
A robust and self-healing elastomer achieved by a thio-β-diketone-Cu(II) coordination and H-bonding dual crosslinked system†
Xiaoming
An
a,
Jie
Liu
a,
Jia-Han
Zhang
b,
Xinxin
Huang
a,
Tangsong
Zhu
a,
Hongping
Yan
c,
Xudong
Jia
*ad and
Qiuhong
Zhang
 *a
*a
aDepartment of Polymer Science and Engineering, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing, 210023, P. R. China. E-mail: jiaxd@nju.edu.cn; chemzqh@nju.edu.cn
bCollaborative Innovation Center of Advanced Microstructures, School of Electronic Science and Engineering, Nanjing University, Nanjing, 210093, China
cDepartment of Chemical Engineering, Stanford University, Stanford, CA 95403, USA
dState Key Laboratory of Coordination Chemistry, Nanjing University, Nanjing, 210023, P. R. China
First published on 13th May 2022
Abstract
Elastomers possessing good mechanical strength and self-healing capability are showing great importance in stretchable electronics, since they can play the role of robust substrates for devices, and prolong the service life of devices. However, it is hard to balance the trade-off between the high mechanical strength and self-healing ability. Here we propose the synergetic strategy via combining thio-β-diketone-Cu2+ metal–ligand (M–L) coordination with hydrogen bonds in one system, through which high mechanical strength, good elasticity and self-healing ability are achieved. The elastomer displays excellent mechanical performances (with a fracture strength of 4.35 MPa and a fracture strain of 3400%). Meanwhile, the elastomer can realize a high self-healing efficiency (94%) within only 3 hours at 80 °C. X-ray absorption fine structure (XAFS) analysis demonstrates the reconfiguration of M–L coordination at the molecular level, and small angle X-ray scattering (SAXS) during stretching demonstrates the microphase structure change of the polymer at the nanoscale level. Based on the elastomer, a self-healable pressure sensor and a self-healable strain sensor are fabricated.
Introduction
High mechanical strength is a desirable property for those materials used in wearable devices,1 electronic skins2 and human–machine interactions.3 These stretchable electronic devices are expected to possess sensing capacity.4,5 Generally, a series of wearable devices were designed to detect the tiny change in the capacitance6 or resistance7 of materials, implementing the sensing function ingeniously. Commercial elastomers used in wearable electronics such as Sylgard 184 (polydimethylsiloxane) usually possess relatively low strength because of their aligned chains,8 so novel synthetic elastomers are needed and exploited for the application of stretchable devices. In practice, these electronics are susceptible to either ruptures or scratches during our daily body movements. In order to prolong the service life of devices, it is imperative to endow materials with the self-healing properties. Traditionally, an extrinsic strategy such as use of a microencapsulated healing agent9 has been utilized to build systems equipped with self-healing ability. However, there still exists a challenge to achieve elastomers with both robust mechanical properties and self-healing performance simultaneously.Recently, some approaches have been developed to fabricate elastomers for satisfying the exclusive demands, including interpenetrating double networks10,11 and nanocomposites.12 Besides, reversible covalent bonds (for instance, D–A reactions,13,14 disulfide,15 trithiocarbonate,16 pinacol,17 thiol–anhydride,18 olefin metathesis,19 and boronic ester20) and reversible non-covalent bonds (such as host–guest interactions,21 topological transformation,22 molecular zipper,23 mechanical interlock,24 π–π stacking,25 electron donor–acceptors,26 ion interactions,27 dipole interactions,28 hydrogen bonds29 and metal–ligand coordination30) are usually introduced into dynamic networks to combine self-healing and pronounced mechanical properties, due to their spontaneous and rapid bond-reformation abilities. Among these dynamic interactions, M–L coordination has potential for its unique features: the strength of coordination bonds could be tuned around a rather broad range from van der Waals interactions to covalent bonds; meanwhile, some M–L interactions could possess thermodynamic stability and kinetic lability, which were beneficial to strengthen the mechanical properties and self-healing ability.31 To date, various ligands such as salicylic acid,32 Schiff bases,33 N-heterocyclic carbenes,34 nitrile,35 ferrocene,36 catechols,37,38 imidazoles,39 carboxylates,40,41 phosphates,42 dimethylglyoxime,43 thiolates,44 bipyridine,45 terpyridine,46 pyridinecarboxamine,47 boron–nitrogen coordination48 as well as metal–organic cages49 have been explored to achieve the expected mechanical strength or desired self-healing capability for target materials. However, most of these ligands were usually trapped in the trade-offs between strong coordination and weak interactions when introduced in polymers. The strong coordination bonds often possess high binding affinity which would decrease the healing efficiency, while the weak interactions would result in weak mechanical properties. Therefore, the rational design of ligands with promising binding affinity and certain dynamicity simultaneously is likely to provide a route to reach the balance.
In our previous work,50 the diketone ligand was firstly used in self-healing polymers. Based on the research, we designed primary thio-β-diketone as the coordinating ligand which contained both sulphur and oxygen coordinating sites in one individual ligand. The union of strong and weak sites bestowed dynamic features on this ligand: the strong site provided strong and stable associations in polymers, while the weak one, which could break and re-form feasibly, guaranteed the healing ability of the polymer. Therefore, the thio-β-diketone ligand might possess the potential to enhance the mechanical strength of polymers, when it was introduced into polymer chains as coordination crosslinks. To realize the promotion of both the mechanical properties and self-healing ability, here we proposed a dual dynamic crosslinked system, combining the thio-β-diketone-Cu2+ coordinating structure with hydrogen bonds in a urethane-linked elastomer, termed as PU–SO–Cu (Scheme 1). The strain at break of the material was as high as over 3400%, the stress at break reached 4.35 MPa, and the elastomer possessed high toughness (81 MJ m−3) and considerable fracture energy (44.65 kJ m−2). Meanwhile, the material could display nearly complete self-healing (efficiency up to 94%) under heating conditions in only 3 hours. Moreover, we fabricated a capacitive pressure sensor through depositing a gold conductive layer onto the as-prepared elastomer. And a resistive strain sensor was produced by spray-coating silver nanowires (AgNWs) onto the elastomer. These sensors were capable of monitoring pressures and human movements with considerable sensitivity. These devices could also self-repair and nearly restore the sensing ability after being wounded.
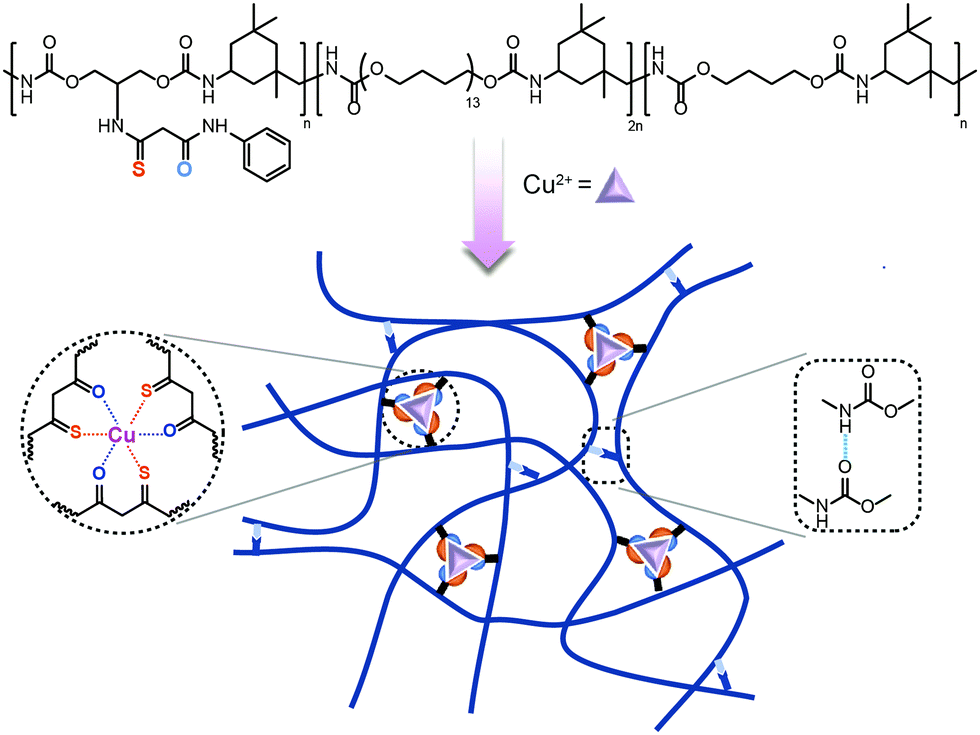 | ||
| Scheme 1 Schematic diagram of the stretchable and self-healing elastomer PU–SO–Cu. | ||
Experimental
Materials
N,N-Dimethylformamide (DMF) and methanol (MeOH) were supplied by General-Reagent. Dimethyl sulfoxide (DMSO), polytetramethylene glycol (PTMG, Mn = 1000), isophorone diisocyanate (IPDI), dibutyltin dilaurate (95%) and 1,4-butanediol (BDO) were supplied by Sigma-Aldrich (USA) and dried under vacuum before use. Aniline (99%), sulfur (98%), malonyl dichloride (96%) and copper(II) chloride (CuCl2·2H2O, 99.9%) were supplied by Aladdin Chemical. Deuterated solvents were purchased from Cambridge Isotope Laboratory (Andover, MA). Sliver nanowires (55–75 nm in diameter and 20–40 μm in length) were purchased from Zhejiang Kechuang Advanced Materials Technology LTD.Synthetic procedures
The chelating ligand (termed as SO) was prepared by following the literature.51 Phenylamine (1 mmol, 93 mg) and DMSO (0.3 mL, 4.5 equiv.) were added into a 10 mL tube containing anhydride maleic (1 mmol, 98 mg). The resulting mixture was shaken vigorously with a vortex mixer (5 min) to give a viscous pale yellow. Then 2-amino-1,3-propanediol (1 mmol, 91 mg), sulfur (1.25 mmol, 40 mg) and a magnetic stirrer bar were added. The tube was closed with a septum and the mixture was stirred at 50 °C for 16 h. After reaction, the reactant was purified by column chromatography on silica gel (eluent CH2Cl2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EtOAc 1:0 to 20:1), yielding a yellow solid (187 mg, 76% yield). The NMR spectra of the SO ligand are shown in Fig. S1, ESI.†
:EtOAc 1:0 to 20:1), yielding a yellow solid (187 mg, 76% yield). The NMR spectra of the SO ligand are shown in Fig. S1, ESI.†
PU–SO was prepared by a one-pot polymerization. Subsequently, IPDI (4 mmol) and a catalytic amount of dibutyltin dilaurate (DBTDL) were added into a three-necked flask equipped with a mechanical stirrer under nitrogen, followed by adding PTMG (Mn = 1000, 2 mmol) into the prepolymer and stirring at 80 °C for 5 h. Then, 1,4-butanediol (BDO, 1 mmol) and SO (1 mmol) were added into the prepolymer. The mixture was stirred at 80 °C for 5 h and the reactant was poured into a polytetrafluoroethylene (PTFE) plate. The plate was put in an oven at 80 °C for 24 h, and then the PU–SO film was prepared.
PU–SO (2 g) and CuCl2·2H2O (23 mg, Cu:SO = 1:3) were dissolved in 20 mL DMF. Then the reactants were stirred at 80 °C for 8 h and then concentrated. The concentrated solution was poured into a PTFE plate. The plate was dried at 80 °C for 24 h. The PU–SO–Cu film was finally prepared. The synthetic procedures of control samples PU–BD and PU–BD–Cu are provided in the ESI.†
Fabrication of devices
Preparation of a PU–SO–Cu based capacitive pressure sensor: the elastomer PU–SO–Cu was put on the steel plate and fixed by polyimide insulating tape. A PTFE mask (10 mm × 10 mm) was made and stuck onto the surface of the elastomer. Then, the gold layer was deposited on the substrate by thermal vacuum deposition.Preparation of a PU–SO–Cu based resistive strain sensor: the elastomer PU–SO–Cu was put on the steel plate and fixed by polyimide insulating tape. A PTFE mask was made and stuck onto the surface of the elastomer. Then, AgNWs (10 mg) were dispersed in 50 mL isopropyl alcohol, which was spray coated on the elastomer to form the resistive strain sensor.
Results and discussion
Since the SO molecule was considered as the chelating ligand to the Cu2+ cation, the UV-vis spectra of a series of samples with different stoichiometry were recorded. The coordination structure with a stoichiometry of 3:1 (SO to CuCl2) showed a minimum peak at ∼280 nm (Fig. 1a). This was attributed to the formation of the specific SO:Cu (3:1) coordination complex. The Fourier transform infrared spectroscopy (FTIR) spectra of these samples showed that none of the NCO groups (∼2260 cm−1) were left, indicating the full reaction of the isocyanate (Fig. S2, ESI†). Then the 1H NMR spectrum of PU–SO showed multiple peaks around 7.0 ppm, indicating that the thio-β-diketone moiety was incorporated into polymer chains (Fig. S3, ESI†). The gel permeation chromatography (GPC) measured average molecular weights of PU–SO were Mw = 173 kDa and Mn = 43 kDa. After coordination of CuCl2 in DMF solution, the linear chains evolved into polymer networks crosslinked by coordination bonds (termed as PU–SO–Cu). As shown in Fig. 1b, the appearance of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O peak at 1620 cm−1 in the FTIR spectrum could also indicate the existence of the SO moiety in the PU–SO polymer chain, while the peak disappeared in the control sample PU–BD with no SO moiety. Moreover, along with the increase of Cu2+ contents, this peak tended to decrease, which might have originated from the association of Cu2+ and ligand SO. The results of wide-angle X-ray diffraction (WXRD) presented that the broad peak in polymers shifted when Cu2+ was added, implying the association of Cu2+ with the ligand as well (Fig. S4, ESI†). To determine the valence state of the Cu cation in polymers, we used X-ray photoelectron spectroscopy (XPS) to measure the surface chemistry of PU–SO–Cu. The peaks at 932.7 eV and 952.6 eV were attributed to the Cu2+ coordination complex without any redox process,52 and the shake-up peaks at 940–944 eV indicated the presence of partly coordinated copper cations (Fig. 1c). The morphology of the coordinated network PU–SO–Cu was characterized by transmission electron microscopy (TEM), from which we could observe that the aggregation of about 4–6 nm from Cu-complexation was distributed in the polymer matrix (Fig. 1d).
O peak at 1620 cm−1 in the FTIR spectrum could also indicate the existence of the SO moiety in the PU–SO polymer chain, while the peak disappeared in the control sample PU–BD with no SO moiety. Moreover, along with the increase of Cu2+ contents, this peak tended to decrease, which might have originated from the association of Cu2+ and ligand SO. The results of wide-angle X-ray diffraction (WXRD) presented that the broad peak in polymers shifted when Cu2+ was added, implying the association of Cu2+ with the ligand as well (Fig. S4, ESI†). To determine the valence state of the Cu cation in polymers, we used X-ray photoelectron spectroscopy (XPS) to measure the surface chemistry of PU–SO–Cu. The peaks at 932.7 eV and 952.6 eV were attributed to the Cu2+ coordination complex without any redox process,52 and the shake-up peaks at 940–944 eV indicated the presence of partly coordinated copper cations (Fig. 1c). The morphology of the coordinated network PU–SO–Cu was characterized by transmission electron microscopy (TEM), from which we could observe that the aggregation of about 4–6 nm from Cu-complexation was distributed in the polymer matrix (Fig. 1d).
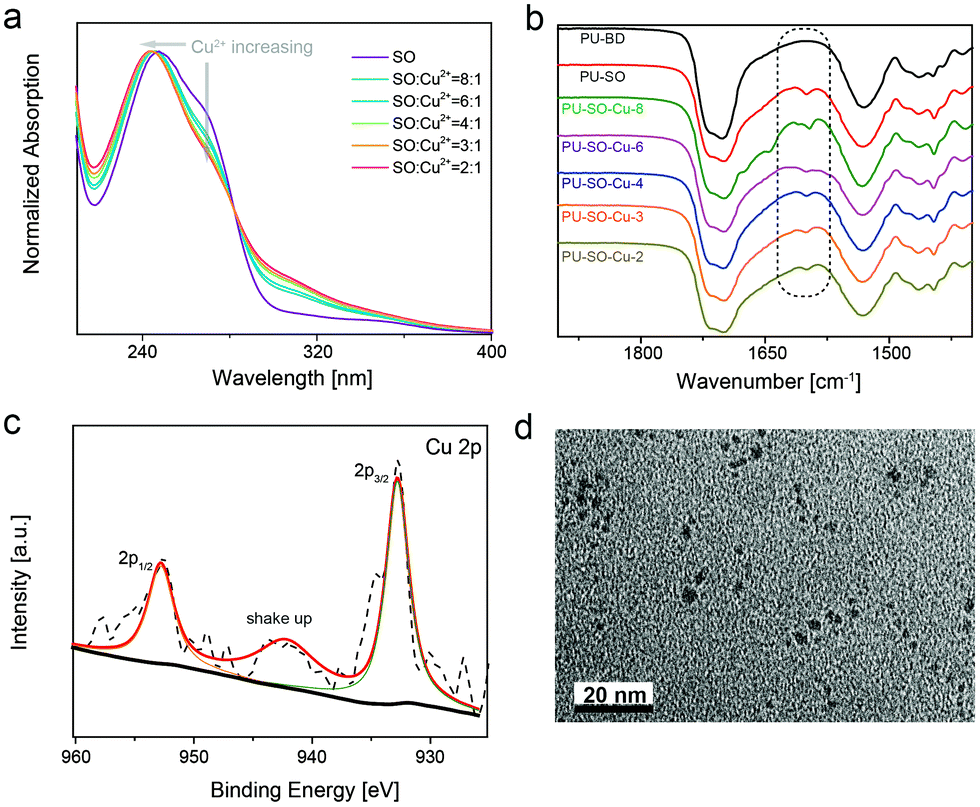 | ||
| Fig. 1 Structural characterization studies of the PU–SO–Cu elastomer. (a) UV-Vis spectra of samples with different SO:Cu2+ ratios in MeOH. The curves were normalized using the absorption band at about 250 nm. (b) FT-IR spectra of different samples. (c) The XPS spectrum of the Cu 2p region in PU–SO–Cu. (d) TEM image of a dynamic interaction system in the PU–SO–Cu substrate. Scale bar: 20 nm. | ||
The binding affinity53 (thermodynamic stability) and ligand exchange rate54 (kinetic lability) were equally important parameters influencing the properties of materials. According to the isothermal calorimetry titration (ITC) research on Cu2+–SO coordination, the binding affinity K was measured as 5.91 × 104, which was quite higher than the strength of the hydrogen bond55 (Fig. 2a and Table S1, ESI†). The strong binding affinity of coordination would result in strong crosslinks among polymers, increasing the modulus of materials. Meanwhile, the 1H NMR spectrum of the CuCl2–(SO)3 complex in MeOH at −40 °C displayed two sets of signal peaks around δ = 3.6, respectively, corresponding to methylene in associated and dissociated SO ligands. When the temperature increased (0 °C and 20 °C), these signals gradually turned into a double peak, indicating a rapid ligand exchange process between associated and dissociative ligands at ambient temperature (Fig. 2b). It was well-known that the dissociation and exchange of coordination dominated the dynamic mechanical properties of the networks.56 As expected, the frequency-dependent modulus of PU–SO–Cu was much higher than the moduli of PU–SO, PU–BD and PU–BD–Cu, measured by rheology (Fig. 2c). Besides, G′ > G′′ at all frequencies for the coordinated PU–SO–Cu, while other control samples (PU–SO, PU–BD and PU–BD–Cu) showed crossover (G′ = G′′), suggesting that a crosslinked network was generated by Cu–SO coordination. Furthermore, tensile tests of PU–SO–Cu, PU–SO, PU–BD and PU–BD–Cu were performed as shown in Fig. 2d, from which we could see that the mechanical properties of PU–SO–Cu were obviously higher than those of other samples (stress at break 4.25 ± 0.16 MPa, strain at break 3410 ± 190%, Young's modulus 1.92 ± 0.22 MPa and toughness 81 ± 6.5 MJ m−3, all collected detailed data are summarized in Table S2, ESI†). Considering that neither PU–SO, PU–BD nor PU–BD–Cu contained the Cu2+–SO coordination, herein we could further confirm that it was the stable and labile M–L coordination that improved the mechanical properties of the bulk material greatly.
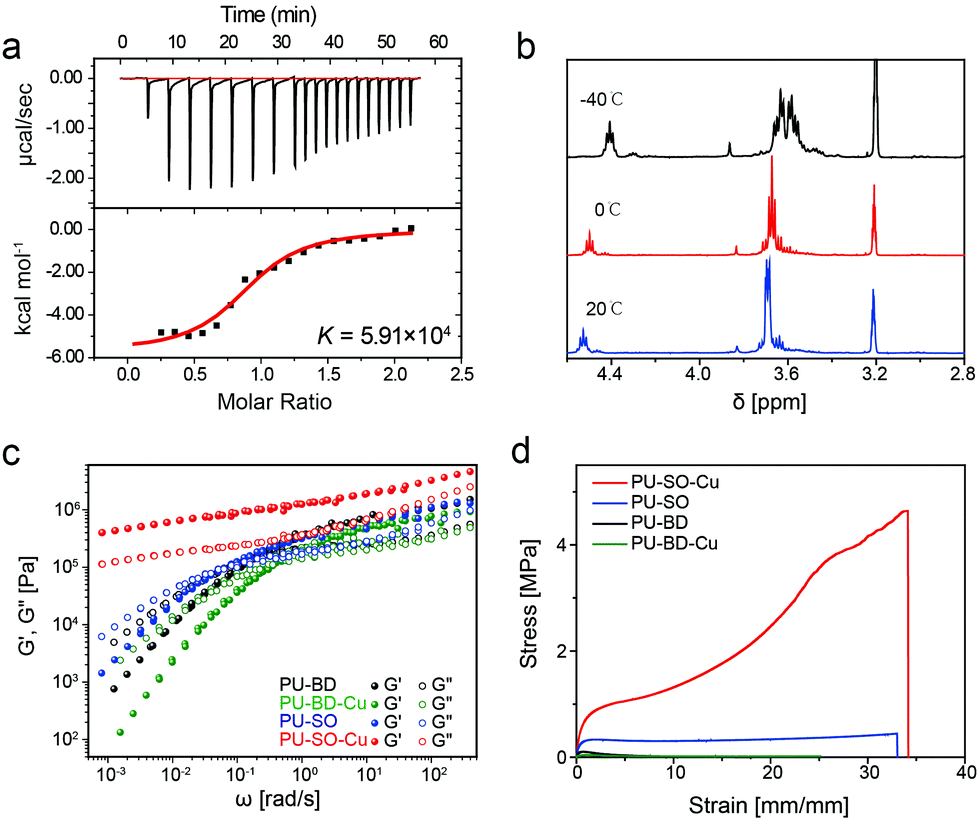 | ||
| Fig. 2 (a) The ITC titration data of the SO ligand with CuCl2 in anhydrous MeOH at 298 K. (b) Variable-temperature 1H nuclear magnetic resonance (1H NMR) for PU–SO–Cu. (c) Master curves of PU–BD, PU–BD–Cu, PU–SO and PU–SO–Cu scaled by time–temperature superposition (TTS) at a reference temperature of 20 °C. (d) Typical stress–strain curves of different samples including PU–BD, PU–BD–Cu, PU–SO and PU–SO–Cu, with a strain rate of 20 mm min−1. | ||
Tensile tests were also performed on films with different Cu2+ contents at room temperature. All the coordination-crosslinked materials showed typical rubber-like behaviours (Fig. 3a). In particular, the sample PU–SO–Cu (SO:Cu2+ = 3:1) possessed the highest tensile stress, which was consistent with the coordination stoichiometry of SO–Cu2+. Then the PU–SO–Cu system was chosen as the typical sample for further research. The Young's modulus of the sample increased as the tensile rate increased (Fig. 3b). Significantly, PU–SO–Cu showed excellent stretchability that the elongation at break approached up to 3400% (Fig. 3c). Moreover, we have noticed that PU–SO–Cu possessed good elastic and recovery performances. The cyclic tensile test is shown in Fig. 3d, and the sample possessed hysteresis and Mullins effects as derived from the volume fraction of the soft segment increased during the initial cycle. The sample was stretched to 6× its initial length and then unloaded, repeated for 10 cycles. The calculated energy dissipation and hysteresis proportions involving each cycle are presented in Fig. 3e, and the energy density dissipated within the loop of the first cycle was estimated to be about 2.83 MJ m−3. With this value arbitrarily set as 100%, 47% of the energy dissipation was observed to be lost in cycle 2, and then the dissipation decreased slightly with an increase in the number of cycles. After relaxing for 1 h, the sample fully restored to its original state during cycle 11, and the dissipation and hysteresis of cycle 11 were almost equal to those of the first cycle (Fig. 3d), demonstrating good self-recovery properties of the material. We conjectured that it was due to the reconfigurable Cu–SO coordination, as well as the reformation of hydrogen bonds. The cyclic tensile test curves of the material with increased maximum strains are also shown in Fig. S5, ESI.† Besides, the fracture energy of the film was 44.65 kJ m−2, calculated from fracture toughness experiments (Fig. S6, ESI†), which was comparable to those of the recently reported polyurethane elastomers (Fig. S7, ESI†). These observed mechanical behaviours indicated that our material was endowed with good mechanical strength as well as excellent elastic performances, by combining hydrogen bond and Cu–SO coordination interactions in the system. Furthermore, we performed stress-relaxation measurements for different samples,57 as shown in Fig. S8, ESI.† PU–SO–Cu released stress much slower than PU–SO did. The slower stress relaxation of PU–SO–Cu might be attributed to the network structure formed by Cu–SO coordination, which would restrict the mobility of polymer chains.
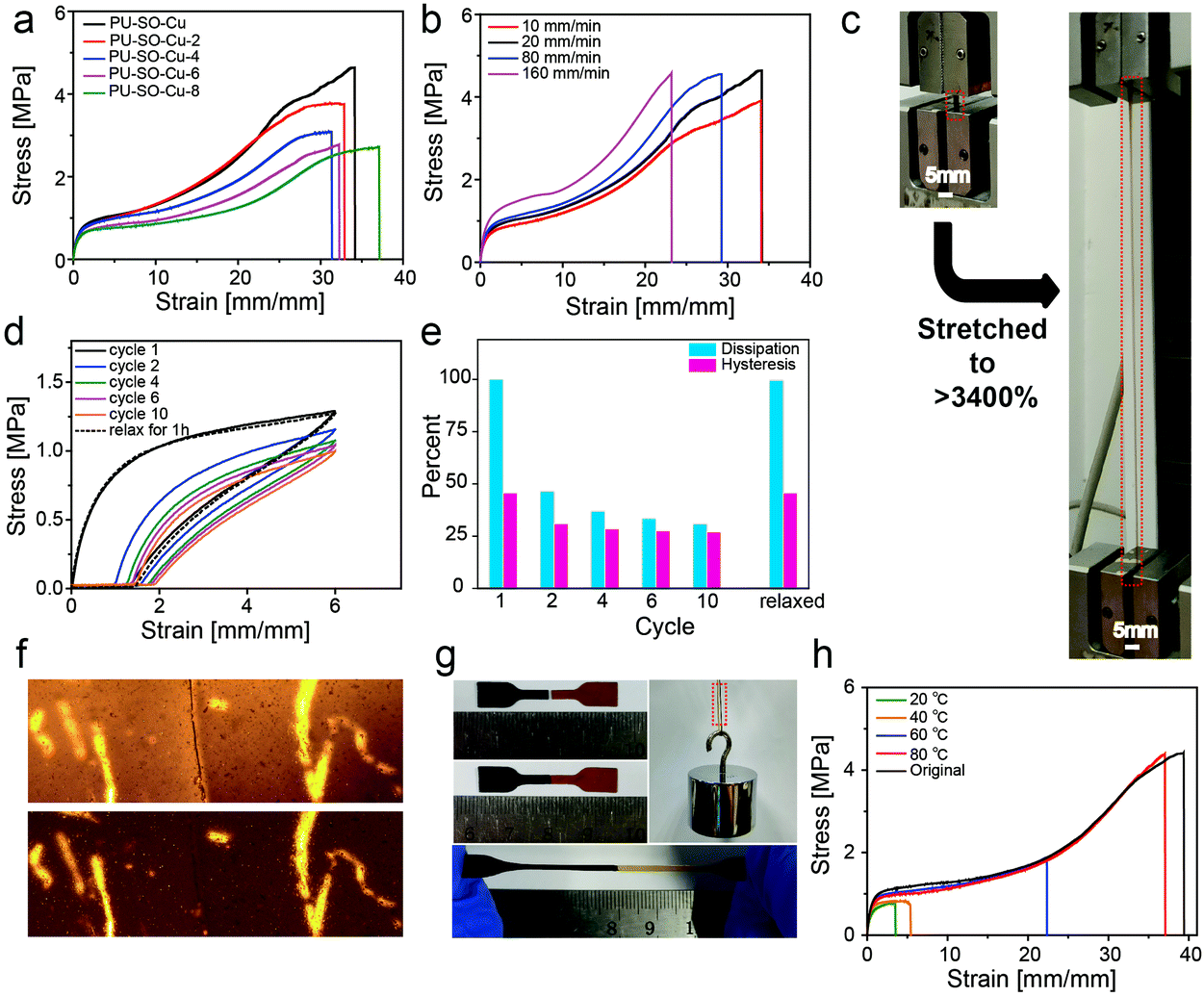 | ||
| Fig. 3 Mechanical properties and self-healing capability of the elastomer. (a) Stress–strain curves of films prepared with different molar ratios of SO to Cu2+ with a strain rate of 20 mm min−1. (b) Engineering stress–strain curves of PU–SO–Cu stretched at different rates, for a sample of width 5 mm, thickness 0.8 mm, and gauge length 5 mm. (c) The photographs of the PU–SO–Cu strip (marked by a red rectangle) before and after being stretched showed its high stretchability. (d) Recovery and cyclic loading of the PU–SO–Cu film. The samples were loaded (600% strain), unloaded, and immediately reloaded ten times (tensile rate: 20 mm min−1), and then the samples were allowed to rest for 1 hour at 25 °C after the release of the strain and stress again (cycle 11). (e) The dissipation and hysteresis data in different cyclic loadings of PU–SO–Cu. (f) Optical image of PU–SO–Cu before and after healing at 80 °C for 3 h. Cut depth: 20–30% of its thickness. (g) The cut pieces were aligned and joined together for healing at 80 °C for 3 h. Upon stretching the healed film manually, no cracking or breaking occurred, and the robustness to withstand weight restored. (h) Typical stress–strain curves of PU–SO–Cu after different healing times. The samples were cut into completely separate pieces using a razor blade and they were aligned and healed at 80 °C for different times. Tensile tests were carried out with a tensile rate of 20 mm min−1. | ||
Since the coordination bonds were regarded as tuneable interactions, we thought that the SO ligand could coordinate with other metals, hence we used MnCl2 to replace CuCl2 in the system, fabricating the PU–SO–Mn elastomer. First, we measured the binding affinity K of Mn2+–SO coordination through ITC to be as high as 1.65 × 104 (Fig. S9a, ESI†). The binding ability of Mn2+–SO coordination was much lower than that of Cu2+–SO. Then the tensile test of PU–SO–Mn was performed (Fig. S9b, ESI†), from which we could conclude that the Mn–SO coordination also promoted the mechanical properties compared with PU–SO, and the strength as well as toughness of PU–SO–Mn were lower than those of PU–SO–Cu due to the relatively low binding ability, as shown in Table S3 (ESI†). Besides, the cyclic tensile test curves of PU–SO–Mn were similar to those of PU–SO–Cu, implying that PU–SO–Mn possessed good elasticity as well (Fig. S9c, ESI†).
Additionally, our polymer network was endowed with self-healing properties, benefitting from dynamic crosslinks consisting of M–L coordination bonds and hydrogen bonds. Here the self-healing performance of PU–SO–Cu was characterized in detail. The surface scratch healing experiment was performed as shown in Fig. 3f. After healing at 80 °C for 3 h, the notch on the surface of PU–SO–Cu became barely distinguishable under the microscope, indicating that the damage was repaired in the micro level. In addition, we stained one of the two pieces of the pending-repair sample with black dye for easy identification in a typical self-healing experiment. After healing at 80 °C for 3 h, we observed that the cut sample was repaired as before, which was able to endure a large strain and withstand a weight of about 0.5 kg once again (Fig. 3g). Nevertheless, the material presented lower self-healing efficiency at lower temperature, as shown in Fig. 3h. Only when healed at 80 °C, the sample could realize a significant self-healing efficiency as high as 94% within 3 h, in terms of strain at break (Fig. S10, ESI†). In Fig. S11, ESI,† the temperature-dependent loss modulus of PU–SO–Cu exhibited a peak at around 70 °C, indicating a phase transition around here, which belonged to the disassociation of the hard segment. The elastomer possessed a large amount of H-bonds and Cu–SO coordination bonds, whose dynamicity would be increased at high temperature.45,58 And this process would contribute to the self-healing efficiency. For comparison, we enumerated the mechanical properties and self-healing temperature of recently reported elastomers in the ESI,† Fig. S12. Herein we could conclude that our system acquired excellent mechanical properties, as well as respectable self-healing efficiency.
The mechanical properties of the material were affected by structural features in multi scales, from the molecular to nanoscale level. To investigate the M–L coordination change at the molecular level, X-ray absorption fine structure (XAFS) analysis is an efficient measurement, considering that X-ray absorption spectroscopy has been proved to be useful to monitor the reconfiguration of M–L coordination in polymeric systems.59 The dynamics of coordination in PU–SO–Cu during stretching were investigated through the technique, as exhibited in Fig. 4a and b. The X-ray absorption near edge structure (XANES) results of PU–SO–Cu under different strains are shown in Fig. 4a. When stretched to different strains, the symmetry in the coordination structure of PU–SO–Cu changed gradually, which could restore to the pristine state after releasing the strain. Fig. 4b displays the extended X-ray absorption fine structure (EXAFS) results of the relative sample. Similarly, the EXAFS spectra of samples underwent an apparent shift, implying that coordination bonds in Cu2+–SO changed during stretching. Once the strain released back to original, the curve was completely coincident to the pristine result, indicating that the broken bonds re-formed and the coordination structure was totally reconfigurable. Furthermore, in situ small angle X-ray scattering (SAXS) was utilized for intensive research on the microphase in our sample. In 2D SAXS images (Fig. 4c), the circular scattering halo was observed in the pristine state, implying that the microphase was randomly oriented. Once stretched, the scattering halo was gradually oriented in the stretching direction when the strain reached 150% and 250%. For PU–SO–Cu in the pristine state, it showed no intensity change as azimuthal distributions; however, when it was stretched to 250%, it showed obvious change of the orientation; the changes in intensities integrated in horizontal and vertical directions were apparently different during stretching (Fig. 4d and Fig. S13, ESI†). Fig. 4e exhibits the relevant scattering profiles of PU–SO–Cu under different strains, from which the scattering peaks could be observed under all strains obviously. There were two broad scattering peaks located at 0.22 Å−1 and 0.024 Å−1. These scattering peaks might be attributed to the coordination-derived and H-bond-derived microphase separation, and the average distances between the microphases were calculated to be 3 nm and 28 nm respectively. During the stretching (from 0 to 250% strain), the intensity of the peak at 0.024 Å−1 gradually decreased, indicating the relative decrease of microphase separated structures derived by H-bonds. Herein we could conclude that the coordination structure and H-bond-derived microphase were the key to influence the mechanical performances of materials, and the dynamic evolution process of coordination and microphases were detected by XAFS and SAXS techniques.
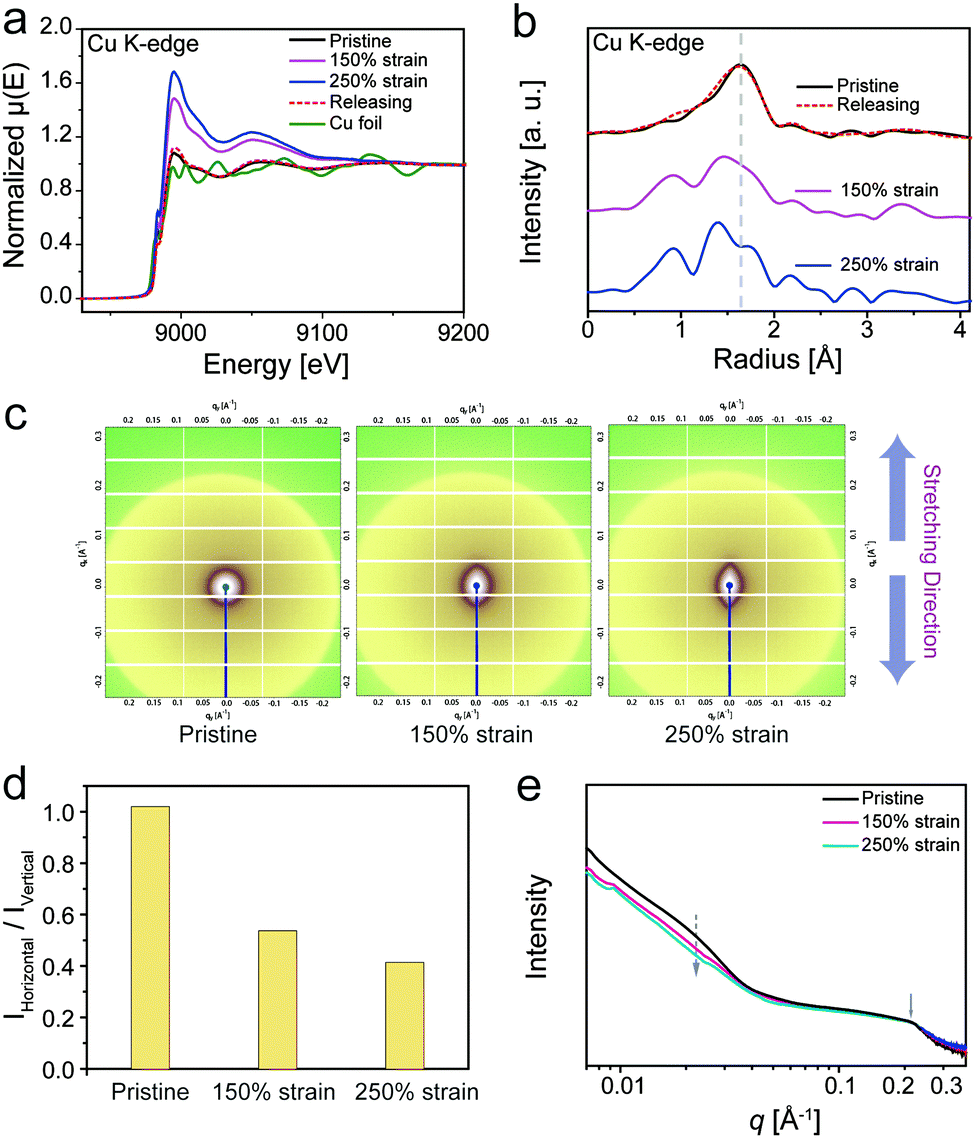 | ||
| Fig. 4 (a) XANES spectra of PU–SO–Cu under different strains. (b) EXAFS spectra of PU–SO–Cu under different strains. (c) 2D SAXS images of PU–SO–Cu with different extension strains during the uniaxial stretching process. (d) Ratio of intensities integrated in different directions (horizontal and vertical) under different strains. All intensities were calculated at q = 0.024 Å−1. (e) 1D scattering profiles of PU–SO–Cu integrated from 2D SAXS patterns under different strains. | ||
Since our elastomer was robust, highly stretchable and self-healable, various types of flexible devices could be fabricated based on this promising elastic substrate. First, the conductive gold layers were deposited on the upper and lower surfaces of the elastomer after thermal vacuum deposition, forming a flexible capacitor which could play the role as a capacitive pressure sensor60 (Fig. 5a). Fig. 5b shows the capacitance change of the sensor under changing pressure, in which the sensitivity (slope of the capacitance change–pressure curve) was 2.1 × 10−3 kPa−1. Moreover, once the sensor was wounded, the pressure sensitivity was nearly restored after healing at 80 °C for 3 h, as shown in Fig. 5c.
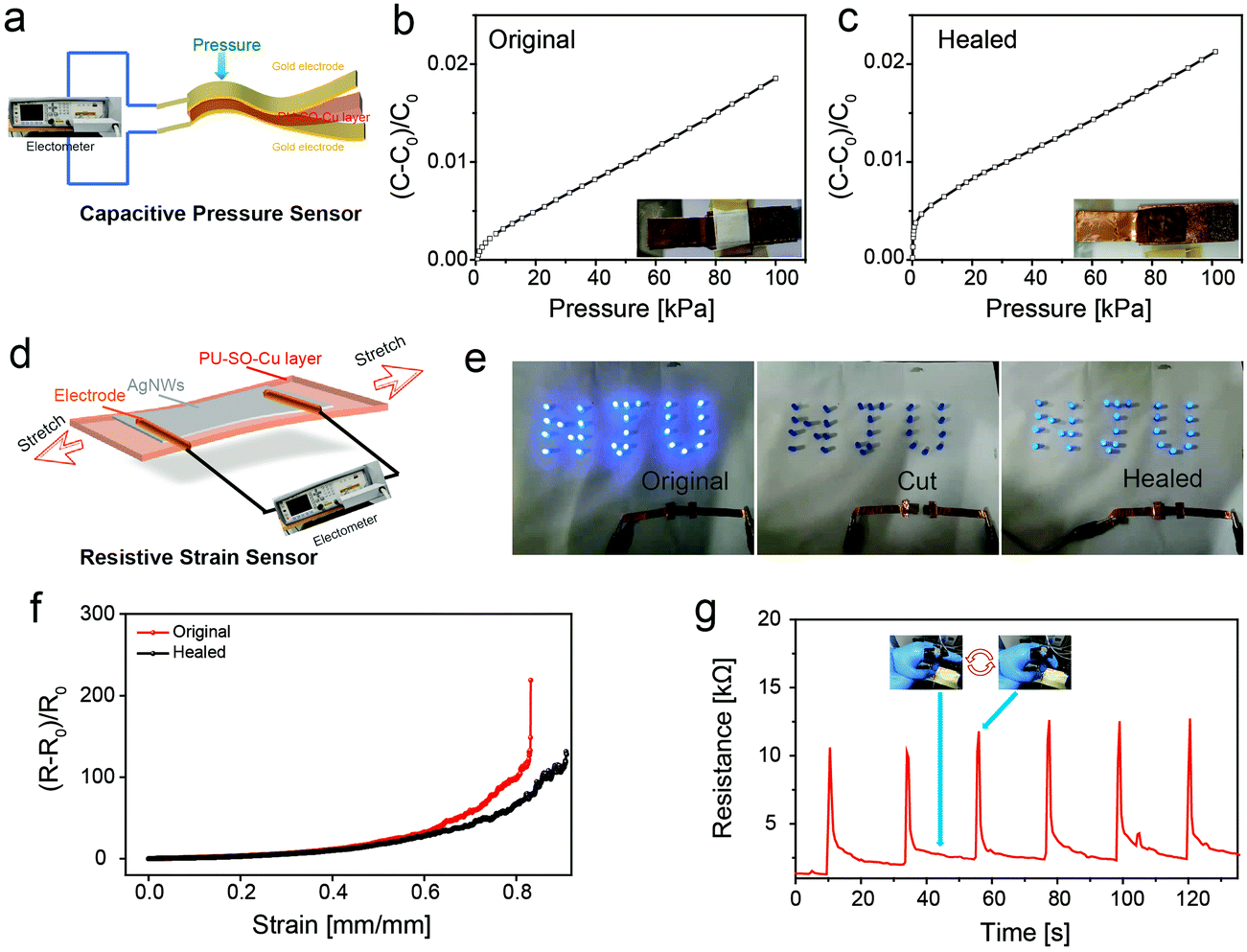 | ||
| Fig. 5 (a) Illustration of the capacitive pressure sensor device. (b) Capacitance changes of the sensor under different pressures, tested on the original sensor. (c) Capacitance changes of the sensor under different pressures, tested on the cut then healed sensor. (d) Illustration of the resistive strain sensor device. (e) Demonstration of the healing process for a circuit with a conductor based on PU–SO–Cu. The conductor was cut into completely separate pieces, and then they were aligned and the circuit could recover to work. (f) The relative resistance changes of the original and self-healed conductor. (g) Repeating resistance changes of the sensor when monitoring the finger bending several times. | ||
Then the strain sensor was fabricated by spray-coating AgNWs as a conductive layer onto the PU–SO–Cu substrate61 (Fig. 5d). The strain sensor could be still conductive when stretched to over 170% with a gauge factor of 6885.8, and the relative changes of resistance with tensile strain could be perfectly fitted into an exponential equation (Fig. S14, ESI†). In order to prove the self-healing ability of the strain sensor, we used our sensor and LED to build a circuit, on which a 3.0 V voltage was applied. The LED turned off when the sensor was cut. And after splicing the two pieces of conductors together and healing at 80 °C for 3 h, the LED was observed to turn on again (Fig. 5e). Moreover, we also measured the resistance change during stretching before and after healing (Fig. 5f). This showed that the stretchable conductor could also be extended over 80% strain after healing. Besides, the strain sensor was able to detect mild human movement as designed. As shown in Fig. 5g, the sensor would export good and stable results of the resistance response when adopting repeating finger bending movements.
Conclusions
To summarize, we presented the design for constructing a dual-dynamic crosslinked system with thio-β-diketone-Cu2+ M–L coordinating interaction and hydrogen bonds in polyurethane. The unique design realized the enhancement of mechanical properties, including the stress at break (4.35 MPa), high toughness (81 MJ m−3), considerable fracture energy (44.65 kJ m−12) and high stretchability (over 3400%). The self-healing capability of the elastomer was guaranteed as well, which could be completely repaired at 80 °C within 3 hours.The thermodynamic stability and kinetic lability of the SO–Cu2+ coordination was proved by ITC and variable-temperature NMR measurements respectively. Moreover, the break and reconfiguration of the coordination and change of microphases in elastomers were demonstrated through XAFS and SAXS techniques, which played an irreplaceable role in effecting the mechanical properties of materials. Furthermore, we fabricated a self-healable capacitive pressure sensor and resistive strain sensor based on the self-healable elastomer. These sensors displayed considerable efficiency in detecting pressures and monitoring human movements successfully. Herein this highly stretchable elastomer definitely exhibited potential in applications of stretchable electronic devices.
Author contributions
Xiaoming An, Xudong Jia andQiuhong Zhang: conceptualization, designs, synthesis, measurements and writing the article. Jie Liu: FT-IR, UV and mechanical tests. Jiahan Zhang: fabrication and measurement of the capacitive pressure sensor. Xinxin Huang: fabrication and measurement of the resistive strain sensor. Tangsong Zhu: XAFS tests. Hongping Yan: SAXS analysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by the National Natural Science Foundation of China (Grant 22075130), as well as the Fundamental Research Funds for the Central Universities. We acknowledge Shanghai Synchrotron Radiation Facility (SSRF) for the beam time on Beamlines BL14W1 and BL16B1, respectively, used for XAFS and SAXS measurements.Notes and references
- X. Wang, Y. Zhang, X. Zhang, Z. Huo, X. Li, M. Que, Z. Peng, H. Wang and C. Pan, A Highly Stretchable Transparent Self-Powered Triboelectric Tactile Sensor with Metallized Nanofibers for Wearable Electronics, Adv. Mater., 2018, 30, e1706738 CrossRef PubMed.
- X. Di, C. Hang, Y. Xu, Q. Ma, F. Li, P. Sun and G. Wu, Bioinspired tough, conductive hydrogels with thermally reversible adhesiveness based on nanoclay confined NIPAM polymerization and a dopamine modified polypeptide, Mater. Chem. Front., 2020, 4, 189–196 RSC.
- Z. Liu, J. Liu, J. Zhang, B. Zheng, X. Ren, Y. Long, L. Fang, R. Ou, T. Liu and Q. Wang, Highly compressible hydrogel sensors with synergistic long-lasting moisture, extreme temperature tolerance and strain-sensitivity properties, Mater. Chem. Front., 2020, 4, 3319–3327 RSC.
- S. Wang, Z. Xu, T. Wang, T. Xiao, X. Y. Hu, Y. Z. Shen and L. Wang, Warm/cool-tone switchable thermochromic material for smart windows by orthogonally integrating properties of pillar[6]arene and ferrocene, Nat. Commun., 2018, 9, 1737 CrossRef PubMed.
- X. Liu, C. Lu, X. Wu and X. Zhang, Self-healing strain sensors based on nanostructured supramolecular conductive elastomers, J. Mater. Chem. A, 2017, 5, 9824–9832 RSC.
- Y. Jiang, Z. Liu, Z. Yin and Q. Zheng, Sandwich structured dielectrics for air-stable and flexible low-voltage organic transistors in ultrasensitive pressure sensing, Mater. Chem. Front., 2020, 4, 1459–1470 RSC.
- X. Fan, N. Wang, J. Wang, B. Xu and F. Yan, Highly sensitive, durable and stretchable plastic strain sensors using sandwich structures of PEDOT:PSS and an elastomer, Mater. Chem. Front., 2018, 2, 355–361 RSC.
- S. Park, K. Mondal, R. M. Treadway, 3rd, V. Kumar, S. Ma, J. D. Holbery and M. D. Dickey, Silicones for Stretchable and Durable Soft Devices: Beyond Sylgard-184, ACS Appl. Mater. Interfaces, 2018, 10, 11261–11268 CrossRef CAS PubMed.
- S. R. White, N. R. Sottos, P. H. Geubelle, J. S. Moore, M. R. Kessler, S. R. Sriram, E. N. Brown and S. Viswanathan, Autonomic Healing of polymer Composites, Nature, 2001, 409, 794–797 CrossRef CAS PubMed.
- J. Y. Sun, X. Zhao, W. R. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Highly stretchable and tough hydrogels, Nature, 2012, 489, 133–136 CrossRef CAS PubMed.
- G. Li, H. Zhang, D. Fortin, H. Xia and Y. Zhao, Poly(vinyl alcohol)-Poly(ethylene glycol) Double-Network Hydrogel: A General Approach to Shape Memory and Self-Healing Functionalities, Langmuir, 2015, 31, 11709–11716 CrossRef CAS PubMed.
- J. Duan, X. Liang, J. Guo, K. Zhu and L. Zhang, Ultra-Stretchable and Force-Sensitive Hydrogels Reinforced with Chitosan Microspheres Embedded in Polymer Networks, Adv. Mater., 2016, 28, 8037–8044 CrossRef CAS PubMed.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, A Thermally Re-mendable Cross-Linked Polymeric Material, Science, 2002, 295, 1698–1701 CrossRef CAS PubMed.
- J. Zhao, R. Xu, G. Luo, J. Wu and H. Xia, A self-healing, re-moldable and biocompatible crosslinked polysiloxane elastomer, J. Mater. Chem. B, 2016, 4, 982–989 RSC.
- U. Lafont, H. van Zeijl and S. van der Zwaag, Influence of cross-linkers on the cohesive and adhesive self-healing ability of polysulfide-based thermosets, ACS Appl. Mater. Interfaces, 2012, 4, 6280–6288 CrossRef CAS PubMed.
- Y. Amamoto, J. Kamada, H. Otsuka, A. Takahara and K. Matyjaszewski, Repeatable photoinduced self-healing of covalently cross-linked polymers through reshuffling of trithiocarbonate units, Angew. Chem., Int. Ed., 2011, 50, 1660–1663 CrossRef CAS PubMed.
- Z. P. Zhang, M. Z. Rong and M. Q. Zhang, Mechanically Robust, Self-Healable, and Highly Stretchable “Living” Crosslinked Polyurethane Based on a Reversible C-C Bond, Adv. Funct. Mater., 2018, 28, 1706050 CrossRef.
- M. Podgorski, S. Mavila, S. Huang, N. Spurgin, J. Sinha and C. N. Bowman, Thiol-Anhydride Dynamic Reversible Networks, Angew. Chem., Int. Ed., 2020, 59, 9345–9349 CrossRef CAS PubMed.
- Y. X. Lu and Z. Guan, Olefin metathesis for effective polymer healing via dynamic exchange of strong carbon-carbon double bonds, J. Am. Chem. Soc., 2012, 134, 14226–14231 CrossRef CAS PubMed.
- W. A. Ogden and Z. Guan, Recyclable, Strong, and Highly Malleable Thermosets Based on Boroxine Networks, J. Am. Chem. Soc., 2018, 140, 6217–6220 CrossRef CAS PubMed.
- E. Zhang, J. Shi, L. Xiao, Q. Zhang, M. Lu, B. Nan, K. Wu and M. Lu, A highly efficient bionic self-healing flexible waterborne polyurethane elastic film based on a cyclodextrin–ferrocene host–guest interaction, Polym. Chem., 2021, 12, 831–842 RSC.
- G. Li, L. Wang, L. Wu, Z. Guo, J. Zhao, Y. Liu, R. Bai and X. Yan, Woven Polymer Networks via the Topological Transformation of a [2]Catenane, J. Am. Chem. Soc., 2020, 142, 14343–14349 CrossRef CAS PubMed.
- C. Y. Shi, Q. Zhang, C. Y. Yu, S. J. Rao, S. Yang, H. Tian and D. H. Qu, An Ultrastrong and Highly Stretchable Polyurethane Elastomer Enabled by a Zipper-Like Ring-Sliding Effect, Adv. Mater., 2020, 32, e2000345 CrossRef PubMed.
- D. Zhao, Z. Zhang, J. Zhao, K. Liu, Y. Liu, G. Li, X. Zhang, R. Bai, X. Yang and X. Yan, A Mortise-and-Tenon Joint Inspired Mechanically Interlocked Network, Angew. Chem., Int. Ed., 2021, 60, 16224–16229 CrossRef CAS PubMed.
- S. Burattini, B. W. Greenland, D. H. Merino, W. Weng, J. Seppala, H. M. Colquhoun, W. Hayes, M. E. Mackay, I. W. Hamley and S. J. Rowan, A Healable Supramolecular Polymer Blend Based on Aromatic π–π Stacking and Hydrogen-Bonding Interactions, J. Am. Chem. Soc., 2010, 132, 12051–12058 CrossRef CAS PubMed.
- W. B. Ying, G. Wang, Z. Kong, C. K. Yao, Y. Wang, H. Hu, F. Li, C. Chen, Y. Tian, J. Zhang, R. Zhang and J. Zhu, A Biologically Muscle-Inspired Polyurethane with Super-Tough, Thermal Reparable and Self-Healing Capabilities for Stretchable Electronics, Adv. Funct. Mater., 2021, 31, 2009869 CrossRef CAS.
- C. Chen, W. B. Ying, J. Li, Z. Kong, F. Li, H. Hu, Y. Tian, D. H. Kim, R. Zhang and J. Zhu, A Self-Healing and Ionic Liquid Affiliative Polyurethane toward a Piezo 2 Protein Inspired Ionic Skin, Adv. Funct. Mater., 2021, 32, 2106341 CrossRef.
- Y. Zhang, M. Li, B. Qin, L. Chen, Y. Liu, X. Zhang and C. Wang, Highly Transparent, Underwater Self-Healing, and Ionic Conductive Elastomer Based on Multivalent Ion–Dipole Interactions, Chem. Mater., 2020, 32, 6310–6317 CrossRef CAS.
- D. Wang, J. Xu, J. Chen, P. Hu, Y. Wang, W. Jiang and J. Fu, Transparent, Mechanically Strong, Extremely Tough, Self-Recoverable, Healable Supramolecular Elastomers Facilely Fabricated via Dynamic Hard Domains Design for Multifunctional Applications, Adv. Funct. Mater., 2019, 30, 1907109 CrossRef.
- M. Burnworth, L. Tang, J. R. Kumpfer, A. J. Duncan, F. L. Beyer, G. L. Fiore, S. J. Rowan and C. Weder, Optically healable supramolecular polymers, Nature, 2011, 472, 334–337 CrossRef CAS PubMed.
- C. H. Li and J. L. Zuo, Self-Healing Polymers Based on Coordination Bonds, Adv. Mater., 2020, 32, e1903762 Search PubMed.
- D. Braun and H. Boudevska, Reversible cross-linking by complex formation. Polymers containing 2-hydroxybenzoic acid residues, Eur. Polym. J., 1976, 12, 525–528 CrossRef CAS.
- D. Yu, X. Zhao, C. Zhou, C. Zhang and S. Zhao, Room Temperature Self-Healing Methyl Phenyl Silicone Rubbers Based on the Metal-Ligand Cross-Link: Synthesis and Characterization, Macromol. Chem. Phys., 2017, 218, 1600519 CrossRef.
- A. O. Razgoniaev, L. M. Glasstetter, T. B. Kouznetsova, K. C. Hall, M. Horst, S. L. Craig and K. J. Franz, Single-Molecule Activation and Quantification of Mechanically Triggered Palladium-Carbene Bond Dissociation, J. Am. Chem. Soc., 2021, 143, 1784–1789 CrossRef CAS PubMed.
- J. Kee, H. Ahn, H. Park, Y. S. Seo, Y. H. Yeo, W. H. Park and J. Koo, Stretchable and Self-Healable Poly(styrene-co-acrylonitrile) Elastomer with Metal-Ligand Coordination Complexes, Langmuir, 2021, 37, 13998–14005 CrossRef CAS PubMed.
- Y. Zhang, Z. Wang, T. B. Kouznetsova, Y. Sha, E. Xu, L. Shannahan, M. Fermen-Coker, Y. Lin, C. Tang and S. L. Craig, Distal conformational locks on ferrocene mechanophores guide reaction pathways for increased mechanochemical reactivity, Nat. Chem., 2021, 13, 56–62 CrossRef CAS PubMed.
- B. P. Lee and S. Konst, Novel hydrogel actuator inspired by reversible mussel adhesive protein chemistry, Adv. Mater., 2014, 26, 3415–3419 CrossRef CAS PubMed.
- S. Kim, A. U. Regitsky, J. Song, J. Ilavsky, G. H. McKinley and N. Holten-Andersen, In situ mechanical reinforcement of polymer hydrogels via metal-coordinated crosslink mineralization, Nat. Commun., 2021, 12, 667 CrossRef CAS PubMed.
- D. Mozhdehi, S. Ayala, O. R. Cromwell and Z. Guan, Self-healing multiphase polymers via dynamic metal-ligand interactions, J. Am. Chem. Soc., 2014, 136, 16128–16131 CrossRef CAS PubMed.
- C. Xing, H. Wu, R. Du, Q. Zhang and X. Jia, Extremely tough and healable elastomer realized via reducing the crystallinity of its rigid domain, Polym. Chem., 2021, 12, 4778–4784 RSC.
- Y. Deng, Q. Zhang, B. L. Feringa, H. Tian and D. H. Qu, Toughening a Self-Healable Supramolecular Polymer by Ionic Cluster-Enhanced Iron-Carboxylate Complexes, Angew. Chem., Int. Ed., 2020, 59, 5278–5283 CrossRef CAS PubMed.
- T. Sato, M. Ebara, S. Tanaka, T. A. Asoh, A. Kikuchi and T. Aoyagi, Rapid self-healable poly(ethylene glycol) hydrogels formed by selective metal-phosphate interactions, Phys. Chem. Chem. Phys., 2013, 15, 10628–10635 RSC.
- L. Zhang, Z. Liu, X. Wu, Q. Guan, S. Chen, L. Sun, Y. Guo, S. Wang, J. Song, E. M. Jeffries, C. He, F. L. Qing, X. Bao and Z. You, A Highly Efficient Self-Healing Elastomer with Unprecedented Mechanical Properties, Adv. Mater., 2019, 31, e1901402 CrossRef PubMed.
- H. Qin, T. Zhang, H.-N. Li, H.-P. Cong, M. Antonietti and S.-H. Yu, Dynamic Au-Thiolate Interaction Induced Rapid Self-Healing Nanocomposite Hydrogels with Remarkable Mechanical Behaviors, Chemistry, 2017, 3, 691–705 CrossRef CAS.
- X. Wang, S. Zhan, Z. Lu, J. Li, X. Yang, Y. Qiao, Y. Men and J. Sun, Healable, Recyclable, and Mechanically Tough Polyurethane Elastomers with Exceptional Damage Tolerance, Adv. Mater., 2020, 32, e2005759 CrossRef PubMed.
- Q. Zhu, K. Vliet, N. Holten-Andersen and A. Miserez, A Double-Layer Mechanochromic Hydrogel with Multidirectional Force Sensing and Encryption Capability, Adv. Funct. Mater., 2019, 29, 1808191 CrossRef.
- C. H. Li, C. Wang, C. Keplinger, J. L. Zuo, L. Jin, Y. Sun, P. Zheng, Y. Cao, F. Lissel, C. Linder, X. Z. You and Z. Bao, A highly stretchable autonomous self-healing elastomer, Nat. Chem., 2016, 8, 618–624 CrossRef CAS PubMed.
- X. Zhang, S. Wang, Z. Jiang, Y. Li and X. Jing, Boronic Ester Based Vitrimers with Enhanced Stability via Internal Boron-Nitrogen Coordination, J. Am. Chem. Soc., 2020, 142, 21852–21860 CrossRef CAS PubMed.
- A. V. Zhukhovitskiy, M. Zhong, E. G. Keeler, V. K. Michaelis, J. E. Sun, M. J. Hore, D. J. Pochan, R. G. Griffin, A. P. Willard and J. A. Johnson, Highly branched and loop-rich gels via formation of metal-organic cages linked by polymers, Nat. Chem., 2016, 8, 33–41 CrossRef CAS PubMed.
- Q. Zhang, S. Niu, L. Wang, J. Lopez, S. Chen, Y. Cai, R. Du, Y. Liu, J. C. Lai, L. Liu, C. H. Li, X. Yan, C. Liu, J. B. Tok, X. Jia and Z. Bao, An Elastic Autonomous Self-Healing Capacitive Sensor Based on a Dynamic Dual Crosslinked Chemical System, Adv. Mater., 2018, 30, 1801435 CrossRef PubMed.
- L. A. Nguyen, P. Retailleau and T. B. Nguyen, Elemental Sulfur/DMSO-Promoted Multicomponent One-pot Synthesis of Malonic Acid Derivatives from Maleic Anhydride and Amines, Adv. Synth. Catal., 2019, 361, 2864–2869 CrossRef CAS.
- Z. Hussain, M. A. Salim, M. A. Khan and E. E. Khawaja, X-Ray Photoelectron and Auger Spectroscopy Study of Copper-sodium-Germamate Glasses, J. Non-Cryst. Solids, 1989, 110, 44–52 CrossRef CAS.
- R. Tepper, S. Bode, R. Geitner, M. Jager, H. Gorls, J. Vitz, B. Dietzek, M. Schmitt, J. Popp, M. D. Hager and U. S. Schubert, Polymeric Halogen-Bond-Based Donor Systems Showing Self-Healing Behavior in Thin Films, Angew. Chem., Int. Ed., 2017, 56, 4047–4051 CrossRef CAS PubMed.
- W. C. Yount, D. M. Loveless and S. L. Craig, Small-Molecule Dynamics and Mechanisms Underlying the Macroscopic Mechanical Properties of Coordinatively Cross-Linked Polymer Networks, J. Am. Chem. Soc., 2005, 127, 14488–14496 CrossRef CAS PubMed.
- E. A. Archer and M. J. Krische, Duplex Oligomers Defined via Covalent Casting of a One-Dimensional Hydrogen-Bonding Motif, J. Am. Chem. Soc., 2002, 124, 5074–5083 CrossRef CAS PubMed.
- Q. Zhang, X. Zhu, C.-H. Li, Y. Cai, X. Jia and Z. Bao, Disassociation and Reformation Under Strain in Polymer with Dynamic Metal–Ligand Coordination Cross-Linking, Macromolecules, 2019, 52, 660–668 CrossRef CAS.
- R. Du, Z. Xu, C. Zhu, Y. Jiang, H. Yan, H. C. Wu, O. Vardoulis, Y. Cai, X. Zhu, Z. Bao, Q. Zhang and X. Jia, A Highly Stretchable and Self-Healing Supramolecular Elastomer Based on Sliding Crosslinks and Hydrogen Bonds, Adv. Funct. Mater., 2019, 30, 1907139 CrossRef.
- Z. Li, Y. L. Zhu, W. Niu, X. Yang, Z. Jiang, Z. Y. Lu, X. Liu and J. Sun, Healable and Recyclable Elastomers with Record-High Mechanical Robustness, Unprecedented Crack Tolerance, and Superhigh Elastic Restorability, Adv. Mater., 2021, 33, e2101498 CrossRef PubMed.
- H. C. Wu, F. Lissel, G. J.-N. Wang, D. M. Koshy, S. Nikzad, H. Yan, J. Xu, S. Luo, N. Matsuhisa, Y. Cheng, F. Wang, B. Ji, D. Li, W. C. Chen, G. Xue and Z. Bao, Metal–Ligand Based Mechanophores Enhance Both Mechanical Robustness and Electronic Performance of Polymer Semiconductors, Adv. Funct. Mater., 2021, 31, 2009201 CrossRef CAS.
- G. D. Tabi, J. S. Kim, B. Nketia-Yawson, D. H. Kim and Y.-Y. Noh, High-capacitance polyurethane ionogels for low-voltage operated organic transistors and pressure sensors, J. Mater. Chem. C, 2020, 8, 17107–17113 RSC.
- M. Zhao, D. Li, J. Huang, D. Wang, A. Mensah and Q. Wei, A multifunctional and highly stretchable electronic device based on silver nanowire/wrap yarn composite for a wearable strain sensor and heater, J. Mater. Chem. C, 2019, 7, 13468–13476 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2qm00259k |
| This journal is © the Partner Organisations 2022 |