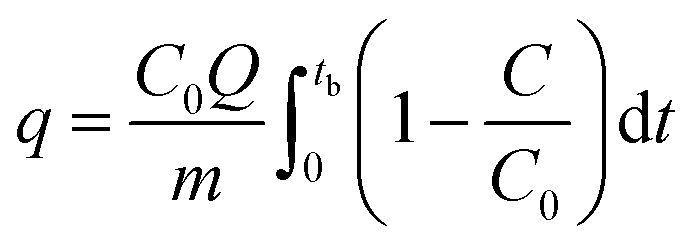DOI:
10.1039/D0RA09017D
(Paper)
RSC Adv., 2021,
11, 4890-4900
Chemisorption of hydrogen sulfide over copper-based metal–organic frameworks: methanol and UV-assisted regeneration†
Received
23rd October 2020
, Accepted 19th January 2021
First published on 27th January 2021
Abstract
Three copper-based metal–organic frameworks (MOFs) with different organic linkers were synthesized for the removal of H2S gas at room temperature. The synthesized MOFs were characterized by microscopic and spectroscopic techniques to understand their structural, functional, and optical properties. The H2S adsorption capacity of MOFs calculated by column studies followed the trend: 105.6 mg g−1 (CuBDC) > 27.1 mg g−1 (CuBTC) > 1.3 mg g−1 (CuBDC-N) in dry conditions. The adsorption capacity increased in moist conditions due to an easy dissolution and dissociation of H2S in a film of water. X-ray photoelectron spectroscopy confirmed the presence of sulfur bound to Cu-sites and sulfate ions. The spent MOFs were regenerated by the successive effect of methanol and low power UV-C radiation. The regenerated CuBTC showed an exceptionally high adsorption capacity of 95.6 mg g−1 in the second cycle, which was linked to the reactivation of Cu-sites and improved surface area and porosity. The regeneration process developed in this study is a cost-effective method to recycle chemisorbed MOFs without compromising with their structural and functional integrity.
1. Introduction
Hydrogen sulfide (H2S) is a toxic gas with a characteristic rotten egg smell and arises from many anthropogenic sources like crude oil refineries, coal gasification plants, food processing units, livestock farms, and municipal sewage treatment facilities. H2S emission has been linked to the formation of acid rain, which, in turn, affects human health and the environment. Acute exposure to H2S at levels of 100–200 ppm can be lethal and have resulted in death.1 Therefore, the removal of H2S from sources has been prioritized to enhance the quality of life and, at the same time, prevent catastrophic events like acid rain.2 In this regard, numerous physical/chemical/biological techniques like adsorption, scrubbing, electrochemical decomposition, catalytic oxidation, and bio-filtration have been explored for the removal of H2S from biogas, feed gases, and flue gases.3,4 Currently, adsorption using porous adsorbents is deemed as a lucrative option due to its simplicity, low operational cost, and high performance. Porous adsorbents like activated carbon, metal oxides, zeolites, and composites have been evaluated for H2S removal and have been shown to have disadvantages like low adsorption capacity and poor reusability.1,4
In the domain of porous adsorbents, a sub-class of organic-inorganic hybrids, i.e., metal–organic frameworks (MOFs), is in the limelight for their exceptional surface and pore properties with the easy tuning of physicochemical properties. MOFs are constructed through coordinative interactions between organic linkers and metal ions to form a highly ordered framework with uniform porosity. MOFs are one of the potential candidates for the capture and storage of carbon dioxide,5 adsorptive removal of toxic gases,6 and catalytic conversion of gases into value-added products.7 In the literature, Cu-based MOFs have been extensively explored for the removal of H2S gas under different experimental conditions to evaluate the adsorption capacity and underlying adsorption mechanism. Though the published works have confirmed Cu-based MOFs as superior adsorbents, among porous materials, the reusability of these MOFs is still a serious concern. Moreover, the literature has been saturated with the studies related to CuBTC (also known as HKUST-1 or MOF-199).8–12 The need to explore other Cu-based MOFs and the lack of proper investigation on the regeneration of spent Cu-based MOFs have motivated us to develop methanol and UV-assisted regeneration method.
In this study, we have fabricated three copper-based MOFs with 1,4-benzene dicarboxylic acid (H2BDC), 2-amino-1,4-benzene dicarboxylic acid (H2BDC-NH2), and benzene-1,3,5-tricarboxylic acid (H3BTC) via ultrasonication method13 to study their applicability in the removal of H2S gas. The synthesized MOFs were characterized by several microscopic and spectroscopic techniques to understand their structural, functional, and optical properties. The MOFs were studied for the adsorptive removal of H2S in both dry and moist conditions with a focus on understanding the adsorptive mechanism. In this study, attempts were made to regenerate spent MOFs using methanol and UV-C treatment. The effect of regeneration on the structural and functional stability of Cu-MOFs was studied by X-ray diffraction (XRD) and X-ray photoelectron spectroscopy (XPS).
2. Materials and methods
2.1 Chemicals and reagents
Copper(II) nitrate trihydrate (Cu(NO3)2·3H2O), H2BDC, H2BDC-NH2, and H3BTC were procured from Sigma Aldrich, Germany. N,N-dimethyl formamide (DMF), ethanol, methanol, and sodium hydroxide (NaOH) were purchased from Samchun Pure Chemicals, Korea. All the chemicals were of analytical grade and used without any further purification. Highly pure H2S gas (500 ppm balanced with N2 gas) was procured from Union gas, Korea.
2.2 Synthesis of Cu-MOFs
The fresh copper hydroxide was prepared by adding NaOH solution (82.5 mL, 1.0 mol L−1 in deionized water) into an aqueous solution of Cu(NO3)2·3H2O (10.0 g in 75 mL of deionized water). To it, an H2BDC solution (6.64 g in 190 mL DMF) was added under ultrasonication (Sonics Vibra-cell 500 W, 20 kHz, 44% amplitude). After 20 min of ultrasonication, the precipitate was collected by centrifugation and washed thoroughly with ethanol. It was followed by drying at 50 °C for 10 days in a hot air oven. The synthesized product was named as CuBDC. Using the same protocol, CuBDC-N was prepared with an H2BDC-NH2 solution (7.24 g in 190 mL DMF). CuBTC was synthesized using an H3BTC solution (8.50 g in 190 mL DMF). The physical appearance and yield are listed in S. Table 1.† The Cu-MOFs were finely grounded and stored at 70 °C in glass vials.
2.3 Instruments
The surface morphology of Cu-MOFs was analyzed by field emission scanning electron microscopy (FE-SEM) (Hitachi S-4300, Japan). Dried samples were coated with a gold-platinum alloy by ion-sputtering (E-1048 Hitachi ion sputter). The transmission electron microscope (TEM) images were recorded with a field emission TEM (FE-TEM, JEM-2010F, JEOL, Japan). The two dimensional (2D) elemental mapping was done by energy-dispersive X-ray spectroscopy (EDS) (X-Maxn 80 T, Oxford, UK). Thermal gravimetric analysis (TGA) was done using a Thermogravimetric Analyzer (TG 209 F3, NETZSCH). For Brunauer–Emmett–Teller (BET) analysis, N2 adsorption–desorption isotherm was measured at −196 °C in a Gemini series Micromeritics 2360 instrument after degassing at 200 °C for 6 h. The diffraction patterns were obtained using Ultima IV (Rigajku, Japan) X-ray diffractometer with Cu Kα and a Ni filter where the scanning speed was set to 3° min−1. Fourier-transform infrared (FTIR) spectra of samples were recorded using KBr pellets over an FTIR spectrometer (Cary670, Agilent). The recording was done with a single-beam spectrometer with 60 added scans. Ultraviolet-visible light-diffuse reflectance spectroscopy (UV-Vis DRS) spectra of photocatalysts were obtained using a SCINCO S-4100 spectrometer equipped with a photodiode array detector and a diffuse reflectance attachment. The electron spin resonance (ESR) measurement was carried out on a JEOL JES-FE1C X-band spectrometer. For XPS analysis, a K-alpha XPS instrument (Thermo Scientific Inc., UK) with a monochromatic Al Kα X-ray source was used where the pressure was fixed to 4.8 × 10−9 mbar. Spectra have been charge corrected to the main line of the carbon 1s spectrum (aromatic carbon) set to 284.7 eV. Spectra were analyzed using CasaXPS software (version 2.3.14). GL(p) = Gaussian/Lorentzian product formula where the mixing is determined by m = p/100, GL(100) is a pure Lorentzian while GL(0) is pure Gaussian. We have used GL(30).
2.4 Breakthrough experiments
The adsorption experiments were performed by taking 250 mg of a powdered MOF in a Pyrex tube (height: 50 cm, diameter: 1 cm) at 25 °C. The H2S gas (500 ppm) was passed through it at a flow rate of 0.3 L min−1. The outgoing gas was analyzed by a Gas analyzer (GSR-310, Sensoronic, Korea) with the detection limit of 0.1 ppm. The H2S concentration was measured every 15 s until the effluent concentration reached 400 ppm. The schematic illustration of the H2S adsorption system is shown in Fig. 1. For evaluating the effect of moisture, moist air (relative humidity ∼60%) was blown through the MOF packed in the reactor at a flow rate of 0.2 L min−1 for 30 min before passing H2S gas.
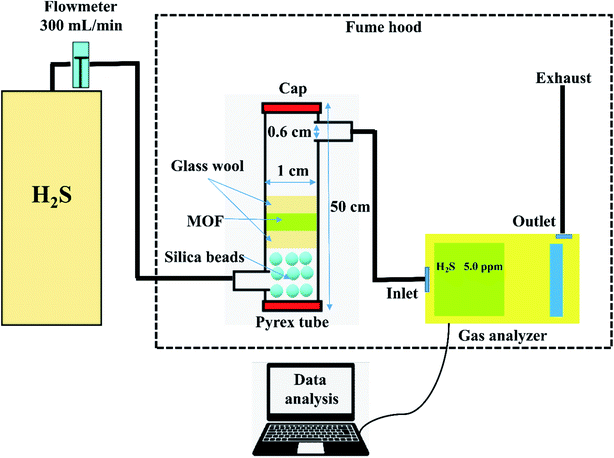 |
| | Fig. 1 Schematic illustration of the H2S adsorption system. | |
For the regeneration of spent MOF, exactly 200 mg of the spent MOF was stirred in 10 mL of methanol for 24 h at 300 rpm, followed by separation of MOF by centrifugation and drying at 70 °C for 24 h. The dried MOF was irradiated in a 1.0 L Pyrex tube fitted in an acryl reactor with 4 UV lamps (19 W, UV-C, Imax ∼ 254 nm) for 4 h in the N2 environment. The adsorption capacity (q, mg g−1) at the breakthrough point (where the effluent concentration reached 400 ppm) was calculated by integration of the area above the breakthrough curve.
| |
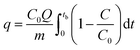 | (1) |
where
C0 – initial concentration (500 ppm or 0.697 mg L
−1),
Q – flowrate,
m – the mass of MOF (g), and
tb – breakthrough time.
3. Results and discussions
3.1 Characterization of Cu-MOFs
The morphology of Cu-MOFs was investigated using SEM (Fig. 2a–c) and HR-TEM (Fig. 2d–f). CuBDC was homogeneously distributed as square sheets of size 0.2–0.8 μm and 75–100 nm thickness (Fig. 2a), which was further confirmed by thin sheet-like structures in the HR-TEM image (Fig. 2d). CuBDC-N had a large number of prismatic-shape microcrystallites (1–5 μm) with some nanocrystals (Fig. 2b and e). CuBTC crystals of varying dimensions were observed with no well-defined morphology (Fig. 2c and f). The 2D elemental mappings of Cu-MOFs confirmed uniform distribution of Cu in MOFs (S. Fig. 1†).
 |
| | Fig. 2 (a–c) SEM images; (d–f) HR-TEM images of Cu-MOFs. | |
The surface and pore properties of Cu-MOFs were evaluated by N2 adsorption–desorption isotherms, as shown in Fig. 3a and b and Table 1. Both CuBDC and CuBTC exhibited type II isotherms with the presence of both micropores and mesopores in the MOFs. The CuBDC-N exhibited low N2 uptakes at relative pressure less than p/p0 ∼ 0.2 with hysteresis loops at p/p0 ∼ 0.45–0.90. These isotherms are described as type IV with characteristics of mesoporous materials.14 The surface area of CuBDC and CuBTC was significantly higher than CuBDC-N. The formation of comparatively large size particles of CuBDC-N was responsible for its low surface area.15
 |
| | Fig. 3 (a) N2 adsorption–desorption isotherms at −196 °C; (b) pore size distribution; (c) XRD patterns; (d) TGA patterns of Cu-MOFs. | |
Table 1 The surface and pore properties of Cu-MOFs
| MOF |
SBET (m2 g−1) |
Vt (cm3 g−1) |
Dp,avg (nm) |
Dmicro (nm) |
| CuBDC |
217.8 |
0.117 |
2.14 |
0.60 |
| CuBDC-N |
38.0 |
0.068 |
7.18 |
1.90 |
| CuBTC |
317.0 |
0.152 |
1.92 |
0.60 |
The XRD patterns of Cu-MOFs are shown in Fig. 3c, and assignments are listed in S. Tables 2–4. The diffraction peaks of CuBDC at 10.16°, 12.08°, 13.58°, 17.12°, 17.70°, 20.40°, and 24.82° were assigned to 110, 020, 11−1, −201, 111, 220, and 131 Bragg reflections of CuBDC, respectively. The MOF had monoclinic symmetry (a = 11.37 Å, b = 14.62 Å, c = 7.72 Å, β = 108.49°) with C2/m space group.16 The CuBDC-N was crystallized in the monoclinic symmetry with a slight variation in the lattice parameters due to the presence of amine functionality (S. Table 5†). The XRD pattern of CuBTC matched well with the HKUST-1, where diffraction peaks at 9.46°, 11.62°, 13.42°, 19.02°, and 25.96° were due to 022, 222, 004, 044, and 355 reflections, respectively. CuBTC crystallized in the cubic symmetry (a = 26.36 Å) with Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group.17
m space group.17
Fig. 3d presents the weight loss of Cu-MOFs when heated from 30 to 900 °C at a heating rate of 10 °C min−1 in the N2/O2 atmosphere. In TGA profiles, the first weight-loss stage (stage I) occurring between 30–110 °C was due to the evaporation of physically adsorbed water. In stage II (110–250 °C), the weight loss was due to the release of chemically bonded water on the metal sites and physically adsorbed solvent (DMF/ethanol). These two losses were significant for CuBDC and CuBTC due to large surface areas and a more open structure of these two MOFs. In stage III (250–370 °C), a 50–60% loss was recorded for all Cu-MOFs, which was attributed to the breakdown of coordination bonds between metal ions and the organic linkers. Around 350 °C, the MOFs were fully decomposed to metal oxides (Cu2O and CuO).18
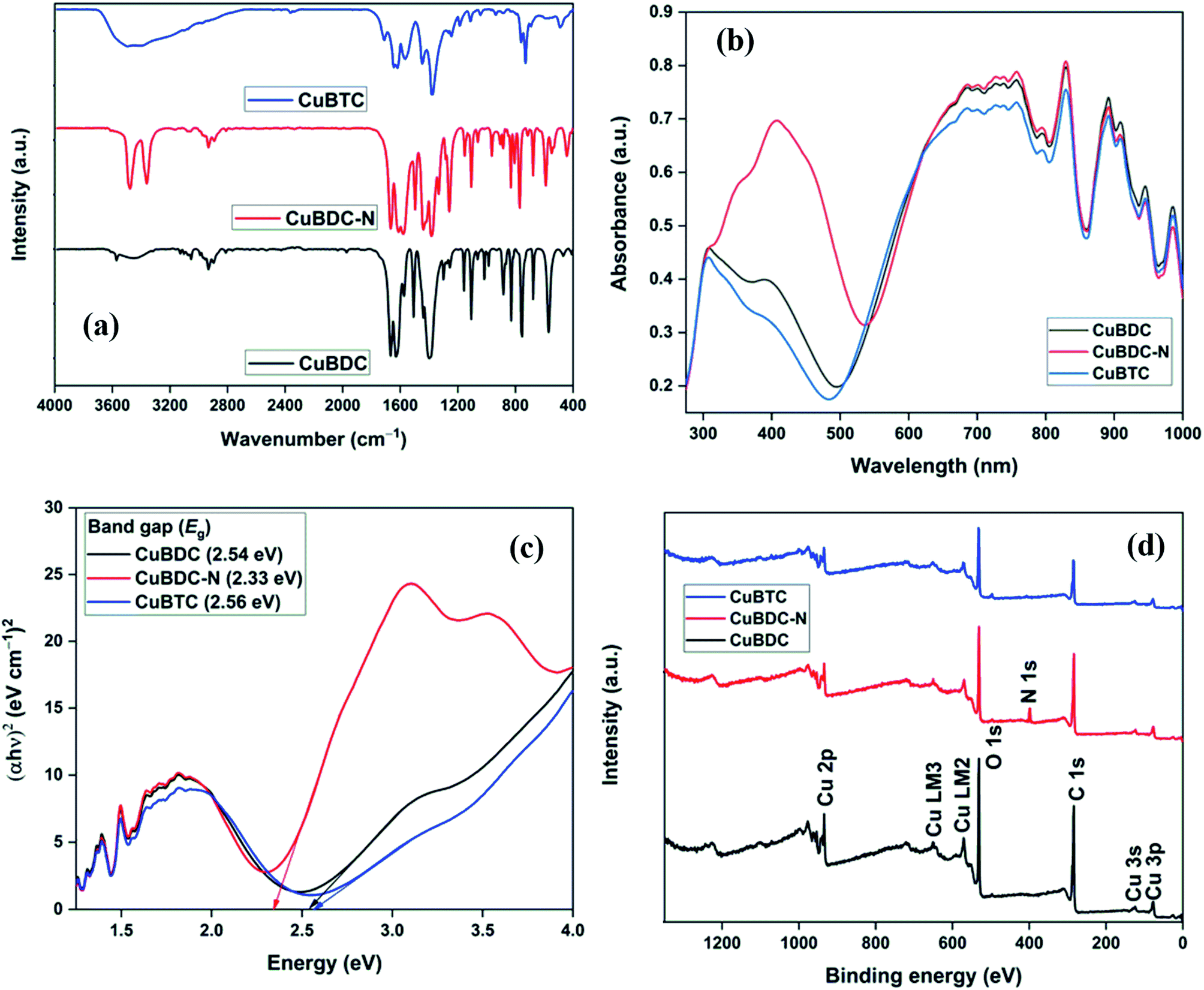
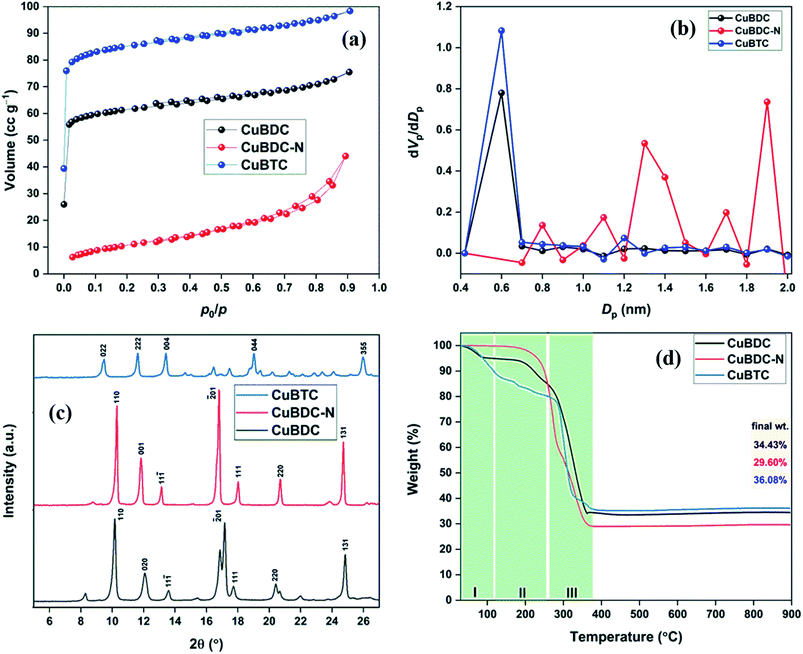
Fig. 4a shows the FTIR spectra of Cu-MOFs. For CuBDC and CuBTC, the band at 3446 cm−1 was due to the O–H stretching vibrations of physically adsorbed water molecules, which was more prominent in CuBTC. Two low-intensity bands at 3052 and 2932 cm−1 were attributed to C–H vibrations of the aromatic skeleton, which were masked by the broad O–H stretching band in CuBTC. Multiple bands in the range of 1200–1000 and 1000–700 cm−1 were assigned to the in-plane and out-of-plane C–H bending modes. For CuBDC, acid C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching at 1672 cm−1, CC at 1511 cm−1, and the symmetric and asymmetric O–C–O stretching at 1396 and 1576 cm−1, respectively, were observed.19 For CuBTC, the bands at 1710 cm−1 for carboxyl groups was observed along with the bands at 1636 and 1386 cm−1 for asymmetric and symmetric stretching of –CO, respectively.20 The Cu–O stretching at 468 cm−1 was observed as a low-intensity band.21 For CuBDC-N, distinct bands for N–H (stretching: 3360 and 3476 cm−1; bending: 1579 cm−1) and aromatic C–N vibrations (stretching: 1259 and 1334 cm−1) of BDC-NH2 linkers were observed.22
O stretching at 1672 cm−1, CC at 1511 cm−1, and the symmetric and asymmetric O–C–O stretching at 1396 and 1576 cm−1, respectively, were observed.19 For CuBTC, the bands at 1710 cm−1 for carboxyl groups was observed along with the bands at 1636 and 1386 cm−1 for asymmetric and symmetric stretching of –CO, respectively.20 The Cu–O stretching at 468 cm−1 was observed as a low-intensity band.21 For CuBDC-N, distinct bands for N–H (stretching: 3360 and 3476 cm−1; bending: 1579 cm−1) and aromatic C–N vibrations (stretching: 1259 and 1334 cm−1) of BDC-NH2 linkers were observed.22
 |
| | Fig. 4 (a) FTIR spectra; (b) UV-Vis DRS spectra; (c) Direct bang gap energy; (d) XPS survey of Cu-MOFs. | |
The optical properties of Cu-MOFs were evaluated by UV-Vis DRS analysis (Fig. 4b). All the MOFs absorbed in the entire UV-Vis-NIR region with multiple peaks originating from the ligand-to-metal charge transfer (LMCT) and d–d transitions around the metal centres. The first band in the 270–500 nm could be assigned to the charged transfer from the oxygen in carboxylate to Cu2+ ions. The variation in the λmax for Cu-MOFs was due to the presence of different organic linkers with varying HOMO energy.23 The second absorption band at a wavelength greater than 500 nm was due to the d–d transition around the distorted octahedral Cu2+ centres. For perfect octahedral geometry, d–d transitions are dipole-forbidden, which leads to weak intensity. The high-intensity d–d band similar to the LMCT band was observed in the present study. A significant distortion in the octahedral geometry due to the ligands heterogeneity around Cu-sites led to multiple peaks and high intensity of the d–d transition band that should be otherwise forbidden.24,25 An insignificant change in the spectra (500–1000 nm) suggested a very similar chemical environment around the metal centres in Cu-MOFs.26,27 The direct bandgap could be estimated using the Tauc equation.
where
α,
hv,
A, and
Eg represent the absorption coefficient, photon energy (eV), a constant, and bandgap energy, respectively. The bandgap energy of Cu-MOFs, as evaluated from the plot of (
αhν)
2 versus hν (
Fig. 4c), was 2.54, 2.33, and 2.56 eV for CuBDC, CuBDC-N, and CuBTC, respectively.
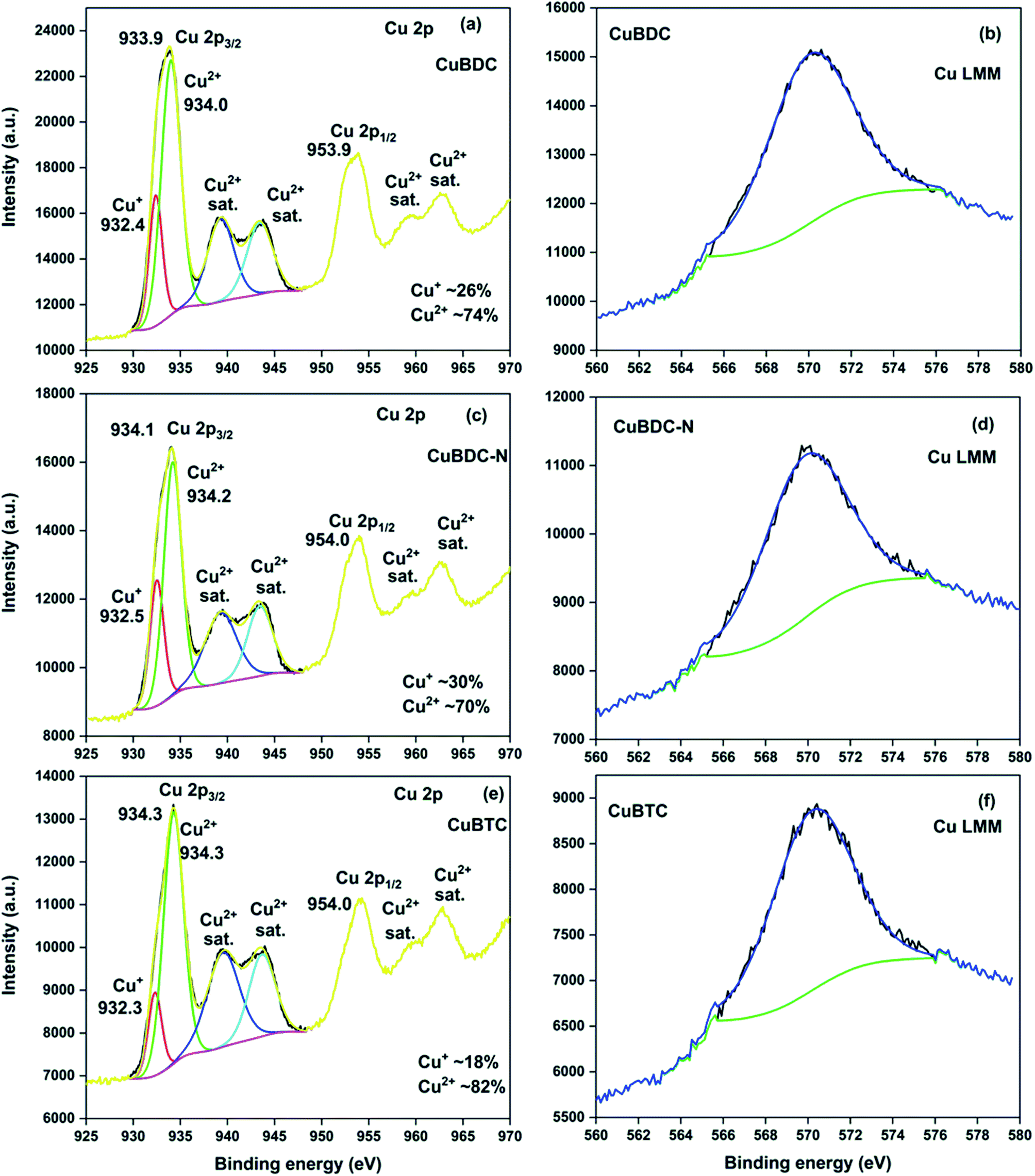
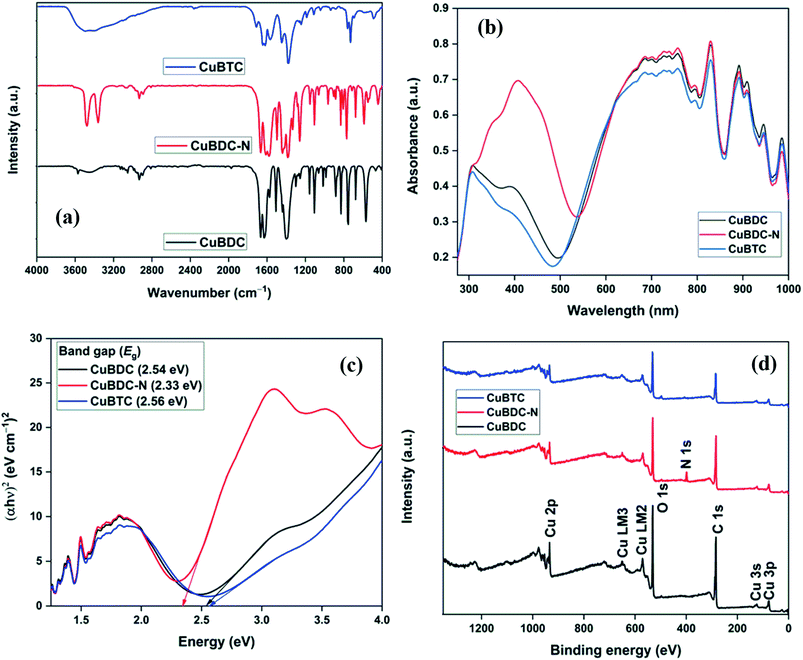
The full scan XPS survey of Cu-MOFs are shown in Fig. 4d. The peaks for C 1s, O 1s, and Cu 2p at their respective binding energy were observed for all the three Cu-MOFs with one additional N 1s peak recorded for CuBDC-N at ∼400 eV. The HRXPS O 1s spectra of Cu-MOFs have two peaks at 531.2 and 531.8 eV, corresponding to Cu–O/CO and C–O bond, respectively19,28 (S. Fig. 2b, d and f†). The HRXPS C 1s spectra of CuBDC had four peaks at 284.7, 286.2, 288.4, and 291.0 eV, which were assigned to CC, C–O/C–H, O–CO, and –COOH, respectively (S. Fig. 2a†). For CuBDC-N (S. Fig. 2c†) and CuBTC (S. Fig. 2e†), the similar observations were made with variations in the binding energy (S. Table 6†). The HRXPS Cu 2p spectra displayed two main peaks at ∼934 (Cu 2p3/2) and ∼954 eV (Cu 2p1/2). The Cu 2p3/2 peak of CuBDC had two contributions at 932.4 and 934.0 eV for Cu+ and Cu2+, respectively. The shake-up satellite peaks observed in the range of 935–945 eV further confirmed the presence of copper as Cu2+ (Fig. 5a).29 For CuBDC-N (Fig. 5c) and CuBTC (Fig. 5e), the Cu+ and Cu2+ peaks were observed at similar binding energies (Table 2). From the HRXPS Cu LMM spectra of Cu-MOFs (Fig. 5b, d, f and S. Fig. 3†), the Cu+ species were further confirmed with the peak at ∼570.6 eV, where Cu0 species remained absent30 (S. Table 7†).
 |
| | Fig. 5 HRXPS Cu 2p spectra of (a) CuBDC; (c) CuBDC-N; (e) CuBTC; Cu LMM spectra of (b) CuBDC; (d) CuBDC-N; (f) CuBTC. | |
Table 2 The peak-fitting results of Cu 2p3/2 high-resolution signals of CuBDC, CuBDC-N, CuBTC
| Samples |
Assignment |
BE (eV) |
FWHM (eV) |
At% |
| CuBDC |
Cu2p3/2 Cu+ |
932.4 |
1.6 |
26.2 |
| Cu2p3/2 Cu2+ |
934.0 |
2.4 |
73.8 |
| Satellite Cu2+ |
939.3 |
3.3 |
— |
| Satellite Cu2+ |
943.4 |
3.2 |
— |
| CuBDC-N |
Cu2p3/2 Cu+ |
932.5 |
1.7 |
30.0 |
| Cu2p3/2 Cu2+ |
934.2 |
2.2 |
70.0 |
| Satellite Cu2+ |
939.3 |
4.0 |
— |
| Satellite Cu2+ |
943.4 |
3.2 |
— |
| CuBTC |
Cu2p3/2 Cu+ |
932.3 |
1.7 |
18.4 |
| Cu2p3/2 Cu2+ |
934.3 |
2.5 |
81.6 |
| Satellite Cu2+ |
939.5 |
4.0 |
— |
| Satellite Cu2+ |
943.7 |
3.3 |
— |
The ESR spectra of Cu-MOFs recorded at 25 °C are shown in S. Fig. 4.† Two signals, one low-intensity signal at the magnetic field (H) ∼ 320 mT (g = 2.06–2.08) and the other high-intensity signal at H ∼ 470 mT (g = 1.42–1.52) were commonly observed in CuBDC and CuBDC-N (S. Fig. 4a and b†). In these MOFs, Cu2+ ions were present in two distinctly different coordination environments. The low-intensity signal (g = 2.06–2.08) was probably due to the presence of monomeric Cu2+ complexes or uncoupled Cu2+ pairs in the MOFs. The high-intensity signal at the higher magnetic field was assigned to the bridged carboxylate-bound two Cu2+ coupled by antiferromagnetic interaction, generating an excited paramagnetic state with total spin S = 1 in the MOF.31 Another low-intensity signal at H = 440 mT (g = 1.42) observed for CuBDC might be due to distortions in the MOF structure. The ESR signal of CuBTC at H = 325 mT (g = 2.13) with a peak-to-peak width of about 100 mT matched well with the reported signal for HKUST-1 (S. Fig. 4c†). The signal observed in the spectrum is attributed to Cu2+ (S = 1/2) paramagnetic centres, presumably due to the monomeric ions complexes ([Cu(H2O)6]2+) or uncoupled Cu2+ pairs in incomplete or stressed paddle-wheels.31
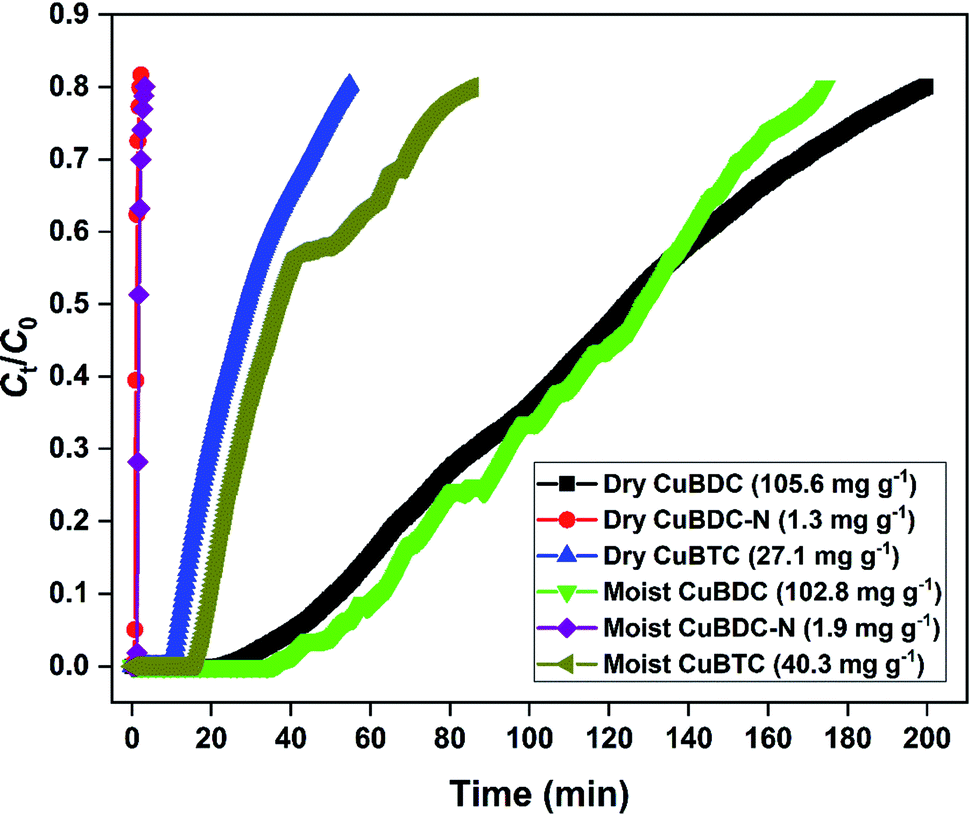
3.2 H2S breakthrough curves
The H2S removal performance of Cu-MOFs was evaluated at 25 °C in a fixed-bed reactor. The obtained breakthrough curves using dry and moist Cu-MOFs are shown in Fig. 6. The CuBDC showed the best adsorption performance among all the MOFs with an adsorption capacity of 105.6 mg g−1. The CuBDC-N showed the lowest adsorption capacity. The trend followed by adsorption capacity was not in line with the surface area. Moreover, the presence of amine-functionalized linkers disfavored the adsorption process as opposed to the reported claim of increased H2S removal over amine-analogues of MOFs.32 Zhang and coworkers have demonstrated that the presence of amine functionalities beyond a certain ratio in MOF-199 disfavored the H2S adsorption process.8 Since the adsorption process of H2S over Cu-MOFs is governed by the formation of strong Cu–S bond at the expense of weak Cu–O bond,9 the adsorption capacity of MOFs is directly influenced by the Cu-site density and nature of Cu-sites. So, the highest copper content of CuBDC could be one of the factors responsible for its exceptional adsorption capacity besides surface area (S. Tables 8 and 9). The adsorption performance of MOFs improved after the addition of moisture for CuBDC-N and CuBTC with a slight loss in the capacity for CuBDC. The presence of water molecules in the MOF structure promoted the adsorption process by the dissolution of H2S molecules. The formed HS− species interacted strongly with the Cu-sites by forming Cu–S bonds.10
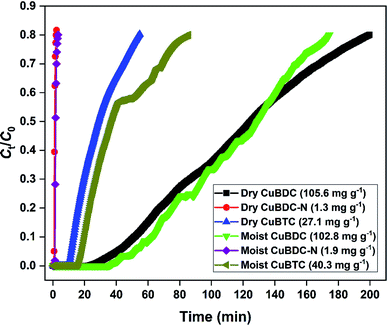 |
| | Fig. 6 H2S breakthrough curve of dry and moist Cu-MOFs. | |
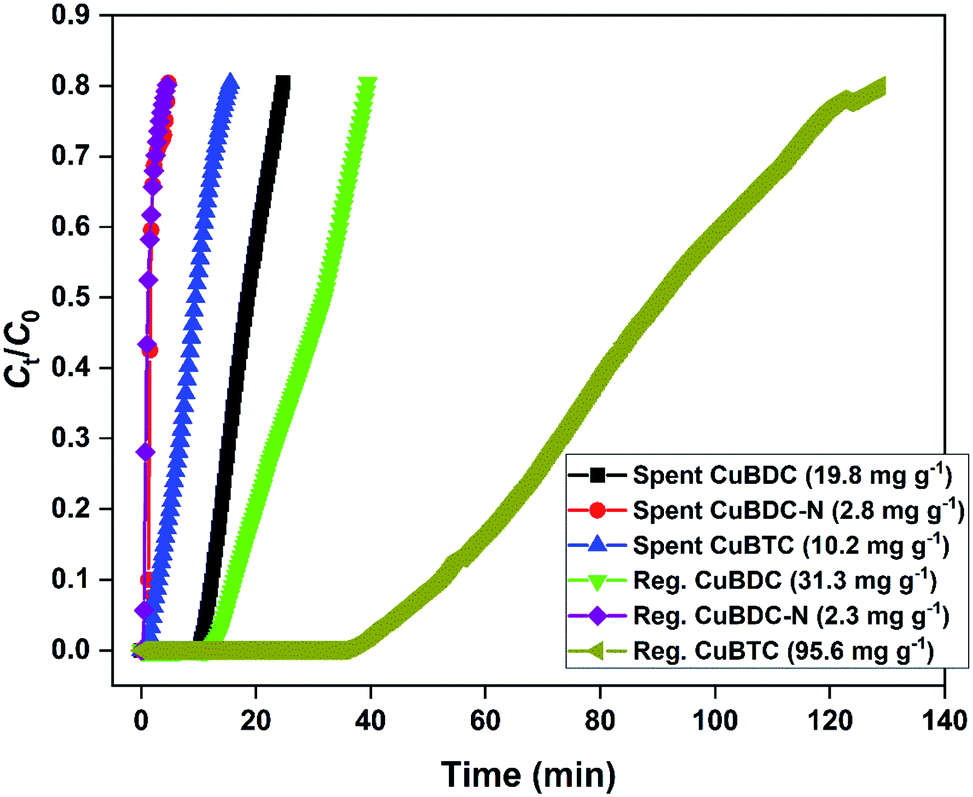
3.3 Regeneration of spent MOFs
The H2S breakthrough curves of spent and regenerated Cu-MOFs are shown in Fig. 7. The spent MOFs have residual adsorption capacity arising from the unsaturated zones formed during the column studies. The presence of ∼20–40% adsorption capacity in the second cycle could lead to an overestimation of regeneration efficiency and has been taken into consideration in the present study. Though the regeneration of H2S-loaded Cu-MOFs is difficult due to the irreversible nature of the chemisorption process, efforts were made to remove physically adsorbed H2S and generate new binding sites by adopting methanol and UV-assisted regeneration method. It has been reported that in the presence of UV irradiation, MOFs undergo a photolytic-decarboxylation process, which increases their basic nature and favours the adsorption of acidic gases.33 In the present study, CuBDC-N could not be regenerated by the process. The regenerated CuBDC showed an increased H2S adsorption capacity of 31.3 mg g−1 as compared to the spent CuBDC (19.8 mg g−1). Though the improved performance was recorded for CuBDC, it was only 32% of the fresh CuBDC, suggesting that most of the Cu-sites were bound to sulfide and could not be regenerated. The regenerated CuBTC showed exceptional adsorption capacity where the capacity increased from 10.2 mg g−1 (spent CuBTC) to 95.6 mg g−1. The regenerated adsorption capacity of CuBTC was 3.5 times the fresh CuBTC. It could be due to the increased surface area and availability of unoccupied Cu-sites, which were inaccessible in the first cycle.
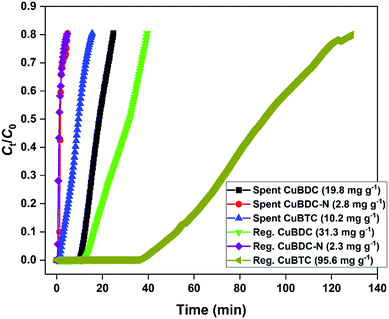 |
| | Fig. 7 H2S breakthrough curve of spent and regenerated Cu-MOFs in the second cycle. | |
3.4 Adsorption and regeneration mechanism
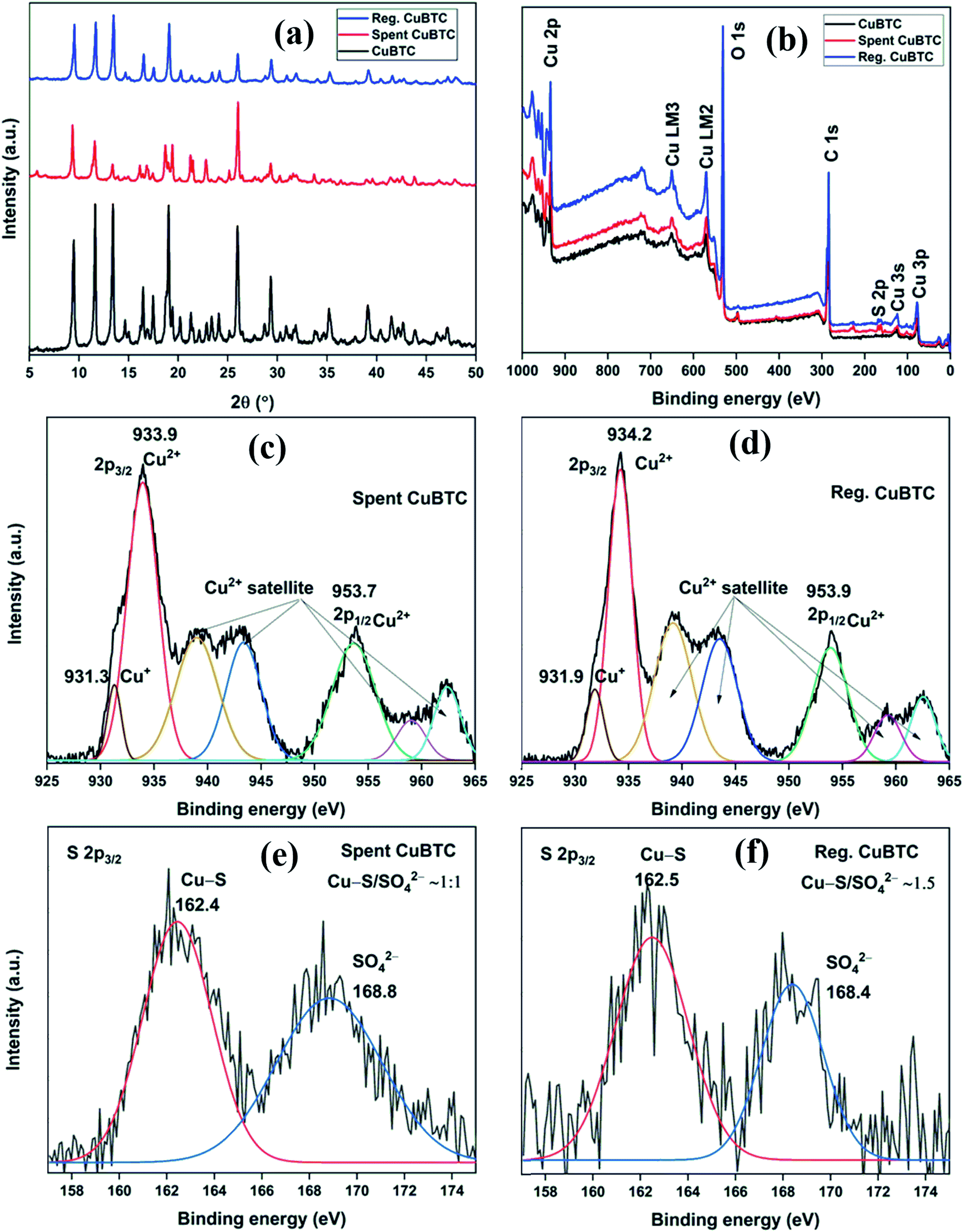
The XRD patterns of fresh, spent, and regenerated CuBTC are shown in Fig. 8a. The reactive adsorption of H2S over CuBTC leads to the breaking of Cu–O bonds with the formation of Cu–S bonds, which decreases the crystallinity of the MOF to an extent that most of the diffraction peaks are lost in the XRD pattern.10 In the present study, the relative intensity ratio of low angle peaks at 2θ = 9.5°, 11.6°, and 13.4° in spent CuBTC changed significantly as compared to the fresh CuBTC. Moreover, the absence of peaks at 2θ = 46.2° (Cu2S) and 31.8° (CuS) suggested that the Cu-sites remained intact in the MOF.34 Thus, the presence of many of the diffraction peaks in the spent MOF suggested that even after H2S adsorption, the MOF stability was partially compromised. After the regeneration of spent CuBTC, the relative intensity ratio of low angle peaks was found very similar to that of the fresh MOF. Moreover, all the major peaks were observed in the diffraction pattern of the regenerated MOF, suggesting that chemisorbed Cu-MOFs could be regenerated by the successive effect of methanol and UV-C irradiation.
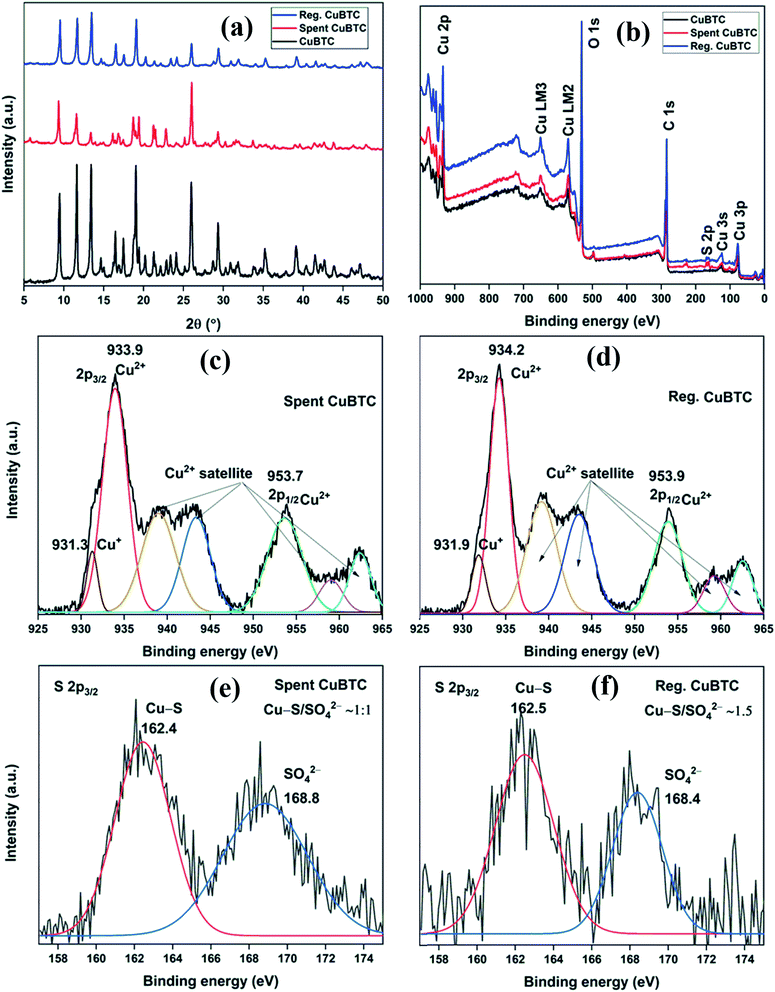 |
| | Fig. 8 (a) XRD patterns; (b) XPS full scan survey of fresh, spent, and regenerated CuBTC; HRXPS Cu 2p spectrum of (c) spent CuBTC (moist); (d) regenerated CuBTC; HRXPS S 2p3/2 spectrum of (e) spent CuBTC; (f) regenerated CuBTC. | |
The XPS full scan survey of spent CuBTC confirmed the presence of sulfur in the MOF as a doublet peak at ∼160–170 eV (Fig. 8b), which accounted for 3.91% of MOF atomic composition (S. Table 10†). The HRXPS Cu 2p spectrum of spent CuBTC showed a shift in peaks for Cu2+ (934.3 → 933.9 eV) and Cu+ (932.3 → 931.3 eV) sites (Fig. 8c). The redshift in the binding energy of Cu peaks confirmed the interaction of S2− with the Cu-sites in both +1 and +2 oxidation states.35 The HRXPS S 2p spectrum of spent CuBTC had two peaks at 162.4 and 168.8 eV, which corresponded to Cu–S and SO42− ions, respectively36 (Fig. 8e). The analysis confirmed an equal proportion of Cu–S and SO42− ions in CuBTC. A large proportion of SO42− ions in MOF formed due to the oxidation of HS− or S2− by adsorbed O2 to elemental S, which was further oxidized to SO42− ions.37 The XPS full scan survey of regenerated CuBTC showed a low-intensity peak for S, which accounted for 1.49% of MOF atomic composition (Fig. 8b, S. Table 10†). The decreased S content and repositioning of Cu peaks in the regenerated CuBTC validated the applicability of the regeneration process. The HRXPS Cu 2p spectrum of regenerated CuBTC showed the repositioning of Cu2+ (933.9 → 934.2 eV) and Cu+ (931.3 → 931.9 eV) peaks, which were found close to that of the fresh CuBTC (Fig. 8d). The HRXPS S 2p spectrum of regenerated CuBTC showed the presence of both kinds of sulfur species, but the proportion of Cu–S was higher than that of SO42− ions (Fig. 8f). The Cu–S presence in regenerated CuBTC could be assigned to the sulfur bound to the Cu+ sites (the Cu+ peak was not fully repositioned in the regenerated CuBTC). A decrease in S content and reactivation of Cu-sites achieved through the regeneration process was responsible for an excellent H2S adsorption capacity of regenerated CuBTC.
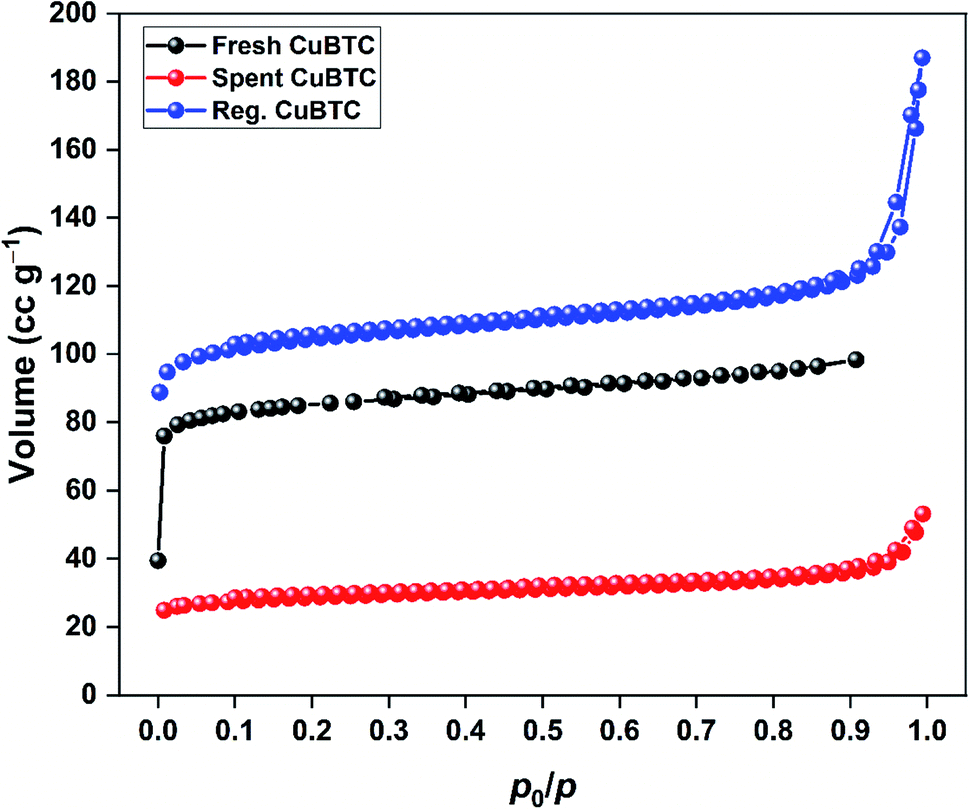
The N2 adsorption–desorption isotherms of spent and regenerated CuBTC are shown in Fig. 9. The surface area and pore characteristics of spent and regenerated CuBTC are listed in Table 3. After H2S adsorption, the surface area and pore volume of CuBTC decreased by 65 and 49%, respectively. These structural changes were linked to the cleaving of Cu–carboxylate bonds upon H2S interactions with the Cu-sites. Such changes are well-documented for stable MOFs like ZIF-8 after H2S exposure.38 The regenerated CuBTC has a surface area of 410.8 m2 g−1, which was higher than that of the fresh MOF (317.0 m2 g−1). Moreover, the pore volume significantly improved after the regeneration process. Similar observations have been reported for the UV-assisted regeneration of iron-incorporated ZSM-5 zeolite and amorphous silica after the adsorption of volatile organic sulfur compounds.39 These improved surface area and pore properties were largely responsible for the enhanced H2S uptake of regenerated CuBTC.
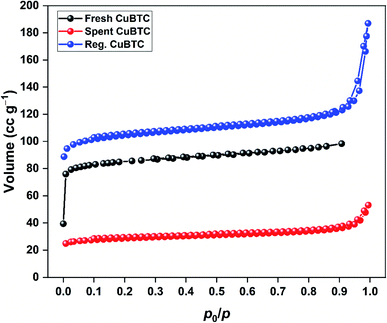 |
| | Fig. 9 N2 adsorption–desorption isotherms of fresh, spent, and regenerated CuBTC. | |
Table 3 The surface and pore properties of fresh, spent, and regenerated CuBTC
| MOF |
SBET (m2 g−1) |
Vt (cm3 g−1) |
Dp,avg (nm) |
| Fresh CuBTC |
317.0 |
0.152 |
1.92 |
| Spent CuBTC |
110.9 |
0.078 |
1.23 |
| Regenerated CuBTC |
410.8 |
0.278 |
1.15 |
4. Conclusions
Copper-based MOFs with different linkers were synthesized by a rapid ultrasonication method for the removal of H2S gas at ambient conditions. The MOFs were characterized by different analytical techniques to understand their physicochemical and optical properties. The adsorption capacity of Cu-MOFs, as calculated for the breakthrough study, followed the trend: 105.6 mg g−1 (CuBDC) > 27.1 mg g−1 (CuBTC) > 1.3 mg g−1 (CuBDC-N) in dry condition. Other than the surface area and pore volume, the Cu+/Cu2+ ratio governed the trend. Stronger interactions of HS− or S2− with Cu2+ sites dominated the adsorption process, and CuBDC with the lowest ratio showed the best adsorption capacity. In the moist condition, the adsorption capacity increased due to the easy dissociation of H2S in water film. The XPS analysis confirmed the formation of Cu–S and SO42− in the moist condition. The XRD pattern of CuBTC showed a partial loss in the crystallinity due to the formation of sulfuric acid and Cu–S bonds. The regenerated Cu-MOFs were possible using the successive effects of methanol and UV-C irradiation. The regenerated CuBTC showed an exceptionally high adsorption capacity of 95.6 mg g−1, which was 3.5 times the fresh CuBTC. Restoration of low-angle peaks in the XRD pattern confirmed the reversibility of the chemisorption process. Moreover, decreased sulfur content of regenerated CuBTC, repositioning of Cu peaks, and increased surface area confirmed that methanol and UV-assisted regeneration is an affordable method to recycle Cu-MOFs with an insignificant loss in the structural and functional integrity.
Author contributions
N. K. G. was solely responsible for conceptualization, formal analysis, software, writing original draft, and review and editing. N. K. G and S. K. were in charge of data curation, methodology, visualization, and validation. J. B. and K. S. K were responsible for funding acquisition, investigation, project administration, resources, and supervision.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors are very grateful for the funds [Project #20200451-001] provided by the “Korea Institute of Civil Engineering and Building Technology” (KICT), Republic of Korea.
References
- A. Georgiadis, N. Charisiou and M. Goula, Catalysts, 2020, 10, 521 CrossRef CAS.
- S. L. Malone Rubright, L. L. Pearce and J. Peterson, Nitric Oxide, 2017, 71, 1–13 CrossRef CAS.
- R. Muñoz, L. Meier, I. Diaz and D. Jeison, Reviews in Environmental Science and Biotechnology, 2015, 14, 727–759 CrossRef.
- M. S. Shah, M. Tsapatsis and J. I. Siepmann, Chem. Rev., 2017, 117, 9755–9803 CrossRef CAS.
- R. Maity, H. D. Singh, A. K. Yadav, D. Chakraborty and R. Vaidhyanathan, Chem.–Asian J., 2019, 14, 3736–3741 CrossRef CAS.
- E. Barea, C. Montoro and J. A. R. Navarro, Chem. Soc. Rev., 2014, 43, 5419–5430 RSC.
- W.-G. Cui, G.-Y. Zhang, T.-L. Hu and X.-H. Bu, Coord. Chem. Rev., 2019, 387, 79–120 CrossRef CAS.
- H.-Y. Zhang, C. Yang, Q. Geng, H.-L. Fan, B.-J. Wang, M.-M. Wu and Z. Tian, Appl. Surf. Sci., 2019, 497, 143815 CrossRef CAS.
- M.-H. Lee, K. Vikrant, S. A. Younis, J. E. Szulejko and K.-H. Kim, J. Cleaner Prod., 2020, 250, 119486 CrossRef CAS.
- C. Petit, B. Mendoza and T. J. Bandosz, ChemPhysChem, 2010, 11, 3678–3684 CrossRef CAS.
- N. Bhoria, G. Basina, J. Pokhrel, K. S. Kumar Reddy, S. Anastasiou, V. V. Balasubramanian, Y. F. AlWahedi and G. N. Karanikolos, J. Hazard. Mater., 2020, 394, 122565 CrossRef CAS.
- C. Petit, B. Levasseur, B. Mendoza and T. J. Bandosz, Microporous Mesoporous Mater., 2012, 154, 107–112 CrossRef CAS.
- W. Xu, G. Li, W. Li and H. Zhang, RSC Adv., 2016, 6, 37530–37534 RSC.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CAS.
- C. Bueno-Ferrer, S. Parres-Esclapez, D. Lozano-Castelló and A. Bueno-López, J. Rare Earths, 2010, 28, 647–653 CrossRef CAS.
- M. Shete, P. Kumar, J. E. Bachman, X. Ma, Z. P. Smith, W. Xu, K. A. Mkhoyan, J. R. Long and M. Tsapatsis, J. Membr. Sci., 2018, 549, 312–320 CrossRef CAS.
- A. Ahmed, C. M. Robertson, A. Steiner, T. Whittles, A. Ho, V. Dhanak and H. Zhang, RSC Adv., 2016, 6, 8902–8905 RSC.
- A. Aarti, S. Bhadauria, A. Nanoti, S. Dasgupta, S. Divekar, P. Gupta and R. Chauhan, RSC Adv., 2016, 6, 93003–93009 RSC.
- A. C. Elder, S. Bhattacharyya, S. Nair and T. M. Orlando, J. Phys. Chem. C, 2018, 122, 10413–10422 CrossRef CAS.
- C. Petit and T. J. Bandosz, Dalton Trans., 2012, 41, 4027 RSC.
- G. Mahalakshmi and V. Balachandran, Spectrochim. Acta, Part A, 2014, 124, 535–547 CrossRef CAS.
- T. Bunchuay, R. Ketkaew, P. Chotmongkolsap, T. Chutimasakul, J. Kanarat, Y. Tantirungrotechai and J. Tantirungrotechai, Catal. Sci. Technol., 2017, 7, 6069–6079 RSC.
- M. A. Syzgantseva, N. F. Stepanov and O. A. Syzgantseva, ACS Appl. Mater. Interfaces, 2020, 12, 17611–17619 CrossRef CAS.
- C. Prestipino, L. Regli, J. G. Vitillo, F. Bonino, A. Damin, C. Lamberti, A. Zecchina, P. L. Solari, K. O. Kongshaug and S. Bordiga, Chem. Mater., 2006, 18, 1337–1346 CrossRef CAS.
- G. Leofanti, M. Padovan, M. Garilli, D. Carmello, A. Zecchina, G. Spoto, S. Bordiga, G. T. Palomino and C. Lamberti, J. Catal., 2000, 189, 91–104 CrossRef CAS.
- H. K. Kim, W. S. Yun, M.-B. Kim, J. Y. Kim, Y.-S. Bae, J. Lee and N. C. Jeong, J. Am. Chem. Soc., 2015, 137, 10009–10015 CrossRef CAS.
- E. Borfecchia, S. Maurelli, D. Gianolio, E. Groppo, M. Chiesa, F. Bonino and C. Lamberti, J. Phys. Chem. C, 2012, 116, 19839–19850 CrossRef CAS.
- N. A. Travlou, K. Singh, E. Rodríguez-Castellón and T. J. Bandosz, J. Mater. Chem. A, 2015, 3, 11417–11429 RSC.
- M. C. Biesinger, Surf. Interface Anal., 2017, 49, 1325–1334 CrossRef CAS.
- D. P. Oyarzún Jerez, M. López Teijelo, W. Ramos Cervantes, O. E. Linarez Pérez, J. Sánchez, G. del C. Pizarro, G. Acosta, M. Flores and R. Arratia-Perez, J. Electroanal. Chem., 2017, 807, 181–186 CrossRef.
- M. Mazaj, T. Čendak, G. Buscarino, M. Todaro and N. Zabukovec Logar, J. Mater. Chem. A, 2017, 5, 22305–22315 RSC.
- J. Liu, Y. Wei, P. Li, Y. Zhao and R. Zou, J. Phys. Chem. C, 2017, 121, 13249–13255 CrossRef CAS.
- X. Han, Q. Cheng, X. Meng, Z. Shao, K. Ma, D. Wei, J. Ding and H. Hou, Chem. Commun., 2017, 53, 10314–10317 RSC.
- M. Mousavi-Kamazani, Z. Zarghami and M. Salavati-Niasari, J. Phys. Chem. C, 2016, 120, 2096–2108 CrossRef CAS.
- A. Galtayries and J.-P. Bonnelle, Surf. Interface Anal., 1995, 23, 171–179 CrossRef CAS.
- J. Wang, L. Wang, H. Fan, H. Wang, Y. Hu and Z. Wang, Fuel, 2017, 209, 329–338 CrossRef CAS.
- C. Yang, Y. Wang, H. Fan, G. de Falco, S. Yang, J. Shangguan and T. J. Bandosz, Appl. Catal., B, 2020, 266, 118674 CrossRef.
- A. Dutta, N. Tymińska, G. Zhu, J. Collins, R. P. Lively, J. R. Schmidt and S. Vasenkov, J. Phys. Chem. C, 2018, 122, 7278–7287 CrossRef CAS.
- S. Kim, N. K. Gupta, J. Bae and K. S. Kim, Chemosphere, 2020, 128943 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra09017d |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab