
Hydride-induced Meisenheimer complex formation reflects activity of nitro aromatic anti-tuberculosis compounds†
Rui
Liu
a,
Lowell
Markley
a,
Patricia A.
Miller
a,
Scott
Franzblau
b,
Gauri
Shetye
b,
Rui
Ma
b,
Karin
Savková
c,
Katarína
Mikušová
 c,
Bei Shi
Lee
d,
Kevin
Pethe
de,
Garrett C.
Moraski
f and
Marvin J.
Miller
*a
c,
Bei Shi
Lee
d,
Kevin
Pethe
de,
Garrett C.
Moraski
f and
Marvin J.
Miller
*a
aDepartment of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, IN 46556, USA. E-mail: mmiller1@nd.edu
bInstitute for Tuberculosis Research, College of Pharmacy, University of Illinois at Chicago, 833 South Wood Street, Chicago, Il 60612, USA
cDepartment of Biochemistry, Faculty of Natural Sciences, Comenius University in Bratislava, Ilkovičova 6, 84215 Bratislava, Slovakia
dSchool of Biological Sciences, Nanyang Technological University, Singapore 637551
eLee Kong Chian School of Medicine, Nanyang Technological University, Singapore 636921
fDepartment of Chemistry and Biochemistry, Montana State University, Bozeman, MT 59717, USA
First published on 4th January 2021
Abstract
The formation efficiency of hydride-induced Meisenheimer complexes of nitroaromatic compounds is consistent with their anti-TB activities exemplied by MDL860 and benzothiazol N-oxide (BTO) analogs. Herein we report that nitro cyano phenoxybenzenes (MDL860 and analogs) reacted slowly and incompletely which reflected their moderate anti-TB activity, in contrast to the instantaneous reaction of BTO derivatives to quantitatively generate Meisenheimer complexes which corresponded to their enhanced anti-TB activity. These results were corroborated by mycobacterial and radiolabelling studies that confirmed inhibition of the DprE1 enzyme by BTO derivatives but not MDL860 analogs.
Introduction
Nitroaromatic compounds have a rich history chemically, biologically and microbiologically.1 While concerns are often raised about potential toxicity of nitroaromatics, a number are important therapeutic agents.2 Antibiotics like metronidazole (1), nitrofurantoin (2) and chloramphenicol (3) have been listed as essential medicines for treatment of bacterial and fungal infections despite some complications. Others, including nitro furans,3 and nitroimidazoles, like PA824 (4)4 have anti-tuberculosis activity. In fact, PA824, now called pretomanid, has recently been approved by the FDA for use in combination with bedaquiline and linezolid for treatment of select patients with extensively drug resistant tuberculosis.5 Nitrobenzothiazinones like BTZ043 (5) and pBTZ169 (6) have outstanding anti-tuberculosis activity and are currently in advanced clinical trials.6 In both PA824 and the BTZs, the nitro group plays an essential role. Biological hydride from a deazaflavin cofactor attacks the carbon adjacent to the nitro group of PA824 to eventually trigger release of nitric oxide (NO) as the active agent.7 The nitrobenzothiazinones (BTZ043 and pBTZ169) also are prodrugs that are reductively activated to give a nitroso moiety (7) that forms a covalent adduct (8) with an essential cysteine of decaprenylphosphoryl-β-D-ribose 2′oxidase (DprE1) that is essential for cell wall biosynthesis in Mycobacterium tuberculosis (Mtb), the causative agent for tuberculosis.8–10 A number of related electron deficient nitro aromatic compounds have recently been shown to have a similar mode of anti-TB activity.11–15Detailed mechanistic understanding is still needed about exactly how the nitro groups of the nitrobenzothiazinones and related nitro aromatic compounds are reduced to the important nitroso moieties. It is well known that the essential DprE1 enzyme system is coupled to NADH/NAD redox chemistry,8–10 but specifically how that important biological chemistry proceeds remains of great interest. We have previously shown that reactions of BTZ043 (5) with nucleophiles like thiolates, cyanide and hydride proceeds rapidly by cine attack ortho and para to the nitro group to generate intermediate Meisenheimer complexes (9a, 9beqn (1)).16 The fate of the intermediate depends on the nucleophile. Most relevant to anti-tuberculosis activity was the observation that reactions with chemical hydride (NaBH4) produced isolable reduction product 9b that is the same as that obtained metabolically from BTZ043 (5).17 This is reminiscent of the first step in the reductive activation of PA824 through Michael addition of hydride at the carbon adjacent to the nitro group (cine reaction). These Meisenheimer complexes (9a and 9b) require only loss of water to generate the corresponding, putatively active, nitroso moiety 7. Reaction of BTZ043 with NaBD4 generates the corresponding deuterated Meisenheimer complexes and reoxidation results in net deuteration of the aromatic core.18 These results indicate that enzymatically induced biological hydride reactions might proceed in a similar fashion to generate the ultimate nitroso warhead. Thus, consideration of appropriate combination of molecular recognition elements and ability to be chemically susceptible to reductive Meisenheimer complex formation, may be useful for identification and evaluation of important and targeted inhibitors (Fig. 1).
 | ||
| Fig. 1 Nitro aromatic antibiotics and anti-tuberculosis agents. | ||

| (1) |
Recently, compounds based on a benzothiazol N-oxide (BTO) carboxamide scaffold with the meta nitro and CF3 groups (10, n = 1, R = amine), also present and important in BTZ043, are impressively potent anti-TB agents that, again, like BTZ043, target the essential DprE1 enzyme.19 Detailed studies demonstrated that just like BTZ043, the nitro group serves as a prodrug and is first reduced to the corresponding nitroso moiety 11 that then reacts with the key DprE1 cysteine thiolate to generate a semimercaptal (12) that results in covalent inhibition. Although these compounds have remarkable anti-TB activity (MICs as low as 0.02 μg mL−1), the example described had moderate cytotoxicity (IC50 = 24 μM).19 Interestingly, reduction of the N-oxide results in compounds (10, n = 0) that are less toxic (IC50 > 80 μM) and retain good (MICs as low as 0.1 μg mL−1), but not as potent activity.10,19
Nitro group containing MDL860 (13)20 and analogs also have been found to react with an essential cysteine residue that results in antiviral activity.21 MDL860 reportedly reacts enzymatically with a cysteine thiol resulting in covalent modification (14) from a nucleophilic aromatic substitution.22 We were intrigued by the similarity of the aromatic core to that of the nitrobenzothiazinones (BTZ-like), BTO and related anti-TB nitroaromatics. Herein, we report that, like BTZ043, MDL860 and representative benzothiazol N-oxide (BTO) derivatives undergo reversible reaction with hydride and deuteride to form intermediate Meisenheimer complexes (i.e.16a and 16b). The results indicate that while nitro aromatics based on a benzothiazol N-oxide (BTO) scaffold (10) and nitro cyano phenoxybenzenes, (MDL860 analogs) reversibly form Meisenheimer complexes upon reaction with hydride similar to BTZ043, they differ considerably in their anti-TB activity (Schemes 1 and 2).
 | ||
| Scheme 1 Nitro substituted benzothiazol-N-oxide reaction with the DprE1 enzyme. | ||
 | ||
| Scheme 2 Enzymatic reaction and chemical reduction of MDL860 (13). | ||
Results and discussion
Reaction of MDL860, 13,23 with NaBD4 generated a red/orange solution characteristic of Meisenheimer complex formation. However, compared to the same reaction with BTZ043 (5), the reaction was visibly slower and the color less intense. Furthermore, monitoring the reductions by LC/MS revealed that while the reduction of BTZ043 was essentially instantaneous with complete conversion of starting material to the Meisenheimer complex, extended reaction of MDL860 with 5, 10 or even 15 equivalents of NaBD4 gave incomplete conversion to the Meisenheimer complex. As with the Meisenheimer complexes generated from BTZ043, reoxidation of the Meisenheimer complexes (16a, b) from the mixture with starting MDL860 regenerated the starting MDL860, but also with deuterium incorporation (17a, b). In contrast to the reported enzymatic reaction, did not result in displacement of the dichlorophenol 15. Proton NMR analysis after reduction with 5 equivalents of NaBD4 followed by reoxidation revealed only 13% deuterium incorporation at the two carbons ortho to the nitro group. In contrast, our previous studies on complete deuteride reduction followed by reoxidation of BTZ043 resulted in 57% and 24% deuterium incorporation ortho and para, respectively. Repetition of the reduction–reoxidation procedure five times on the same samples of MDL860 and BTZ043 enhanced the amount of deuterium content to 57% and 53% at the two positions ortho to the nitro group in MDL860 and 94% (ortho) and 57% (para) in BTZ. It should be noted that the chemical shifts of the aromatic protons of MDL860 and BTZ043 that are exchanged with deuterium differ. In MDL860, the chemical shifts of the two protons ortho to the nitro group are δ 8.36–8.38 and 8.59–8.60 whereas in BTZ043 they are δ 8.78 (para) and δ 8.84 (ortho) suggesting differences in electron deficiency of the corresponding aromatic carbons. Thus, it became of interest to determine if the inherent difference in reactivity and deuterium incorporation of BTZ043 and analogs of MDL860, especially those with similar peripheral substitution (i.e., compound 26), would have similar or different anti-TB activity.Based on literature precedent,24,25 we also synthesized BTO analogs, including 23 (Scheme 3) which incorporates the same piperidine acetal component of BTZ043. Again, the key step was nucleophilic aromatic substitution of 22 with mercaptoacetamide (21) followed by nitro aldol like cyclization to give the N-oxide (BTO, 23). As expected, since the aromatic core of 23 more closely resembles the BTZ043 scaffold, and the chemical shifts of the aromatic protons (δ 8.82 and 8.84) are essentially identical to those of BTZ043, it reacted completely and visibly more quickly with NaBD4 than did MDL860 to provide BTZ043 comparable vivid orange/red Meisenheimer complexes. Oxidation of the complexes under the same conditions reported previously for BTZ043, regenerated the starting compounds and with deuterium incorporation (24) of 48% and 61% ortho and para, respectively, after one reduction – reoxidation cycle and 78% and 89% after repeating the process five times. These results were thus more similar to that observed with BTZ043 rather than with MDL860.
 | ||
| Scheme 3 Synthesis and reduction reoxidation chemistry of a BTO with the BTZ043 side chain. | ||
This BTZ043-like hydride induced reactivity and Meisenheimer complex formation prompted us to synthesize additional analogs of MDL860, BTOs and the corresponding BT (nitrone reduced) compounds and determine if they have anti-TB activity that was reflected by their reactivity. As shown by the representative example with the piperidine spiro ketal component (19) of BTZ043 (eqn (2)), a set of MDL860 analogs (26–36) was synthesized by direct nucleophilic aromatic substitution reactions of commercially available 4-chloro-3-cyano nitrobenzene (25) with various amines (Table 1).
| Cpd No. | Structure | MIC (μM) | IC50 (μM) | |
|---|---|---|---|---|
| GASc | 7H12d | Vero cell | ||
| a Reaction condition: N-methylmorpholine, DIPEA, 80 °C. b Reaction condition: NaOH, DMF, 100 °C. c GAS: glycerol–alanine salts medium. d 7H12: 7H9 medium + casitone, palmitic acid, albumin, and catalase. | ||||
| 13 | MDL860 | 45 | >50 | >50 |
| 26 |

|
2 | 2.4 | >100 |
| 27 |

|
5 | >5 | >100 |
| 28 |

|
>5 | >5 | >100 |
| 29 |

|
5 | >5 | 29 |
| 30 |

|
>5 | >5 | >100 |
| 31 |

|
>5 | >5 | >100 |
| 32 |

|
>5 | >5 | >100 |
| 33 |

|
5 | >5 | >100 |
| 34 |

|
2 | >5 | >100 |
| 35 |

|
3 | >5 | >100 |
| 36 |

|
>5 | >5 | >100 |
| 4 | PA-824 | 0.2 | 0.08 | >100 |

| (2) |
Reagent and conditions: a. N-methylmorpholine, DIPEA, 80 °C; or b. NaOH, DMF, 100 °C.
Based on Scheme 3, we also prepared a small additional set of BTOs (Scheme 4, 39–41). Subsequent phosphite-mediated reduction gave representative BT compounds (42 and 43, Scheme 4, Table 2).
 | ||
| Scheme 4 Syntheses of BTO and BT compounds. | ||
| Cpd No. | Structure | MIC (μM) | IC50 (μM) | ||
|---|---|---|---|---|---|
| Mtb 7H12a | Mtb GASb | Mv | Vero cell | ||
| MHc | |||||
| Mv = M. vaccae IMET 10670. a 7H12: 7H9 medium + casitone, palmitic acid, albumin, and catalase. b GAS: glycerol–alanine salts medium. c MH: Mueller–Hinton broth. | |||||
| 23 |

|
0.24 | 0.23 | 0.015 | 79.6 |
| 39 |

|
0.01 | 0.01 | <0.001 | 76.0 |
| 40 |
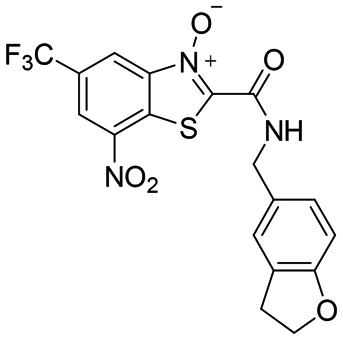
|
0.01 | <0.002 | <0.001 | >100 |
| 41 |
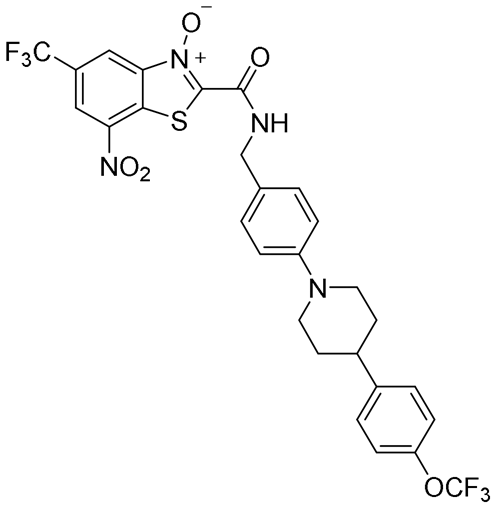
|
0.025 | 0.07 | 0.02 | 81.0 |
| 42 |

|
0.7 | 0.14 | 0.008 | >100 |
| 43 |

|
0.24 | 0.13 | 0.125 | >100 |
| 44 |

|
0.03 | 0.007 | 0.004 | >100 |
| 5 | BTZ043 | <0.004 | <0.004 | <0.001 | >100 |
As noted, the hydride reactions with representatives of the different classes of nitro aromatic compounds described above indicated that the BTO/BT compounds visually reacted more similarly to BTZ043 than did MDL860. This observation corresponded to LC/MS analyses of the reduction reaction mixtures. MDL860 (13, Table 1) itself and the separately synthesized24 BTO ester (45) and unsubstituted amide (46, Scheme 5) had no to low activity against TB (45, MIC > 5 μM in 7H12 and 4.97 μM in GAS and 46, 2.19 μM in 7H12 and 1.2 μM in GAS). This was surprising because amide 46 had demonstrated anticancer activity by inhibition of polo kinase (PLK1) by reversible addition of an essential cysteine (Cys67) in the ATP binding site to give a Meisenheimer complex (47).25 Therefore, it was anticipated that incorporation of substituents to promote molecular recognition by the presumed DprE1 target would provide compounds with activity reflective of their ease of formation of hydride induced Meisenheimer complex formation. The anti-TB assay results shown in Tables 1 and 2 are consistent with these expectations.
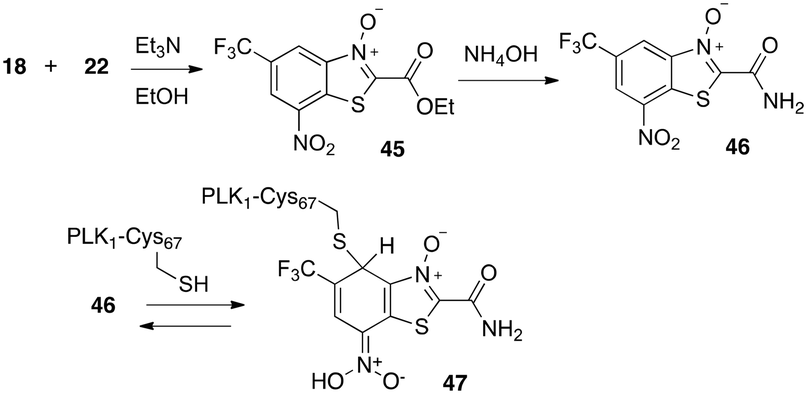 | ||
| Scheme 5 Syntheses of BTO ester and unsubstituted amide and reported reversible formation of Meisenheimer complex with cysteine 67 of oncology target polokinase 1 (PLK1). | ||
Indeed, some substituted MDL860 analogs and all BTO and BT derivatives had moderate to outstanding anti-TB activity. All MDL860 compounds (Table 1) were less active than either the BTO or BT compounds (Table 2) and, as might be expected based on their enhanced electrophilic character, the BTOs were more active than the BT compounds. Still, among the MDL860 compounds, compound 26 with the same piperidine spiro ketal as in BTZ043, was among the most active. Some of the other compounds (27, 29, 33, 34, 35) with hydrophobic substituents akin to pBTZ169 (6) also showed moderate activity. Removal of the Boc protecting group from 35 to give the more hydrophilic free amine 36, also reduced activity. Incorporation of more elaborate amide substituents provided compounds with outstanding anti-TB activity relative to the ester (45) and simple amide (46). Addition of the BTZ043 like piperidine spiro ketal (23, 0.24 μM) or more hydrophobic moieties as in 39 (0.01 μM) and 40 (0.01 μM) or even the large substituent in 41 (0.03 μM), which is the same as in the potent anti-TB agent Q203 (44) resulted in dramatic improvement in activity. Q203 exerts its anti-TB activity as a QcrB inhibitor, not by inhibition of DprE1, but also seems to require significantly hydrophobic substituents, perhaps for permeability as well as binding. Reduction of the N-oxides to give 42 and 43, resulted in more than ten-fold reduction of anti-TB activity, consistent with reduced warhead electrophilicity. Previous studies of BTO and BT type compounds are consistent with these results.13 Those same studies also indicated that while BTO compounds were generally more active than the corresponding BT analogs, the BTOs studied were notably cytotoxic. These results also appear to correlate with the reduction potential of the nitro aromatic compounds studied. This is also consistent with the VERO cell studies also shown in Table 2 (compare 39 with 42 and 41 with 43). However, the reduced VERO cell toxicity of BTO 42 merits further consideration.
While the anti-TB data shown in Tables 1 and 2, seems to reflect the importance of incorporation of both a strongly electron deficient warhead and hydrophobic binding/molecular recognition components to enhance activity, we also considered it important to determine if the compounds actually did target and inhibit the DprE1 in mycobacteria. First we tested the activity of BTZ043 (5), BTO 41, BT 43 and Q203 (44) against additional strains of mycobacteria since it is well known that in order for BTZ type compounds to be active, the target mycobacteria must contain an essential cysteine in the DprE1 active site. As shown in Table 2, not only were all four of these compounds very active against Mtb, but also M. vaccae, which is known to be susceptible to BTZ043. However, Mycobacterium aurum, which is missing the essential DprE1 cysteine, was resistant to all compounds in Table 2 including BTZ043, BT 41 and BTO 43 (MIC = >1 μM, data not shown). This is consistent with all compounds targeting DprE1 in the cells.
To further corroborate this assumption we evaluated the effects of the selected compounds by metabolic labeling of Mtb H37Rv. Analysis of the radiolabeled lipids reveals DprE1 related activity as accumulation of trehalose dimycolates and trehalose monomycolates due to lack of arabinan chains serving as attachment sites for mycolates in the mycobacterial cell wall12 (Fig. 2).
 | ||
| Fig. 2 Evaluation of the effects of the selected compounds on Mtb H37Rv by metabolic radiolabeling. TLC analysis of the lipids from radiolabeled Mtb H37Rv. Mycobacteria were incubated with [14C]-acetate and BTZ043 (5) or target compounds at 100 × MIC for 24 h. TMM, trehalose monomycolates; TDM, trehalose dimycolates; PE – phosphatidylethanolamine; CL – cardiolipin; Con – control (containing DMSO). | ||
The anti-TB data in Tables 1 and 2 along with the radiolabeling studies clearly indicate, that as expected, the BTO compounds (Table 2) are the most active against Mtb H37Rv and, like BTZ043 and pBTZ169, inhibit the putative target DprE1 enzyme in mycobacteria (Fig. 2). Interestingly, BTO 23, with the BTZ043 piperidine spiro ketal moiety is less active than the BTO derivatives with peripheral aromatic substituents. As also reported for related BTO and BT compounds, reduction of the N-oxide to give representative BT compounds (42–43) considerably diminished activity but with retention of DprE1 reactivity (Fig. 2). Although some of the MDL860 analogs have moderate activity, which is consistent with their less electrophilic warhead and visibly slower formation of hydride induced Meisenheimer complexes, they do not show DprE1 activity (Fig. 2). Despite the reduced activity, further studies on the potential mode of activity of the MDL860 analogs is merited, but beyond the scope of this report.
The structural similarity of BTO 41 and BT 43 to Q203 (44) also prompted us to determine if they might be dual DprE1/QcrB inhibitors. Thus, BTO (41) and BT (43) were subjected to an ATP depletion assay in M. bovis BCG wild type and BCG ΔcydAB as described before.26 In this assay, a QcrB inhibitor such as Q203 depleted ATP levels much more in the BCG ΔcydAB background relative to the parental strain (see Fig. 3), while bedaquiline is equally potent in both strains due to its action of the FOF1 ATP synthase (Fig. 3). Conversely, both compounds 41 and 43 deregulated ATP levels at higher doses, which is a characteristic of a drug targeting cell-wall synthesis. Thus, despite their structural similarity to Q203, neither of the two compounds (41 nor 43) appear to target QcrB, or any other components of the oxidative phosphorylation pathway. Thus, consistent with previous reports, the activity of the new BTO and BT compounds is due to inhibition of DprE1.
 | ||
| Fig. 3 Evaluation of compound activity against QcrB. The effect of the test compounds on intracellular ATP levels was tested in M. bovis BCG parental strain (a) and a genetic knockout of cydAB (b). Various concentrations of 41 are shown in filled circles; 43 shown in crosses; Q203 shown in hollow triangles; and bedaquiline (BDQ) shown in asterisks. | ||
Conclusion
Overall, these results further suggest that design of potent nitro aromatic anti-TB agents that target the essential DprE1 enzyme requires incorporation of substituents that promote molecular recognition by the target enzyme as well as strongly electron deficient warheads capable of rapid reaction with hydride to form Meisenheimer complexes. These unique intermediates may serve as precursors to the essential nitroso moiety that inhibits DprE1 by forming covalent adducts.Experimental
General methods
All solvents and reagents were obtained from commercial sources and used without further purification unless otherwise stated. Silica gel (230–400 mesh) was purchased from Silicycle, Quebec City, Canada. All compounds are >98% pure by HPLC analysis. All compounds were analyzed for purity by HPLC and characterized by 1H and 13C NMR using Bruker 400 MHz, 500 MHz NMR spectrometer. Chemical shifts are reported in ppm (δ) relative to the residual solvent peak in the corresponding spectra, and coupling constants (J) are reported in hertz (Hz). The liquid chromatography mass spectrum (LC/MS) analyses were carried out on a Waters ZQ instrument consisting of chromatography module Alliance HT, photodiode array detector 2996, and mass spectrometer Micromass ZQ, using a 3 × 50 mm Pro C18 YMC reverse phase column. Mobile phases: 10 mM ammonium acetate in HPLC grade water (A) and HPLC grade acetonitrile (B). A gradient was formed from 10% to 80% of B in 10 min at 0.7 mL min−1.To a mixture of the trityl acid (1 mmol, 334 mg) and (S)-2-methyl-1,4-dioxa-8-azaspiro[4.5]decane (19, 1 mmol, 157 mg) in 8 mL of DMF was added HATU (1.5 mmol, 570 mg) and DIPEA (1.5 mmol, 276 μL). The solution was stirred at room temperature overnight. The reaction was quenched with H2O and extracted with EtOAc. The EtOAc layer was washed with brine, dried with Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel eluting with hexanes/EtOAc (5/1 to 3/1, v/v) to give 20 as a white solid in 76% yield. 1H-NMR (CDCl3, 500 MHz) δ 1.25 (d, 3H, J = 5.0 Hz), 1.54–1.66 (m, 4H), 2.93 (s, 2H), 3.06–3.11 (m, 2H), 3.40–3.45 (m, 1H), 3.48–3.58 (m, 1H), 3.64–3.69 (m, 1H), 4.02–4.06 (m, 1H), 4.17–4.25 (m, 1H), 7.21–7.24 (m, 3H), 7.28–7.31 (m, 6 H), 7.45–7.47 (m, 6H).
General synthetic method to synthesize MDL860 analogs (26–36)
Biological assays
Cytotoxicity in mammalian cells. Cytotoxicity was tested using the Vero cell (ATCC CRL-81) line as previously described.28 Briefly, eagle's minimum essential medium (MEM) containing 10% fetal bovine serum (FBS) plus penicillin and streptomycin was used for culturing Vero cells. Vero cells were washed with 0.25% trypsin–EDTA 1× solution in Hanks' balanced salt solution (HBSS; pH 7.4), and adjusted to the density of 3 to 5 × 105 cells per ml in MEM. Test compounds were added to the 100 μl of the cell suspension in a 96 well plate and then incubated at 37 °C for 72 h; visual inspection was performed after 24 h. Then 20 μl of 0.6 mM resazurin was added into each well and incubated for 4 h. The fluorescence was measured at excitation/emission wavelengths of 530/590 nm. The concentration of test compound effecting a reduction in fluorescence of 50% relative to untreated cells (IC50) was calculated.
Analysis of the effects of the studied compounds on lipids of M. tuberculosis H37Rv
Mtb H37Rv were grown shaking at 37 °C in GAS medium containing 0.05% tyloxapol until OD = 0.34. Aliquots of 95 μl were transferred into Eppendorf tubes. Subsequently, the drugs or DMSO (final conc. of the drugs was 100 × MIC and DMSO was at 2%) and 0.1 μCi [14C]-acetate [specific activity: 110 mCi mmol−1, American Radiolabeled Chemicals, Inc.] were added to the tubes. After 24 h the whole cultures were transferred into 1.5 ml of chloroform![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :methanol (2:1), and treated for 2 h at 65 °C. The samples were then subjected to biphasic Folch wash, TLC analysis in CHCl3/CH3OH/H2O (20:4:0.5, v/v), and autoradiography (Biomax MR-1 film, Kodak, 5 days at −80 °C).
:methanol (2:1), and treated for 2 h at 65 °C. The samples were then subjected to biphasic Folch wash, TLC analysis in CHCl3/CH3OH/H2O (20:4:0.5, v/v), and autoradiography (Biomax MR-1 film, Kodak, 5 days at −80 °C).
Measurement of the compounds' activity against QcrB in M. bovis BCG
The BacTiter-Glo™ Microbial Cell Viability Assay (Promega, USA) was used for the quantitation of bacterial intracellular ATP content. Parental and ΔcydAB M. bovis BCG were cultivated in 7H9 supplemented with ADS (bovine serum albumin, D-glucose, and NaCl) and 0.05% Tween80 to logarithmic phase and subsequently diluted to OD = 0.05 for the assay. Varying concentrations of drug compounds were added to the cultures and incubated at 37 °C for 20 hours. Following the addition of the Bactiter-Glo™ reagent, the plates were incubated at room temperature for 12 minutes before the measurement was taken. The luminescence was measured using the Cytation-3 cell imaging multiple-mode reader. The experiment was done in triplicates and repeated once.Author contributions
R. L. synthesized compounds and collaborated in drafting the manuscript. L. M. participated in the design of synthesized molecules. P. A. M. performed all non-TB mycobacterial assays and analyzed data. S. F. designed anti-tuberculosis and cytotoxicity assays and oversaw the related studies. G. S. performed anti-TB MIC studies, analyzed and reported data. R. M. performed cytotoxicity studies, analyzed and reported data. K. S. performed radiolabelling DprE1 studies and reported data. K. M. designed and supervised radiolabelling DprE1 studies, analyzed, interpreted, reported data and helped to draft the manuscript. B. S. L. performed target selectivity studies to determine whether the compounds were dual inhibitor and reported data. K. P. designed target selectivity studies to determine whether the compounds were dual inhibitor, analyzed, reported data and also helped to draft the manuscript. G. M. synthesized precursor components to molecules, organized data base for all data and edited the manuscript. M. J. M. conceived of the project, organized the project and oversaw all collaborative interactions, guided the syntheses, interpreted all data and was the primary author of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by NIH grants R01 AI054193 and R37 AI054193 and the University of Notre Dame (George & Winifred Clark professorship, M. J. M). K. M. acknowledges support by Ministry of Education, Science, Research and Sport of the Slovak Republic (contract 0395/2016 for Slovak/Russian cooperation in science 2015-15075/33841:1-15E0) and the Slovak Research and Development Agency (project APVV-15-0515).References
- K.-S. Ju and R. E. Parales, Microbiol. Mol. Biol. Rev., 2010, 74, 250 CrossRef CAS.
- K. Nepauli, H.-Y. Lee and J.-P. Liou, J. Med. Chem., 2019, 62, 2851 CrossRef.
- K. E. Hevener, D. M. Ball, J. K. Buolamwini and R. E. Lee Bioorg, Med. Chem., 2008, 16, 8042 CAS.
- A. J. Lenaerts, V. Gruppo, K. S. Marietta, C. M. Johnson, D. K. Driscoll, N. M. Tompkins, J. D. Rose, R. C. Reynolds and I. M. Orme, Antimicrob. Agents Chemother., 2005, 49, 2294 CrossRef CAS.
- https://www.fda.gov/news-events/press announcements/fda-approves-new-drug-treatment resistant-forms-tuberculosis-affects-lungs .
- V. Makarov, G. Manina, K. Mikusova, U. Möllmann, O. Ryabova, B. Saint-Joanis, N. Dhar, M. R. Pasca, S. Buroni, A. P. Lucarelli, A. Milano, E. De Rossi, M. Belanova, A. Bobovska, P. Dianiskova, J. Kordulakova, C. Sala, E. Fullam, P. Schneider, J. D. McKinney, P. Brodin, T. Christophe, S. Waddell, P. Butcher, J. Albrethsen, I. Rosenkrands, R. Brosch, V. Nandi, S. Bharath, S. Gaonkar, R. K. Shandil, V. Balasubramanian, T. Balganesh, S. Tyagi, J. Grosset, G. Riccardi and S. T. Cole, Science, 2009, 324, 801 CrossRef CAS.
- R. Singh, U. Manjunatha, H. I. M. Boshoff, Y. H. Ha, P. Niyomrattanakit, R. Ledwidge, C. S. Dowd, I. Y. Lee, P. Kim, L. Zhang, S. Kang, T. H. Keller, J. Jiricek and C. E. Barry 3rd, Science, 2008, 322, 1392 CrossRef CAS.
- C. Trefzer, M. Rengifo-Gonzalez, M. J. Hinner, P. Schneider, V. Makarov, S. T. Cole and K. Johnsson, J. Am. Chem. Soc., 2010, 132, 13663 CrossRef CAS.
- A. Richter, I. Rudolph, U. Möllmann, K. Voigt, C.-W. Chung, O. M. P. Singh, M. Rees, A. Mendoza-Losana, R. Bates, L. Ballell, S. Batt, N. Veerapen, K. Fütterer, G. Besra, P. Imming and A. Argyrou, Sci. Rep., 2018, 8, 13473 CrossRef.
- R. V. Chikhale, M. A. Barmade, P. R. Murumkar and M. R. Yadav, J. Med. Chem., 2018, 61, 8563 CrossRef CAS.
- J. P. Caroline, S.-Y. Foo and S. T. Cole, Drug Discovery Today, 2017, 22, 526 CrossRef.
- G. Karabanovich, J. Dušek, K. Savková, O. Pavliš, I. Pávková, I. Korábečný, T. Kučera, H. K. Vlčková, S. Huszár, Z. Konyariková, K. Konečná, O. Jand'ourek, J. Stolaříková, J. Korduláková, K. Vávrová, P. Pávek, V. Klimešová, A. Hrabálek, K. Mikušová and J. Roh, J. Med. Chem., 2019, 62, 8115 CrossRef CAS.
- J. Piton, A. Vocat, A. Lupien, C. S. Foo, O. Riabova, V. Makarov and S. T. Cole, Antimicrob. Agents Chemother., 2018, 62, e00681 CrossRef CAS.
- A. L. de Jesus Lopes Ribeiro, G. Degiacomi, F. Ewann, S. Buroni, M. L. Incandela, L. R. Chiarelli, G. Mori, J. Kim, M. Contreras-Dominguez, Y.-S. Park, S.-J. Han, P. Brodin, G. Valentini, M. Rizzi, G. Riccardi and M. R. Pasca, PLoS One, 2011, 6, e26675 CrossRef CAS.
- R. Tiwari, U. Möllmann, S. Cho, S. G. Franzblau, P. A. Miller and M. J. Miller, ACS Med. Chem. Lett., 2014, 5, 587 CrossRef CAS.
- R. Tiwari, G. C. Moraski, V. Krchnak, P. A. Miller, M. Colon-Martinez, E. Herrero, A. G. Oliver and M. J. Miller, J. Am. Chem. Soc., 2013, 135, 3539 CrossRef CAS.
- F. Kloss, V. Krchnak, A. Krchnakova, S. Schieferdecker, J. Dreisbach, V. Krone, U. Möllmann, M. Hoelscher and M. J. Miller, Angew. Chem., Int. Ed., 2017, 56, 2187 CrossRef CAS.
- R. Liu, V. Krchnak, S. N. Brown and M. J. Miller, ACS Med. Chem. Lett., 2019, 10, 1462–1466 CrossRef CAS.
- S. Landge, A. B. Mullick, K. Nagalapur, J. Neres, V. Subbulakshmi, K. Murugan, A. Ghosh, C. Sadler, M. D. Fellows, V. Humnabadkar, J. Mahadevaswamy, P. Vachaspati, S. Sharma, P. Kaur, M. Mallya, S. Rudrapatna, D. Awasthy, V. K. Sambandamurthy, F. Pojer, S. T. Cole, T. S. Balganesh, B. G. Ugarkar, V. Balasubramanian, B. S. Bandodkar, M. Panda and V. Ramachandran, Bioorg. Med. Chem., 2015, 23, 7694 CrossRef CAS.
- L. D. Markley, Y. C. Tong, J. K. Dulworth, D. L. Steward, C. T. Goralski, H. Johnston, S. G. Wood, A. P. Vinogradoff and T. M. Bargar, J. Med. Chem., 1986, 29, 427 CrossRef CAS.
- I. Nikolovab, I. Slavcheva, M. Ravutsova, M. Dangalova, Y. Nikolovaa, I. Zagranyarskaa, A. Stoyanovab, N. Nikolovab, L. Mukovab, P. Grozdanovb, R. Nikolovac, B. Shivachevc, V. E. Kuz'mind, L. N. Ognichenkod, A. S. Galabovb and G. M. Dobrikova, Bioorg. Chem., 2019, 85, 487 CrossRef.
- M. Arita, G. Dobrikov, G. Pürstinger and A. S. Galabov, ACS Infect. Dis., 2017, 3, 585 CrossRef CAS.
- L. D. Markley, US Pat., 4254144, 1981 Search PubMed.
- A. P. Kozikowski, Y. Chen, A. Gaysin, B. Chen, M. A. D'Annibale, C. M. Suto and B. C. Langley, J. Med. Chem., 2007, 50, 3054 CrossRef CAS.
- R. J. Pearson, D. G. Blake, M. Mezna, P. M. Fischer, N. J. Westwood and C. Mclnnes Cell, Chem. Biol., 2018, 25, 1107 CAS.
- N. P. Kalia, E. J. Hasenoehrl, N. B. AbRahman, V. H. Koh, M. L. T. Ang, D. R. Sajorda, K. Hards, G. Grüber, S. Alonso, G. M. Cook, M. Berney and K. Pethe, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 7426 CrossRef CAS.
- S. Cho, H. S. Lee and S. Franzblau Methods, Mol. Biol., 2015, 1285, 281 CAS.
- M. P. Choules, N. M. Wolf, H. Lee, J. R. Anderson, E. M. Grzelak, Y. Wang, R. Ma, W. Gao, J. B. McAlpine and Y.-Y. Jin, Antimicrob. Agents Chemother., 2019, 63, e02204 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of newly synthesized compounds. See DOI: 10.1039/d0md00390e |
| This journal is © The Royal Society of Chemistry 2021 |