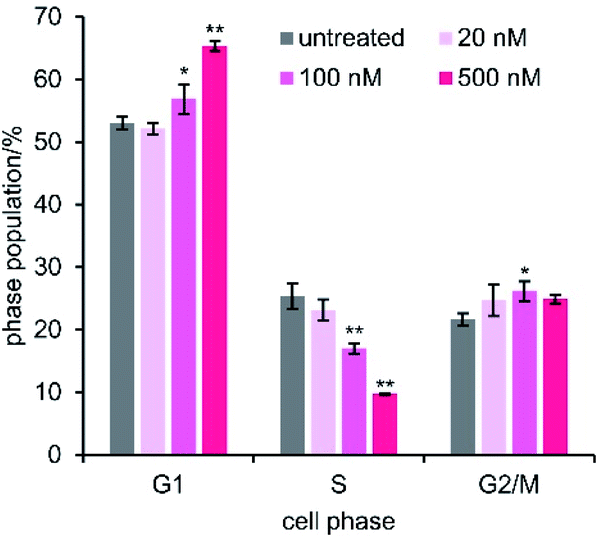 | ||
| Fig. 7 Cell cycle analysis. MCF-7 cells were treated with 1 for 48 h. The values are displayed as mean ± SD of three independent experiments. Asterisks indicate significant differences compared with an untreated control by Dunnett's test. *P < 0.05, **P < 0.01. | ||
Conclusions
The present study on gramicidin A (1) integrated total synthesis, imaging analysis, and functional investigation to decipher the unprecedented spatiotemporal behavior of 1 in MCF-7 cancer cells. We first synthetically prepared 15-mer peptide 1 and its BODIPY-conjugated fluorescent probe 3 as well as inactive analogue 2 and its fluorescent probe 4. Application of the synthetic 1–4 revealed the multiple cellular functions of 1 that ultimately result in the growth inhibition of MCF-7 cells at a nanomolar concentration. Compound 1 quickly depolarized the plasma membrane within 1 h, and then selectively localized to the mitochondria and reduced the H+ gradient of the IMM within 24 h. Consequently, the ion balance across both the plasma and mitochondrial membranes was disrupted, presumably by the facile ion transport though the pore of the head-to-head β6.3-helical dimer of 1. The ion-channel function of 1 in the IMM leads to additional cellular responses: e.g., eradication of the H+ gradient decreases the ATP concentration, and the resultant ATP depletion causes cell cycle arrest at the G1 energetic checkpoint. Moreover, the mitochondrial malfunction triggered elimination of the entire organelle by fusion with the lysosomes through mitophagy. Although how 1 achieves selective localization to the mitochondria remains to be elucidated at the molecular level,71 we uncovered the intricate time-dependent cellular behavior of potent cytostatic agent 1 for the first time. Mitochondria are energy-producing organelles with essential functions in cell biology, and are promising therapeutic targets for treating cancer.72,73 Because 1 has unique mitochondrial function and shows no structural similarity with typical mitochondria-targeting compounds,74,751 and its derivatives should serve as new platform structures for the development of novel mitochondrial modulators and anticancer agents.While 1, inactive analogue 2, and polytheonamide B (5) all share the D,L-alternating sequences and function as ion channels, their cellular behaviors are distinct from each other. Despite a difference in only one substituent (Me for 1, OH for 2), 2 does not accumulate to the mitochondria and possesses 24-fold weaker cytostatic activity against MCF-7 cells. The 48-mer peptide 5 rapidly diminishes the potential across the plasma membrane, localizes to acidic lysosomes, and neutralizes the lysosomal H+ gradient, causing apoptotic cell death of MCF-7 cells. These contrasting behaviors among the three related peptides are likely programed by the side chain structures and the overall sequence of their amino acid components. Therefore, the structural and functional differences of 1, 2, and 5 offer exciting opportunities for potentially controlling their ion channel function, organelle selectivity, anticancer activity, and even antibacterial activity by structural alterations. Such investigation should lead to the development of tailor-made β6.3-helical ion channels for selectively modulating the ion balance of various cells and organelles.
Data availability
The experimental procedures and additional data can be found in the ESI.†Author contributions
H. I. and M. I. conceived and designed the study. Y.-W. X. performed the synthesis of probes and the assays. S. D. performed the cancer panel assay. H. I. and M. I. co-wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by Grant-in-Aids for Scientific Research (S) (JSPS, JP17H06110) to M. I., and for Scientific Research (C) (JSPS, JP21K05286) to H. I. The cancer panel assay was supported by JSPS KAKENHI Grant Number JP16H06276 (AdAMS).Notes and references
- B. C. Pressman, Annu. Rev. Biochem., 1976, 45, 501–530 CrossRef CAS PubMed.
- T. T. Mai, A. Hamaï, A. Hienzsch, T. Cañeque, S. Müller, J. Wicinski, O. Cabaud, C. Leroy, A. David, V. Acevedo, A. Ryo, C. Ginestier, D. Birnbaum, E. Charafe-Jauffret, P. Codogno, M. Mehrpour and R. Rodriguez, Nat. Chem., 2017, 9, 1025–1033 CrossRef CAS PubMed.
- X. Huang, B. Borgström, J. Stegmayr, Y. Abassi, M. Kruszyk, H. Leffler, L. Persson, S. Albinsson, R. Massoumi, I. G. Scheblykin, C. Hegardt, S. Oredsson and D. Strand, ACS Cent. Sci., 2018, 4, 760–767 CrossRef CAS PubMed.
- G. M. Cragg, P. G. Grothaus and D. J. Newman, Chem. Rev., 2009, 109, 3012–3043 CrossRef CAS PubMed.
- A. Henninot, J. C. Collins and J. M. Nuss, J. Med. Chem., 2018, 61, 1382–1414 CrossRef CAS PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2020, 83, 770–803 CrossRef CAS PubMed.
- R. D. Hotchkiss and R. J. Dubos, J. Biol. Chem., 1940, 132, 791–792 CrossRef CAS.
- R. Sarges and B. Witkop, J. Am. Chem. Soc., 1965, 87, 2011–2020 CrossRef CAS PubMed.
- T. Hamada, S. Matsunaga, G. Yano and N. Fusetani, J. Am. Chem. Soc., 2005, 127, 110–118 CrossRef CAS PubMed.
- D. W. Urry, M. C. Goodall, J. D. Glickson and D. F. Mayers, Proc. Natl. Acad. Sci. U. S. A., 1971, 68, 1907–1911 CrossRef CAS PubMed.
- A. S. Arseniev, I. L. Barsukov, V. F. Bystrov, A. L. Lomize and Y. A. Ovchinnikov, FEBS Lett., 1985, 186, 168–174 CrossRef CAS PubMed.
- R. R. Ketchem, W. Hu and T. A. Cross, Science, 1993, 261, 1457–1460 CrossRef CAS PubMed.
- B. A. Wallace, J. Struct. Biol., 1998, 121, 123–141 CrossRef CAS PubMed.
- F. Kovacs, J. Quine and T. A. Cross, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 7910–7915 CrossRef CAS PubMed.
- D. A. Kelkar and A. Chattopadhyay, Biochim. Biophys. Acta, 2007, 1768, 2011–2025 CrossRef CAS PubMed.
- T. Hamada, S. Matsunaga, M. Fujiwara, K. Fujita, H. Hirota, R. Schmucki, P. Güntert and N. Fusetani, J. Am. Chem. Soc., 2010, 132, 12941–12945 CrossRef CAS PubMed.
- V. B. Myers and D. A. Haydon, Biochim. Biophys. Acta, 1972, 274, 313–322 CrossRef CAS.
- M. Iwamoto, H. Shimizu, I. Muramatsu and S. Oiki, FEBS Lett., 2010, 584, 3995–3999 CrossRef CAS PubMed.
- A. Hayata, H. Itoh and M. Inoue, J. Am. Chem. Soc., 2018, 140, 10602–10611 CrossRef CAS PubMed.
- Y.-W. Xue, A. Hayata, H. Itoh and M. Inoue, Chem.–Eur. J., 2019, 25, 15198–15204 CrossRef CAS PubMed.
- For an account of our previous studies on 5 and its analogues, see: H. Itoh and M. Inoue, Acc. Chem. Res., 2013, 46, 1567–1578 CrossRef CAS PubMed.
- J. M. David, T. A. Owens, S. P. Barwe and A. K. Rajasekaran, Mol. Cancer Ther., 2013, 12, 2296–2307 CrossRef CAS PubMed.
- D. Wijesinghe, M. C. M. Arachchige, A. Lu, Y. K. Reshetnyak and O. A. Andreev, Sci. Rep., 2013, 3, 3560 CrossRef PubMed.
- J. M. David, T. A. Owens, L. J. Inge, R. M. Bremner and A. K. Rajasekaran, Mol. Cancer Ther., 2014, 13, 788–799 CrossRef CAS PubMed.
- T. Chen, Y. Wang, Y. Yang, K. Yu, X. Cao, F. Su, H. Xu, Y. Peng, Y. Hu, F. Qian and Z. Wang, Biol. Res., 2019, 52, 57 CrossRef PubMed.
- R.-Q. Wang, J. Geng, W.-J. Sheng, X.-J. Liu, M. Jiang and Y.-S. Zhen, Cancer Cell Int., 2019, 19, 145 CrossRef PubMed.
- J. M. David and A. K. Rajasekaran, J. Kidney Cancer VHL, 2015, 2, 15–24 CrossRef PubMed.
- Y. Takada, H. Itoh, A. Paudel, S. Panthee, H. Hamamoto, K. Sekimizu and M. Inoue, Nat. Commun., 2020, 11, 4935 CrossRef CAS PubMed.
- For a review on the functional modulation of natural products, see: H. Itoh and M. Inoue, Chem. Rev., 2019, 119, 10002–10031 CrossRef CAS PubMed.
- W. C. Chan and P. D. White, Fmoc Solid Phase Peptide Synthesis. A Practical Approach, Oxford University Press, New York, 2000 Search PubMed.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS PubMed.
- F. García-Martín, M. Quintanar-Audelo, Y. García-Ramos, L. J. Cruz, C. Gravel, R. Furic, S. Côté, J. Tulla-Puche and F. Albericio, J. Comb. Chem., 2006, 8, 213–220 CrossRef PubMed.
- H.-M. Yu, S.-T. Chen and K.-T. Wang, J. Org. Chem., 1992, 57, 4781–4784 CrossRef CAS.
- B. Bacsa, K. Hováti, S. Bõsze, F. Andreae and C. O. Kappe, J. Org. Chem., 2008, 73, 7532–7542 CrossRef CAS PubMed.
- S. L. Pedersen, P. Tofteng, L. Malik and K. J. Jensen, Chem. Soc. Rev., 2012, 41, 1826–1844 RSC.
- L. A. Carpino, J. Am. Chem. Soc., 1993, 115, 4397–4398 CrossRef CAS.
- H. Li, X. Jiang, Y.-H. Ye, C. Fan, T. Romoff and M. Goodman, Org. Lett., 1999, 1, 91–94 CrossRef CAS PubMed.
- D. A. Pearson, M. Blanchette, M. L. Baker and C. A. Guindon, Tetrahedron Lett., 1989, 30, 2739–2742 CrossRef CAS.
- J. Coste, D. Le-Nguyen and B. Castro, Tetrahedron Lett., 1990, 31, 205–208 CrossRef CAS.
- T. Yamori, Cancer Chemother. Pharmacol., 2003, 52, 74–79 CrossRef PubMed.
- S. Yaguchi, Y. Fukui, I. Koshimizu, H. Yoshimi, T. Matsuno, H. Gouda, S. Hirono, K. Yamazaki and T. Yamori, J. Natl. Cancer Inst., 2006, 98, 545–556 CrossRef CAS PubMed.
- See ESI† for dose-response curves and GI50 fingerprints of 5.
- V. Vichai and K. Kirtikara, Nat. Protoc., 2006, 1, 1112–1116 CrossRef CAS PubMed.
- Higher concentrations of 1 (1–10 μM) are reported to elicit cytotoxic activity against several cancer cells. See ref. 22 and 25 for examples.
- Gramicidin D, which is a mixture of 1 and its congeners, is widely utilized as a plasma membrane depolarizing agent in biological studies. For example, see: F. D. Virgilio, P. D. Lew, T. Andersson and T. Pozzan, J. Biol. Chem., 1987, 262, 4574–4579 CrossRef PubMed.
- T. Braner, D. F. Hulser and R. J. Strasser, Biochim. Biophys. Acta, 1984, 771, 208–216 CrossRef.
- Localization of probes 3 and 4 to the plasma membrane was not observed, presumably due to the elution of 3 and 4 from the plasma membrane upon replacement of the medium for fluorescent imaging.
- L. V. Johnson, M. L. Walsh, B. J. Bockus and L.-B. Chen, J. Cell Biol., 1981, 88, 526–535 CrossRef CAS PubMed.
- S. V. Costes, D. Daelemans, E. H. Cho, Z. Dobbin, G. Pavlakis and S. Lockett, Biophys. J., 2004, 86, 3993–4003 CrossRef CAS PubMed.
- K. W. Dunn, M. M. Kamocka and J. H. McDonald, Am. J. Physiol.: Cell Physiol., 2011, 300, C723–C742 CrossRef CAS PubMed.
- Mitochondrial localization of 3 was also confirmed by tetramethylrhodamine methyl ester (TMRM). See also Fig. S5a.†.
- M. Koivusalo, C. Welch, H. Hayashi, C. C. Scott, M. Kim, T. Alexander, N. Touret, K. M. Hahn and S. Grinstein, J. Cell Biol., 2010, 188, 547–563 CrossRef CAS PubMed.
- A. I. Ivanov, Methods Mol. Biol., 2008, 440, 15–33 CrossRef CAS PubMed.
- K. Palikaras, E. Lionaki and N. Tavernarakis, Nat. Cell Biol., 2018, 20, 1013–1022 CrossRef CAS PubMed.
- H. Iwashita, S. Torii, N. Nagahora, M. Ishiyama, K. Shioji, K. Sasamoto, S. Shimizu and K. Okuma, ACS Chem. Biol., 2017, 12, 2546–2551 CrossRef CAS PubMed.
- M. L. R. Lim, T. Minamikawa and P. Nagley, FEBS Lett., 2001, 503, 69–74 CrossRef CAS PubMed.
- We confirmed the negligible effect of 1 and 2 on lysosomes by measuring the lysosomal H+ gradient using LysoTracker Red DND-99 (Fig. S6†).
- The mitochondrial activity of 1 has been reported (ref. 22 and 59–61). This study is distinct from previous studies because it deciphered the time-dependent localization of 1 to the mitochondria and the ensuing various cellular responses.
- J. B. Chappell and A. R. Crofts, Biochem. J., 1965, 95, 393–402 CrossRef CAS PubMed.
- H. Rottenberg and R. E. Koeppe II, Biochemistry, 1989, 28, 4355–4360 CrossRef CAS PubMed.
- A. I. Sorochkina, E. Y. Plotnikov, T. I. Rokitskaya, S. I. Kovalchuk, E. A. Kotova, S. V. Sychev, D. B. Zorov and Y. N. Antonenko, PLoS One, 2012, 7, e41919 CrossRef CAS PubMed.
- W. Junge and N. Nelson, Annu. Rev. Biochem., 2015, 84, 631–657 CrossRef CAS PubMed.
- F. B. Rudolph, B. W. Baugher and R. S. Beissner, Methods Enzymol., 1979, 63, 22–42 CAS.
- A. Signorile, L. Micelli, D. De Rasmo, A. Santeramo, F. Papa, R. Ficarella, G. Gattoni, S. Scacco and S. Papa, Biochim. Biophys. Acta, 2014, 1843, 675–684 CrossRef CAS PubMed.
- Oligomycin binds to the Fo domain and inhibits proton translocation: R. J. Devenish, M. Prescott, G. M. Boyle and P. Nagley, J. Bioenerg. Biomembr., 2000, 32, 507–515 CrossRef CAS PubMed.
- Compound 1 was reported to directly inhibit purified mammalian Na+/K+-ATPase. This mode of action was not likely in this assay, however, because the IC50 value of the direct inhibition (8.1 μM) was 120-fold larger than the EC50 value (67 nM) in this study: Y. Takada, K. Matsuo and T. Kataoka, Mol. Cell. Biochem., 2008, 319, 99–103 CrossRef CAS PubMed.
- M. Guppy, P. Leedman, X. Zu and V. Russell, Biochem. J., 2002, 364, 309–315 CrossRef CAS PubMed.
- We recently found that yaku'amide B functions as a potent cytostatic agent by inhibiting mitochondrial FoF1-ATP synthase. K. Kitamura, H. Itoh, K. Sakurai, S. Dan and M. Inoue, J. Am. Chem. Soc., 2018, 140, 12189–12199 CrossRef CAS PubMed.
- S. Sweet and G. Singh, Cancer Res., 1995, 55, 5164–5167 CAS.
- M. Salazar-Roa and M. Malumbres, Trends Cell Biol., 2017, 27, 69–81 CrossRef CAS PubMed.
- Different lipid compositions of organelles are likely to affect the distinct organelle selectivities of 1, 2, and 5: G. van Meer, D. R. Voelker and G. W. Feigenson, Nat. Rev. Mol. Cell Biol., 2008, 9, 112–124 CrossRef CAS PubMed.
- S. Fulda, L. Galluzzi and G. Kroemer, Nat. Rev. Drug Discovery, 2010, 9, 447–464 CrossRef CAS PubMed.
- S. Weinberg and N. S. Chandel, Nat. Chem. Biol., 2015, 11, 9–15 CrossRef CAS PubMed.
- S. Hong and P. L. Pedersen, Microbiol. Mol. Biol. Rev., 2008, 72, 590–641 CrossRef CAS PubMed.
- S. Wisnovsky, E. K. Lei, S. R. Jean and S. O. Kelley, Cell Chem. Biol., 2016, 23, 917–927 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc02024f |
| This journal is © The Royal Society of Chemistry 2022 |