
Itaconate is a covalent inhibitor of the Mycobacterium tuberculosis isocitrate lyase†
Brooke X. C.
Kwai
 a,
Annabelle J.
Collins
a,
Martin J.
Middleditch
bc,
Jonathan
Sperry
*a,
Ghader
Bashiri
*bd and
Ivanhoe K. H.
Leung
*ad
a,
Annabelle J.
Collins
a,
Martin J.
Middleditch
bc,
Jonathan
Sperry
*a,
Ghader
Bashiri
*bd and
Ivanhoe K. H.
Leung
*ad
aSchool of Chemical Sciences, The University of Auckland, Private Bag 92019, Victoria Street West, Auckland 1142, New Zealand. E-mail: j.sperry@auckland.ac.nz; i.leung@auckland.ac.nz
bSchool of Biological Sciences, The University of Auckland, Private Bag 92019, Victoria Street West, Auckland, 1142, New Zealand. E-mail: g.bashiri@auckland.ac.nz
cAuckland Science Analytical Services, The University of Auckland, Private Bag 92019, Victoria Street West, Auckland, 1142, New Zealand
dMaurice Wilkins Centre for Molecular Biodiscovery, The University of Auckland, Private Bag 92019, Victoria Street West, Auckland 1142, New Zealand
First published on 19th October 2020
Abstract
Itaconate is a mammalian antimicrobial metabolite that inhibits the isocitrate lyases (ICLs) of Mycobacterium tuberculosis. Herein, we report that ICLs form a covalent adduct with itaconate through their catalytic cysteine residue. These results reveal atomic details of itaconate inhibition and provide insights into the catalytic mechanism of ICLs.
Isocitrate lyase (ICL) isoforms 1 and 2 are critical for Mycobacterium tuberculosis (Mtb) survival, virulence1,2 and antibiotic tolerance.3 ICLs are key enzymes of the glyoxylate shunt and methylcitrate cycle (Fig. S1†), converting isocitrate/methylisocitrate to glyoxylate/pyruvate and succinate (Fig. 1).4,5 Thus, ICLs enable Mtb to preserve carbon for gluconeogenesis and use odd-chain fatty acids and cholesterol as carbon sources.1,2,4,5 Considering their crucial roles, ICLs are current inhibition targets for the development of new antibiotics to treat tuberculosis,6 with a range of inhibitors, from small molecules7–12 to peptides,7,13 being reported. One of the first reported inhibitors of ICL1 was itaconate (Fig. 1),14–16 a mammalian antimicrobial metabolite that is upregulated in lipopolysaccharide activated macrophages.17–20 Itaconate is an α,β-unsaturated dicarboxylic acid that is structurally analogous to succinate, a product of the ICL-catalysed reaction (Fig. 1). As itaconate is able to access the small, polar binding pocket of ICL,21 it is an attractive lead for the development of effective ICL inhibitors.22
 | ||
| Fig. 1 ICLs catalyse the reversible conversion of isocitrate/methylisocitrate to glyoxylate/pyruvate and succinate. Itaconate acts as an inhibitor of the ICL-catalysed reaction. | ||
ICL possesses a nucleophilic cysteine residue at the active site. The nucleophilicity of the cysteine is enabled by a conserved histidine residue (found on the conserved KKCGH sequence motif), and an unidentified residue in the vicinity of the substrate that could act as a general acid/base.23,24 Recent studies with 3-nitropropionate23,25 and 2-vinyl-D-isocitrate10 demonstrated that they from covalent adducts with the catalytic cysteine residue of ICL1 (Cys191) (Fig. S2†). The Michael acceptors in these examples are propionate-3-nitronate (formed by deprotonation of 3-nitropropionate),25 and 2-vinylglyoxylate (from the ICL-catalysed lysis of 2-vinyl-D-isocitrate).10 Although the mode of inhibition of itaconate has never been reported, itaconate contains an α,β-unsaturated carbonyl moiety that could act as a Michael acceptor.26 Covalent modifications between itaconate and the thiol group of cysteine and glutathione have also been reported previously.27–29 Hence, we sought to investigate whether ICLs might undergo a covalent reaction with itaconate; vital information required to guide the development of antibiotics based on the itaconate scaffold.
We first applied liquid chromatography-mass spectrometry (LC-MS) to conduct intact protein analysis under denaturing conditions. Whereas mass spectrum of Mtb ICL1 shows one peak with a molecular mass of 49![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 642.1 Da (Fig. 2a), matching its calculated molecular mass (49639.3 Da), a 130 Da mass difference was observed upon pre-incubation of ICL1 with excess itaconate and Mg2+ (a molecular mass of 49772.4 Da, Fig. 2b). Given the experiments were conducted under denaturing conditions, any non-covalent interactions would have been disrupted. Hence, the observed mass difference of 130 Da suggests the formation of a covalent ICL1-itaconate adduct through addition at the methylene moiety of itaconate; reaction at any of the carboxylate moieties would result in a different molecular weight.
642.1 Da (Fig. 2a), matching its calculated molecular mass (49639.3 Da), a 130 Da mass difference was observed upon pre-incubation of ICL1 with excess itaconate and Mg2+ (a molecular mass of 49772.4 Da, Fig. 2b). Given the experiments were conducted under denaturing conditions, any non-covalent interactions would have been disrupted. Hence, the observed mass difference of 130 Da suggests the formation of a covalent ICL1-itaconate adduct through addition at the methylene moiety of itaconate; reaction at any of the carboxylate moieties would result in a different molecular weight.
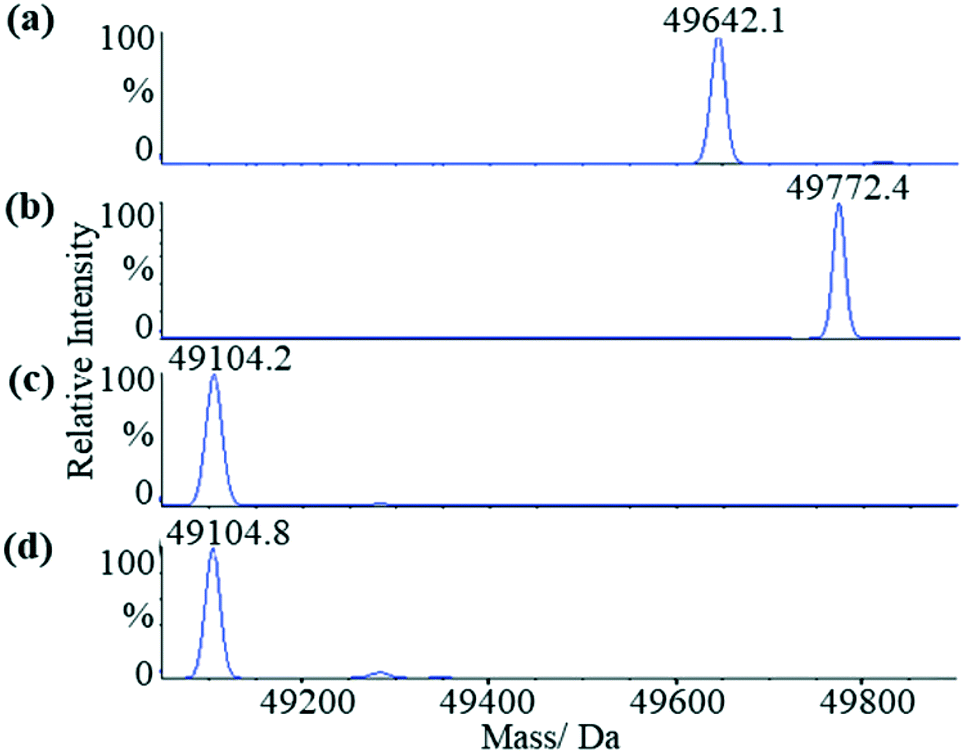 | ||
| Fig. 2 LC-MS analyses of the wild-type and C191S ICL1 constructs. (a) Mass spectrum of the wild-type ICL1. The calculated molecular mass of the intact protein is 49639.3 Da (observed: 49642.1 Da). (b) Mass spectrum of the wild-type ICL1 pre-incubated with itaconate and Mg2+. The calculated molecular mass of the wild-type ICL-itaconate covalent adduct is 49769.4 Da (observed: 49772.4 Da). (c) Mass spectrum of C191S ICL1 construct with a calculated molecular mass 49233.9 Da (observed: 49104.2 Da). The difference between the observed and calculated molecular mass is likely due to cleavage of the N-terminal methionine by the Escherichia coli methionine aminopeptidase.38 (d) Mass spectrum of C191S ICL1 pre-incubated with itaconate and Mg2+. No changes were observed between the samples with or without pre-incubation with itaconate and Mg2+. | ||
To verify the position of the itaconate adduct, trypsin digestion was performed on ICL1 followed by tandem mass spectrometry (MS/MS) analyses. The results showed that upon pre-incubation with itaconate and Mg2+, one ICL1 peptide (Ala170-Lys197) was covalently modified, as indicated by a 130 Da increase in mass when compared to the unmodified peptide (Fig. S3†). This peptide comprises the active site cysteine residue (Cys191) thought to be responsible for the covalent modification. This was subsequently confirmed by conducting similar experiments with a C191S mutant of ICL1 that replaces the active site cysteine with a less nucleophilic serine. Previous report has shown that C191S ICL1 possesses no isocitrate lyase or methylisocitrate lyase activities.30 Comparing the mass spectra of the C191S construct in the presence (Fig. 2c) or absence (Fig. 2d) of itaconate and Mg2+, there were no changes in the observed molecular mass of the intact protein. These results confirm that itaconate covalently modifies the active site cysteine of ICL1.
Encouraged by these results, we conducted similar experiments with Mtb ICL2. We have recently reported that Mtb ICL2 is comprised of two domains that are joined by a flexible linker.22 These two domains include a regulatory C-terminal domain that binds acetyl- or propionyl-coenzyme A, and a catalytic N-terminal domain that structurally resembles ICL1. To investigate whether itaconate may bind ICL2 via a similar covalent adduct formation with the catalytic cysteine, trypsin digestion experiments were performed with ICL2, similar to those described for ICL1. MS/MS analyses revealed that one ICL2 peptide (Cys215-Arg233), containing the catalytic cysteine (Cys215), showed an increase of 130 Da in mass in the sample that was pre-incubated with itaconate and Mg2+ (Fig. S4†). These results collectively demonstrate that the catalytic cysteine residues in both ICL1 and ICL2 are involved in the covalent adduct formation with itaconate.
In order to understand the structural aspects of the itaconate binding, a crystal structure of the ICL1-itaconate adduct was determined and refined to 1.55 Å resolution. One ICL1 tetramer is present in the asymmetric unit with overall root-mean-square differences between four chains of 0.0724–0.1297 Å over 424–426 aligned Cα positions. The structure appeared in a closed conformation, similar to other reported inhibitor-bound ICL1 structures.21 All four monomers showed clear and unambiguous electron density for a magnesium cation and the covalently-modified cysteine residue in the active site. We modelled in the itaconate adduct as 2-methylsuccinate forming a covalent bond with Cys191 (Fig. 3). The new chiral centre at C(2) was found to adapt a (S)-configuration. The Mg2+ ion coordinates with three nearby water molecules (at a distance of 2.2 Å), a carboxylate oxygen from the Asp153 sidechain (at 2.2 Å) and the C4-carboxylate oxygen of the covalently bound itaconate (at 2.4 Å). The other C4-carboxylate oxygen of the covalently bound itaconate forms hydrogen bond interactions with Arg228 (2.8 Å) and a water molecule (2.7 Å). On the C1 end of the 2-methylsuccinate (covalently bound itaconate), Asn313, Ser315 and Thr347 form hydrogen bond interactions with the carboxylate oxygen (2.7–3.0 Å), while the adjacent carboxylate oxygen forms a hydrogen bond with Ser317 (at 2.5 Å). Structures of the C191S ICL1 construct were also determined using crystals grown in the presence of itaconate, Mg2+ and glyoxylate; however, the active site was found to be highly disordered with no electron density observed for the active site loop or any of the ligands (data not shown).
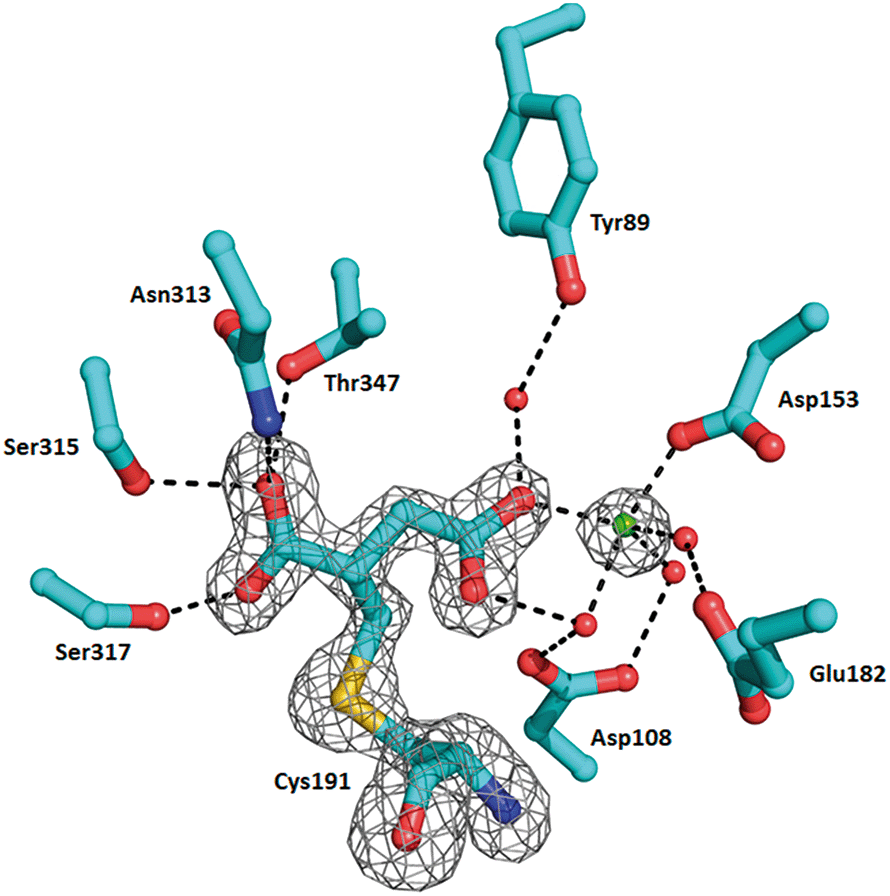 | ||
| Fig. 3 Crystal structure of Mtb ICL1 with covalently bound itaconate. Ball-and-stick representation of the ICL1 active site covalently modified by itaconate, with the cysteine adduct shown in 2Fo–Fc omit density (generated by Phenix),39 contoured at 3.0σ. Atomic colours are as follows: oxygen, red; nitrogen, blue. Mg2+ ion is shown as a green sphere and water molecules as red spheres. Hydrogen bond interactions are shown as dashed lines. Arg228 is not shown for clarity. | ||
A clear understanding of the factors that govern the formation of the ICL-itaconate adduct would inform the development of new ICL covalent inhibitors. ICLs are Mg2+-dependent enzymes31 and it has been proposed that the natural substrates isocitrate or methylisocitrate bind the enzyme by chelating the active site Mg2+via C(1) carboxylate and C(2) hydroxyl groups.21 We therefore investigated the extent to which Mg2+ might impact itaconate binding and hence formation of the ICL1-itaconate covalent adduct. Using LC-MS analyses of the intact protein, we observed no covalent adduct formation between ICL1 and itaconate in the absence of Mg2+ (Fig. S5†). In order to explore this observation further, nuclear magnetic resonance (NMR)-based binding studies were performed to quantify the binding affinity (KD) between itaconate and ICL1. In the presence of Mg2+, itaconate binds to both the wild-type and C191S ICL1 constructs with a similar affinity (KD values of 112 ± 10.7 μM and 155 ± 29.4 μM, respectively) (Fig. S6 and S7†). However, significantly weaker binding was observed in the absence of Mg2+ (KD ≫ 500 μM) (Fig. S8†). These results are intriguing considering that the C191S ICL1 mutant cannot form a covalent adduct with itaconate, suggesting that the covalent adduct formation may not be the only driving force for itaconate binding.
Time course measurements of the ICL1-itaconate adduct formation in the presence of Mg2+ indicated that the formation of the covalent adduct is relatively slow (Fig. S9a†); around 50% of the ICL1-itaconate covalent adduct was formed after about 75 minutes of incubation, with full conversion observed after 5 hours. These observations are intriguing as they infer that not every binding event between itaconate and ICL1 might lead to the formation of a covalent adduct. Electrophilic functional groups need to be at a correct orientation and position (relative to the position of nucleophiles) for covalent interactions to occur.32 Considering that itaconate is a structural analogue of succinate, and that ICL is able to catalyse the back conversion of succinate and glyoxylate to isocitrate, we hypothesised that the addition of glyoxylate may facilitate the formation of the ICL1-itaconate covalent adduct. Repeating the time course experiments between ICL1 and itaconate in the presence of glyoxylate indicated a much faster ICL1-itaconate adduct formation, with a full conversion observed after only 1 minute of incubation (Fig. S9b†). Surprisingly, glyoxylate does not appear to increase the binding affinity of ICL1 to itaconate; the KD value of itaconate binding to ICL1 in the presence of glyoxylate and Mg2+ was determined to be 123 ± 18.0 μM (Fig. S10†), with a decreased binding affinity (KD ≫ 500 μM) obtained in the absence of Mg2+ (Fig. S11†). In addition, negligible amounts of the covalent adduct were observed under the same condition (Fig. S12†), indicating that glyoxylate does not facilitate covalent interaction in the way the Mg2+ would.
Given our crystal structure (Fig. 3) showed the involvement of itaconate carboxylate groups in multiple hydrogen bonding interactions with the active site residues as well as with Mg2+, we subsequently explored the importance of these interactions using synthetic itaconate analogues. Inhibition potency (as reflected by the half-maximal inhibitory concentration, IC50) was used as the read-out, since these compounds were weak binders; their KD values were beyond the detection limit of the binding assay. Our results (Fig. 4 and S13†) showed that changing the carboxylate moieties of itaconate into methyl esters 1/2 significantly reduced the inhibition potency, indicating that hydrogen bonding interactions between the carboxylate moieties and Asn313, Ser315, Ser317, Thr347, Arg228 and Mg2+ (Fig. 3) are essential for itaconate binding. Moreover, addition of hydrophobic 2/4, aromatic 3 and polar 5/6 moieties to the alkene significantly weakens the inhibition potency, inferring that the proximity of Cys191 with the alkene of itaconate is a prerequisite for the covalent interaction to occur. Our results suggest that, in order to maximise inhibition potency and binding affinity, it is important to preserve the interactions between the carboxylate moieties of itaconate with ICL. Modifications of the alkene moiety of itaconate is also undesirable. However, branching out at the C(3) position of itaconate may offer opportunities for future analogue design. Such compounds may fill in the spacious pocket currently occupied by glyoxylate (Fig. S14†), which may enhance the compound's binding affinity/inhibition potency and their ability to form covalent adducts with ICLs. In addition, as the electrophilic nature of itaconate may lead to off target effects in humans,27–29 this strategy of branching out from C(3) position may also improve the selectivity of the analogues.
 | ||
| Fig. 4 IC50 values of itaconate analogues against ICL1. Inhibition experiments contained 20 nM ICL1, 1 mM DL-isocitrate, 1 mM MgCl2, 10 mM phenylhydrazine-HCl and varying concentration of itaconate analogues in 50 mM tris (pH 7.5). Errors represent standard deviations from four separate measurements. | ||
Conclusions
In summary, we have demonstrated that itaconate is a covalent inhibitor of the Mtb ICLs through conjugate addition at the catalytic cysteine residue (Cys191 of ICL1 and Cys215 of ICL2). It appears that itaconate binding is initially facilitated by hydrogen bonding interactions with Mg2+ and the active site residues. The addition of glyoxylate then facilitates the covalent reaction to occur. This is supported by our observation that glyoxylate speeds up the formation of the covalent adduct, but not the binding affinity. It is plausible that in the absence of glyoxylate, itaconate may bind in the ICL active site with multiple orientations, whereas the presence of glyoxylate locks the itaconate into a productive orientation that favours the subsequent covalent adduct formation (Fig. S15†). Moreover, these results provide important insights into the mechanism of ICLs, catalysing the reversible conversion between isocitrate and succinate and glyoxylate. Our results support the proposal that the condensation of succinate and glyoxylate into isocitrate may proceed through an aldol-type reaction.23 The formation of the ICL–Mg2+–succinate–glyoxylate quaternary complex locks succinate into the productive binding conformation. The catalytic cysteine residue of ICLs then deprotonates succinate at the C-2 position (i.e., equivalent to the methylene moiety in itaconate), with the resulting enolate attacking the neighbouring glyoxylate to form isocitrate.The development of targeted covalent inhibitors in medicinal chemistry is undergoing a renaissance, with many covalent drug candidates presently in development.32–37 Covalent inhibitors offer longer-lasting inhibition than non-covalent inhibitors, and there is evidence that suggests covalent drugs may be less likely to promote resistance,32–37 one of the main challenges and obstacles for the control and elimination of tuberculosis. The inhibition of ICL enzymes, with crucial roles in Mtb survival, can be achieved by covalent modification of their active site cysteine residues with itaconate and/or potent itaconate analogues is therefore significant. Our results provide important mechanistic details about the itaconate inhibition mechanism, therefore suggesting that the itaconate scaffold can be utilised as an exciting starting point for the development of novel covalent inhibitors to target the polar actives sites of ICLs and, ultimately, to treat tuberculosis.
We thank the University of Auckland for funding. A. J. C. was supported by a University of Auckland Doctoral Scholarship. G. B. is supported by a Sir Charles Hercus Fellowship (Health Research Council of New Zealand). We thank Dr Michael Schmitz (The University of Auckland) for maintenance of the NMR spectroscopy facility. This research was undertaken in part using the MX2 beamline at the Australian Synchrotron, part of ANSTO, and made use of the Australian Cancer Research Foundation (ACRF) detector. The coordinates and structure factors have been deposited in the Protein Data Bank under accession code 6XPP.
Conflicts of interest
There are no conflicts to declare.Notes and references
- E. J. Muñoz-Elías and J. D. McKinney, Nat. Med., 2005, 11, 638–644 CrossRef.
- J. D. McKinney, K. H. Z. Bentrup, E. J. Muñoz-Elías, A. Miczak, B. Chen, W. T. Chan, D. Swenson, J. C. Sacchettini, W. R. Jacobs Jr and D. G. Russell, Nature, 2000, 406, 735–738 CrossRef CAS.
- M. Nandakumar, C. Nathan and K. Y. Rhee, Nat. Commun., 2014, 5, 4306 CrossRef CAS.
- M. C. Lorenz and G. R. Fink, Eukaryotic Cell, 2002, 1, 657–662 CrossRef CAS.
- E. J. Muñoz-Elías, A. M. Upton, J. Cherian and J. D. McKinney, Mol. Microbiol., 2006, 60, 1109–1122 CrossRef.
- R. P. Bhusal, G. Bashiri, B. X. C. Kwai, J. Sperry and I. K. H. Leung, Drug Discovery Today, 2017, 22, 1008–1016 CrossRef CAS.
- Y. V. Lee, H. A. Wahab and Y. S. Choong, BioMed Res. Int., 2015, 2015, 895453 Search PubMed.
- M. Krátký and J. Vinová, Curr. Med. Chem., 2012, 19, 6126–6137 CrossRef.
- Y.-V. Lee, S. B. Choi, H. A. Wahab, T. S. Lim and Y. S. Choong, J. Chem. Inf. Model., 2019, 59, 2487–2495 CrossRef CAS.
- T. V. Pham, A. S. Murkin, M. M. Moynihan, L. Harris, P. C. Tyler, N. Shetty, J. C. Sacchettini, H.-l. Huang and T. D. Meek, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 7617–7622 CrossRef CAS.
- R. P. Bhusal, K. Patel, B. X. C. Kwai, A. Swartjes, G. Bashiri, J. Reynisson, J. Sperry and I. K. H. Leung, Med. Chem. Commun., 2017, 8, 2155–2163 RSC.
- Y. Liu, S. Zhou, Q. Deng, X. Li, J. Meng, Y. Guan, C. Li and C. Xiao, Tuberculosis, 2016, 97, 38–46 CrossRef CAS.
- X. Liu, Y. Zang, B. Sun and Y. Yin, Med. Chem. Res., 2014, 23, 2543–2553 CrossRef CAS.
- C. R. Rao and B. A. McFadden, Arch. Biochem. Biophys., 1965, 112, 294–303 CrossRef.
- J. W. Rittenhouse and B. A. McFadden, Arch. Biochem. Biophys., 1974, 163, 79–86 CrossRef CAS.
- B. A. McFadden and S. Purohit, J. Bacteriol., 1977, 131, 136–144 CrossRef CAS.
- L. A. J. O'Neill and M. N. Artyomov, Nat. Rev. Immunol., 2019, 19, 273–281 CrossRef.
- H. H. Luan and R. Medzhitov, Cell Metab., 2016, 24, 379–387 CrossRef CAS.
- T. Cordes, A. Michelucci and K. Hiller, Annu. Rev. Nutr., 2015, 35, 451–473 CrossRef CAS.
- C. L. Strelko, W. Lu, F. J. Dufort, T. N. Seyfried, T. C. Chiles, J. D. Rabinowitz and M. F. Roberts, J. Am. Chem. Soc., 2011, 133, 16386–16389 CrossRef CAS.
- V. Sharma, S. Sharma, K. H. Z. Bentrup, J. D. McKinney, D. G. Russell, W. R. Jacobs Jr. and J. C. Sacchettini, Nat. Struct. Biol., 2000, 7, 663–668 CrossRef CAS.
- R. P. Bhusal, W. Jiao, B. X. C. Kwai, J. Reynisson, A. J. Collins, J. Sperry, G. Bashiri and I. K. H. Leung, Nat. Commun., 2019, 10, 4639 CrossRef.
- M. M. Moynihan and A. S. Murkin, Biochemistry, 2014, 53, 178–187 CrossRef CAS.
- N. Jongkon, W. Chotpatiwetchkul and M. P. Gleeson, J. Phys. Chem. B, 2015, 119, 11473–11484 CrossRef CAS.
- S. Ray, D. F. Kreitler, A. M. Gulick and A. S. Murkin, ACS Chem. Biol., 2018, 13, 1470–1473 CrossRef CAS.
- P. A. Jackson, J. C. Widen, D. A. Harki and K. M. Brummond, J. Med. Chem., 2017, 60, 839–885 CrossRef CAS.
- W. Qin, K. Qin, Y. Zhang, W. Jia, Y. Chen, B. Cheng, L. Peng, N. Chen, Y. Liu, W. Zhou, Y.-L. Wang, X. Chen and C. Wang, Nat. Chem. Biol., 2019, 15, 983–991 CrossRef CAS.
- M. Bambouskova, L. Gorvel, V. Lampropoulou, A. Sergushichev, E. Loginicheva, K. Johnson, D. Korenfeld, M. E. Mathyer, H. Kim, L.-H. Huang, D. Duncan, H. Bregman, A. Keskin, A. Santeford, R. S. Apte, R. Sehgal, B. Johnson, G. K. Amarasinghe, M. P. Soares, T. Satoh, S. Akira, T. Hai, C. de Guzman Strong, K. Auclair, T. P. Roddy, S. A. Biller, M. Jovanovic, E. Klechevsky, K. M. Stewart, G. J. Randolph and M. N. Artyomov, Nature, 2018, 556, 501–504 CrossRef CAS.
- J. Yang, Nat. Chem. Biol., 2019, 15, 935–936 CrossRef CAS.
- T. A. Gould, H. van de Langemheen, E. J. Muñoz-Elías, J. D. McKinney and J. C. Sacchettini, Mol. Microbiol., 2006, 61, 940–947 CrossRef CAS.
- K. H. Z. Bentrup, A. Miczak, D. L. Swenson and D. G. Russell, J. Bacteriol., 1999, 181, 7161–7167 CrossRef CAS.
- R. Lonsdale and R. A. Ward, Chem. Soc. Rev., 2018, 47, 3816–3830 RSC.
- T. A. Baillie, Angew. Chem., Int. Ed., 2016, 55, 13408–13421 CrossRef CAS.
- R. Mah, J. R. Thomas and C. M. Shafer, Bioorg. Med. Chem. Lett., 2014, 24, 33–39 CrossRef CAS.
- S. De Cesco, J. Kurian, C. Dufresne, A. K. Mittermaier and N. Moitessier, Eur. J. Med. Chem., 2017, 138, 96–114 CrossRef CAS.
- F. Sutanto, M. Konstantinidou and A. Dömling, RSC Med. Chem., 2020, 11, 876–884 RSC.
- J. Singh, R. C. Petter, T. A. Baillie and A. Whitty, Nat. Rev. Drug Discovery, 2011, 10, 307–317 CrossRef CAS.
- P. T. Wingfield, Curr. Protoc. Protein Sci., 2017, 88, 6.14.11–16.14.13 Search PubMed.
- P. D. Adams, R. W. Grosse-Kunstleve, L. W. Hung, T. R. Ioerger, N. W. A. J. Moriarty, R. J. Read, J. C. Sacchettini, N. K. Sauter and T. C. Terwilliger, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2002, 58, 1948–1954 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, supplementary tables and supplementary figures. See DOI: 10.1039/d0md00301h |
| This journal is © The Royal Society of Chemistry 2021 |