DOI:
10.1039/C5RA21801B
(Paper)
RSC Adv., 2015,
5, 101641-101646
Ammonium persulfate activated DMSO as a one-carbon synthon for the synthesis of methylenebisamides and other applications†
Received
19th October 2015
, Accepted 16th November 2015
First published on 18th November 2015
Abstract
Activation of DMSO to work as an economical and environmentally benign one-carbon synthon has been achieved by using a bench-top reagent ammonium persulfate for general and efficient access to symmetrical methylenebisamides from primary amides. This methodology was used to achieve a three-component Mannich reaction using acetophenone, saccharin and DMSO to furnish a β-amino ketone. It also provided a metal-free synthesis of thiadiazole and bis(phenyl)methane. Effectively, this method uses DMSO as a safer surrogate to formaldehyde. A mechanism for methylenebisamide formation involving radical intermediates has been proposed based on mechanistic studies.
Introduction
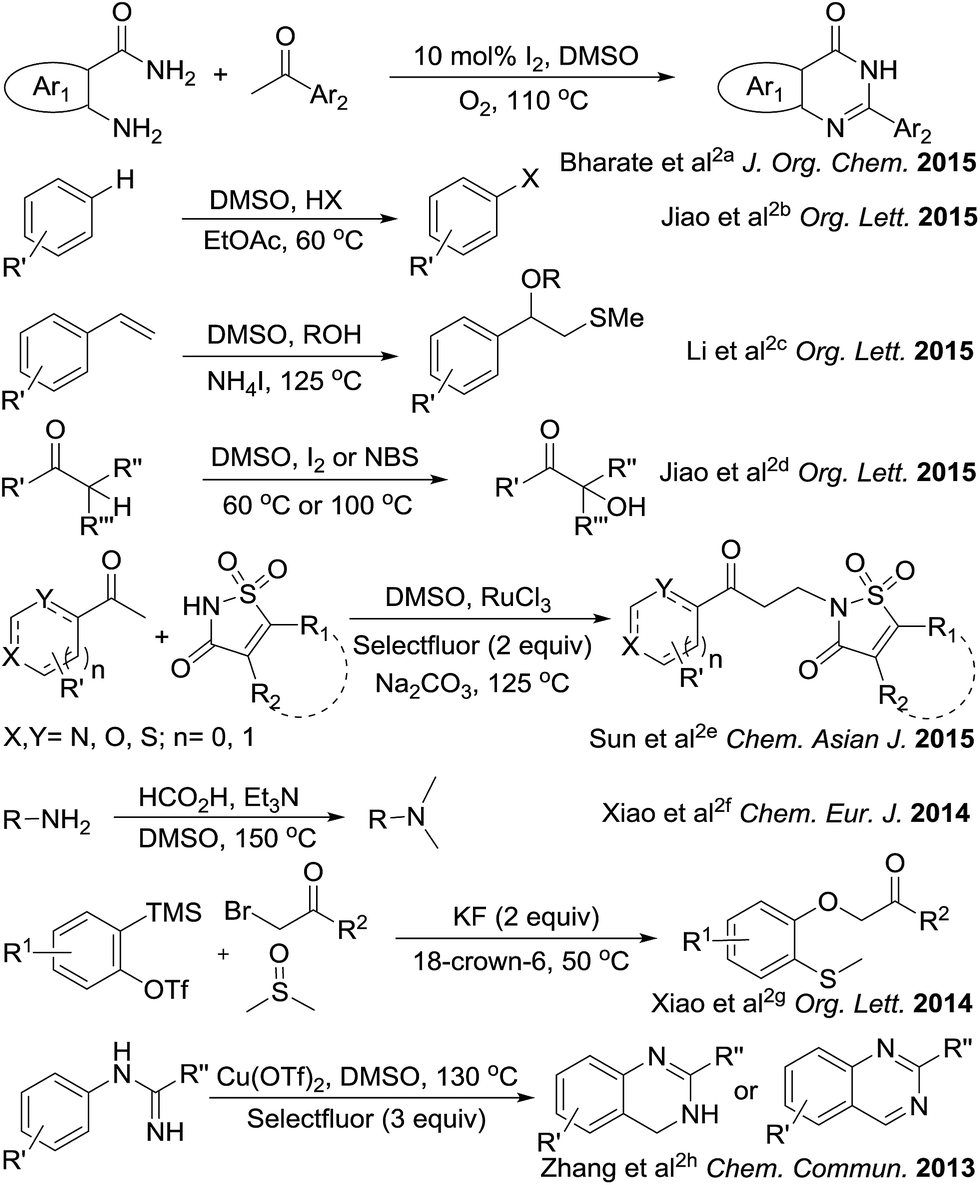
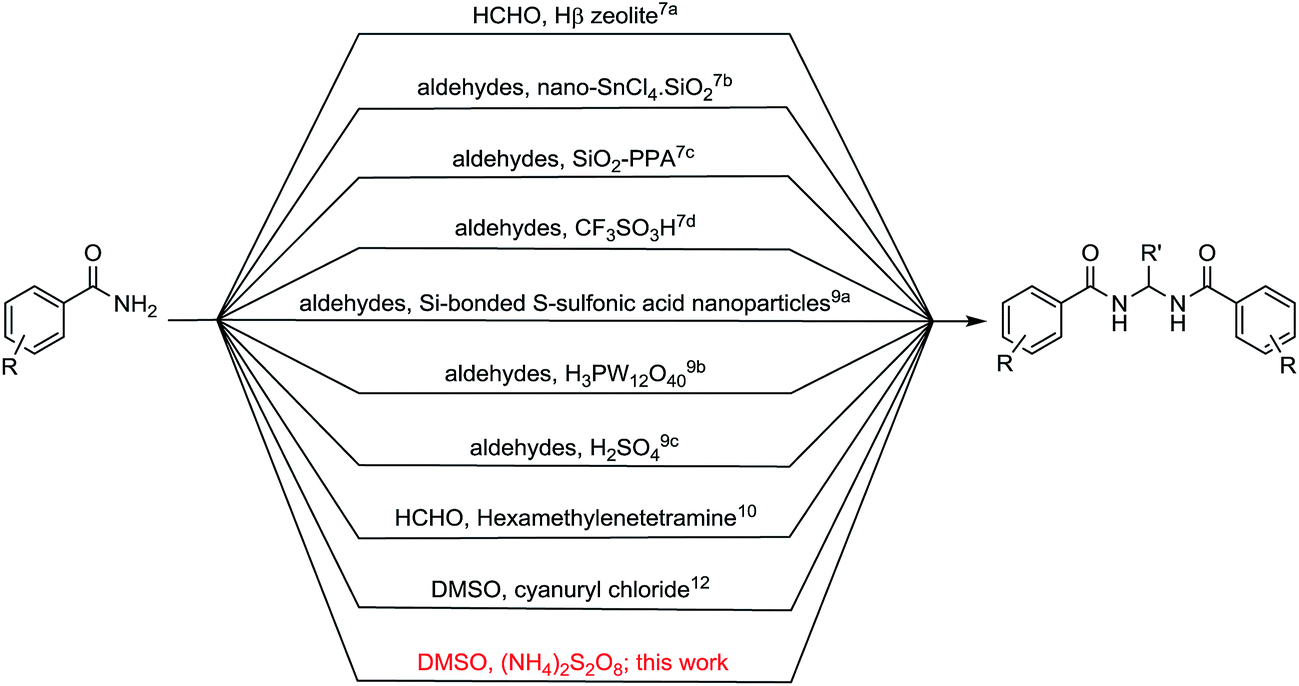
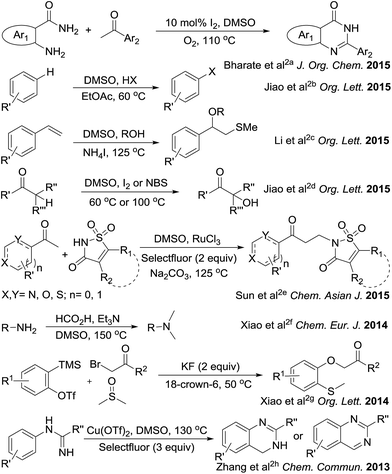
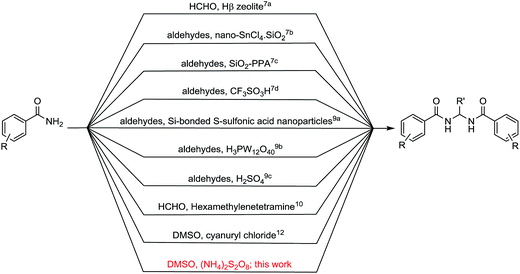
The sulfonium salts generated in situ by the activation of DMSO using various electrophiles for the “Swern oxidation” class of reactions are one of the most important synthetic transformations.1 Similarly activation of DMSO to serve as a one-carbon synthon or for other useful organic transformations has been a subject of contemporary interest (Fig. 1).2 Organic solvents such as CHCl3, DMF or DMSO, are becoming increasingly utilized as a reagent in synthetic transformations.3 We are mainly interested in the transformation of primary amides to methylenebisamides using DMSO as a one-carbon synthon, because bisamides are of considerable interest in the synthesis of peptidomimetic compounds,4 natural products5 and potential key intermediates for the synthesis of bioactive compounds.6,7 In particular, they are key fragments for the introduction of gem-diaminoalkyl residues in retro-inverso pseudopeptide derivatives synthesized for structure–activity relationship studies.8 Generally, methylenebisamides are prepared (Fig. 2) by the reaction of amides or nitriles with formaldehyde using strong acid catalysts.7,9 Hexamethylenetetramine10 and activated DMSO have been also used instead of formaldehyde.11 A review of literature shows that methylenebisamides can be obtained in low yields by activation of DMSO using various electrophiles.11 Li et al. reported the synthesis of methylenebisamides in good to fair yields from amides by using 2,4,6-trichloro[1,3,5]triazine (cyanuric chloride) and 2,4-dichloro-6-methoxy[1,3,5]triazine to activate DMSO.12
 |
| | Fig. 1 Recent advances demonstrating application of activated DMSO. | |
 |
| | Fig. 2 Traditional routes to methylenebisamides and this work. | |
However, the development of a general synthetic process to bisamides is still challenging.13 During the C–H activation studies on primary amide substrates using Pd as catalyst, ammonium persulfate (APS) as an oxidant and small amount of DMSO (5% v/v), we noticed the formation of bisamides. In view of their importance in bioorganic, natural product and medicinal chemistry research and our interest in the development of facile synthetic processes, we planned to explore our observation to establish a general method for the synthesis of bisamides from primary amides using DMSO activated by APS as a new approach. It is an effort to replace formaldehyde, acids and other corrosive/toxic reagents to make a greener process.
Results and discussion
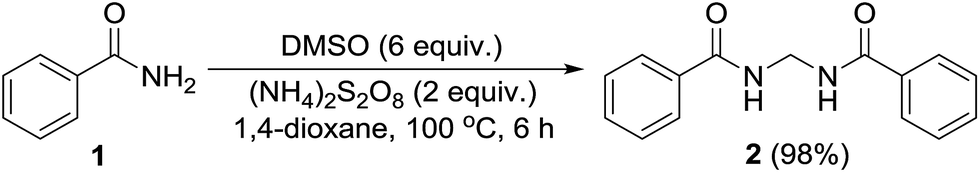
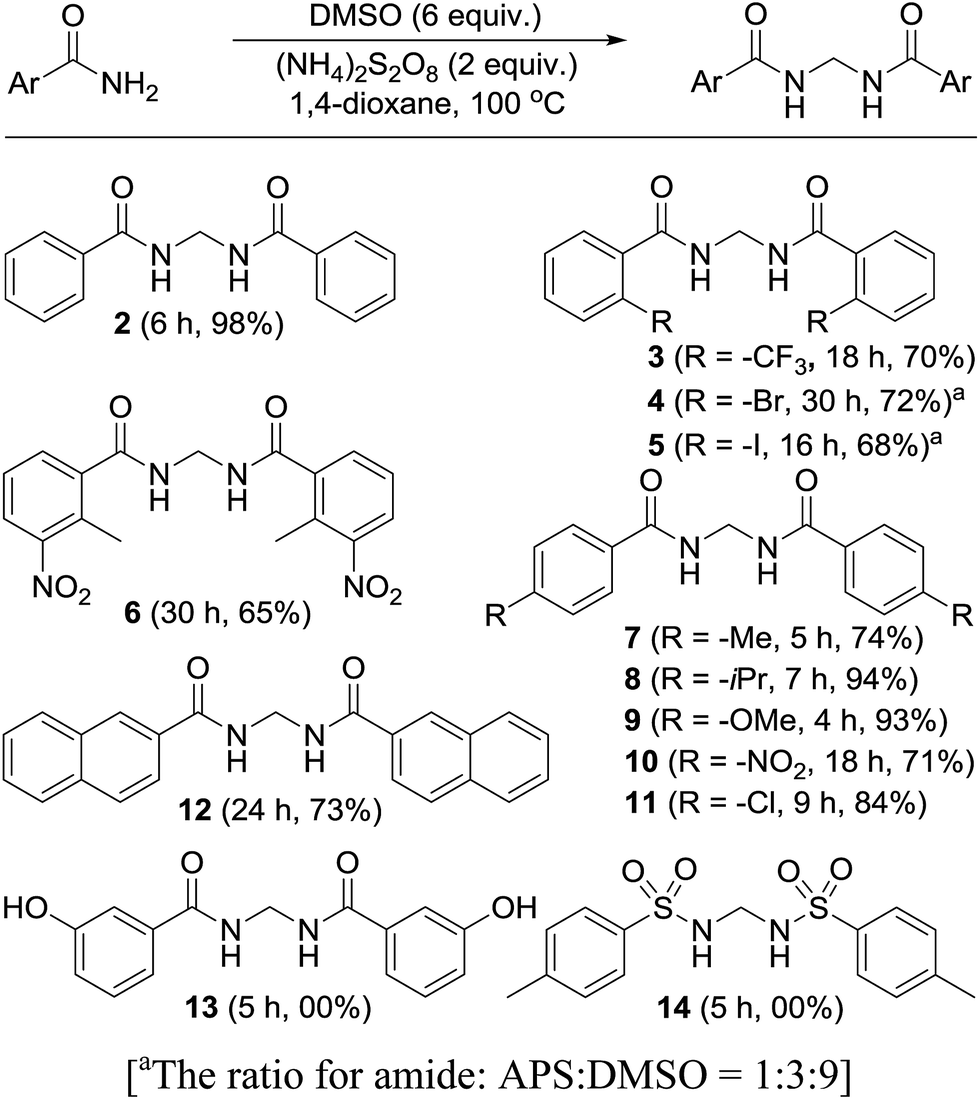
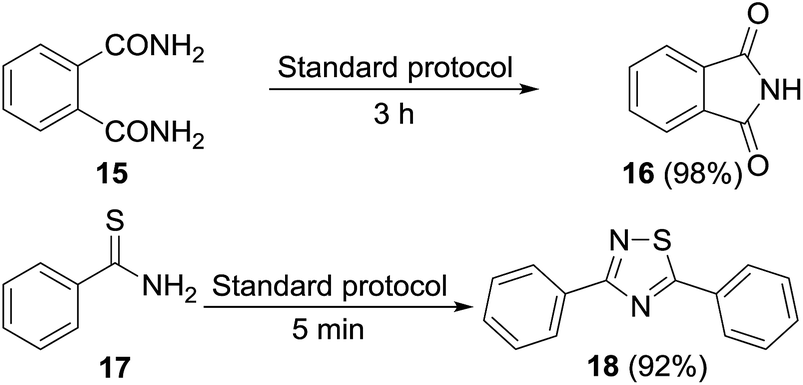
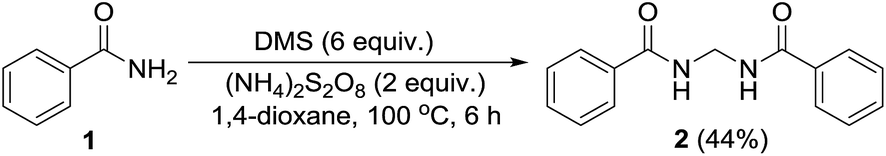
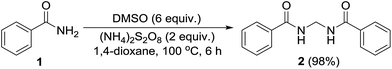
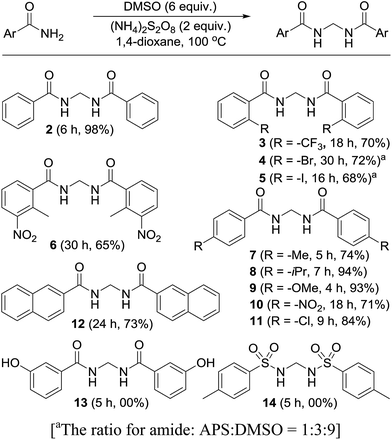
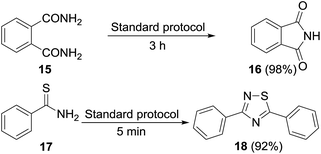
Permutations and combinations of solvents, reagents and their mole ratios at varying temperatures (Table 1) provided the optimum reaction condition, wherein heating the 1,4-dioxane solution of benzamide (1, 1 equiv.) in the presence of APS (2 equiv.) and DMSO (6 equiv.) smoothly furnished the methylenebisamide 2 in quantitative yield (Scheme 1). We planned for the substrate scope study on varyingly substituted aromatic/aliphatic amides. The benzamides substituted with –CF3, –Br and –I at the ortho-position of the amide furnished the corresponding bisamides 3–5 respectively in good yields. The benzamides with electron withdrawing m-NO2 functionality provided methylenebisamide 6 in 65% yield. The effect of electron donating and withdrawing substituents at the para-position was also studied. The alkyl substituted bisamides 7 and 8 were obtained in good to excellent yields and the methoxy substituted bisamide 9 formed equally well. However, the yield of bisamide 10 from the p-NO2 substituted benzamide was similar to the yield of the m-NO2 substituted bisamide 6. These observations suggest that under the developed protocol the reactivity of aromatic amides with electron donating groups is high as compared to the electron withdrawing substituents. p-Chloro benzamide too gave the corresponding bisamide 11 in good yield. The polyaromatic bisamide 12 was obtained in good yield from 2-naphthamide, though in lower yield than bisamide 2. However, 3-hydroxybenzamide and p-toluenesulfonamide remained unreactive and the corresponding bisamides 13 and 14 were not observed at all (Fig. 3). Amides 15 and 17 did not furnish the expected bisamides (Scheme 2). Under the standard protocol phthalamide (15) was converted to pthalimide (16) in quantitative yield. Benzothioamide (17) was converted to diphenylthiadiazole 18 in excellent yield. This example represents an efficient and simple alternative protocol for the formation of thiadiazoles from thioamides.14 Probably, both the reactions followed the APS-DMSO mediated deaminative and desulfurative cyclization pathway respectively, and their mechanism may be similar to our previous report on dehydrative cyclization of amic acids to imides.15
Table 1 Optimization studiesa
|
| Entry |
Oxidant/DMSO (equiv.) |
Solvent (2 mL) |
Temp./time (°C h−1) |
Yieldb (%) |
| All reactions were performed on 50 mg scale of amide 1 in a round bottom flask equipped with a water condenser. Isolated yields. DBPO-dibenzoyl peroxide. |
| 1 |
APS (1)/4 |
Dioxane |
100/18 |
38 |
| 2 |
APS (1)/— |
Dioxane |
100/18 |
00 |
| 3 |
—/4 |
Dioxane |
100/04 |
00 |
| 4 |
APS (1)/excess |
— |
100/18 |
Trace |
| 5 |
APS (2)/excess |
— |
100/10 |
05 |
| 6 |
APS (2)/4 |
Dioxane |
100/18 |
88 |
| 7 |
APS (3)/4 |
Dioxane |
100/18 |
71 |
| 8 |
APS (2)/5 |
Dioxane |
100/09 |
94 |
| 9 |
APS (2)/6 |
Dioxane |
100/06 |
98 |
| 10 |
APS (1)/6 |
Dioxane |
100/12 |
40 |
| 11 |
APS (2)/10 |
Dioxane |
100/06 |
97 |
| 12 |
K2S2O8 (2)/6 |
Dioxane |
100/06 |
35 |
| 13 |
tBuO-OtBu |
Dioxane |
100/06 |
00 |
| 14 |
DBPOc (2)/6 |
Dioxane |
100/06 |
Trace |
| 15 |
PhI(OAc)2 (2)/6 |
Dioxane |
100/06 |
Trace |
| 16 |
Oxone™ |
Dioxane |
100/06 |
17 |
| 17 |
APS (2)/6 |
Toluene |
111/06 |
58 |
| 18 |
APS (2)/6 |
Water |
100/06 |
07 |
 |
| | Scheme 1 Protocol optimized on benzamide (1). | |
 |
| | Fig. 3 Scope of aromatic amides. | |
 |
| | Scheme 2 Different products than expected bisamides. | |
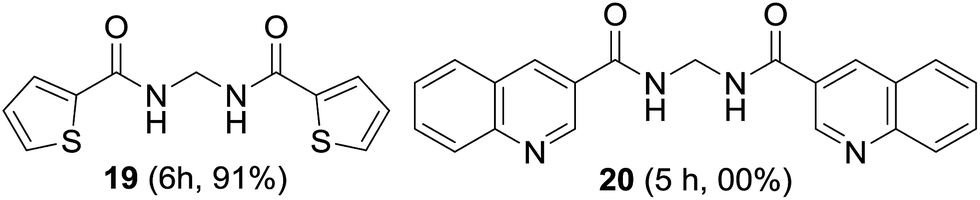
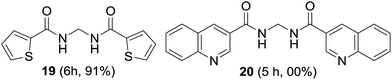
The suitability of symmetrical heteroaromatic methylenebisamides was studied. Thiophene-2-carboxamide provided bisamide 19 in high yield; however, quinoline-3-carboxamide did not furnish bisamide 20, plausibly due to oxidation of the highly basic quinoline nitrogen (Fig. 4).
 |
| | Fig. 4 Heteroaromatic bisamides. | |
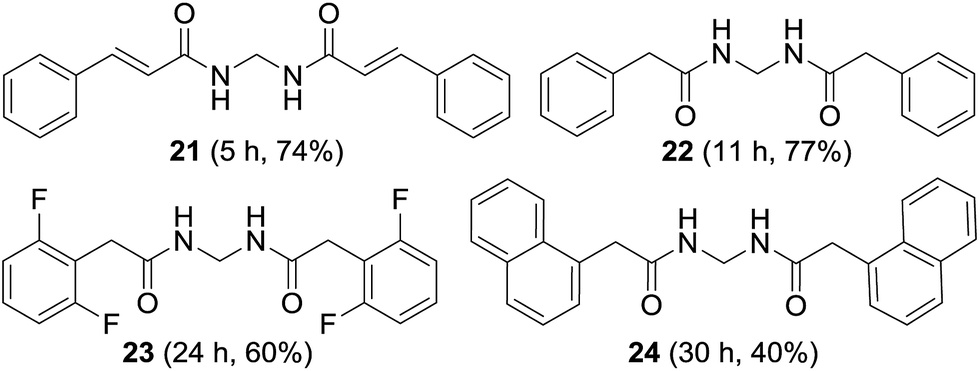
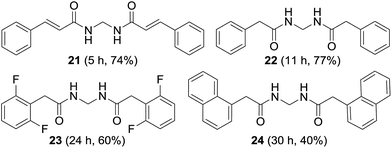
The synthesis of non-aromatic methylenebisamides was examined using the usual procedure (Fig. 5). Cinnamamide, an α,β-unsaturated amide worked well to provide the bisamide 21 in good yield. Formation of various bisamides 22–24 from the corresponding arylacetamides was feasible in good to moderate yields.
 |
| | Fig. 5 Cinnamide and arylacetamides. | |
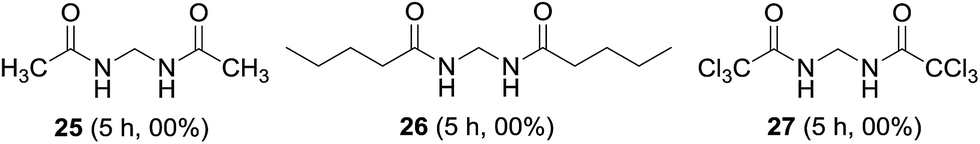
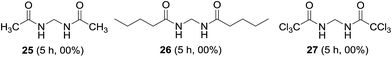
Short and long chain aliphatic amides 25 and 26 were examined as substrates; however, complex mixture of products rather than methylenebisamides was obtained (Fig. 6). We assumed that the instability of the generated radical intermediate might be the reason, hence the reaction was performed on trichloroacetamide, however the expected product 27 was not observed. The exact reason for the failure of aliphatic amides to provide bisamides under these conditions is obscure (Fig. 6). Li et al. also reported difficulties in preparing aliphatic bismethyleneamides from aliphatic amides.12
 |
| | Fig. 6 Aliphatic amides did not work. | |
During the course of our studies Sun et al. reported an interesting multicomponent Mannich reaction of a ketone, saccharine and DMSO using RuCl3 and Selectfluor® (Fig. 1).2e We performed our standard reaction on their substrates, compounds 28 and 29, and found that the desired β-amino ketone 30 was obtained in moderate yield (Scheme 3). Further optimization of our standard protocol might improve the efficiency of this multicomponent reaction. Application of Mannich reaction under similar condition and formation of bisamide with N-methyl benzamide did not work.
 |
| | Scheme 3 Application to a multicomponent Mannich reaction. | |
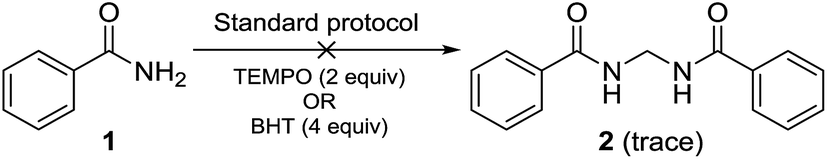
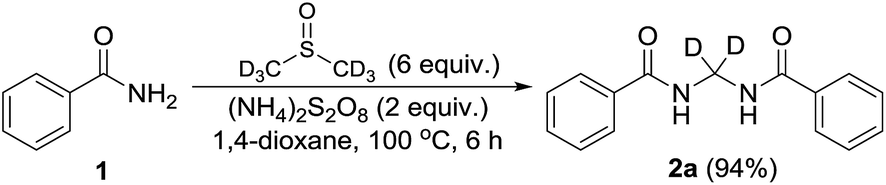
Performing the reaction with amide 1 as substrate in the presence of free radical scavengers such as TEMPO or BHT resulted in only trace amounts of bismethyleneamide 2 (Scheme 4). This result suggests that the reaction proceeds via mechanism involving the formation of free radicals. The source of methylene unit was confirmed to be DMSO by performing isotopic labelling experiment using DMSO-d6 (Scheme 5). The deuterium labelled methylenebisamide 2a was identified by 1H NMR and HRMS-ESI.
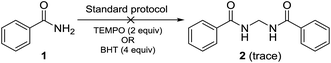 |
| | Scheme 4 Radical trapping experiment. | |
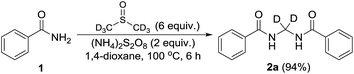 |
| | Scheme 5 Isotopic labelling experiment. | |
Two noteworthy publications appeared during the compilation of this manuscript, wherein the activation of DMSO was achieved using NH4I to synthesize β-alkoxy methyl sulphides by a multicomponent reaction2c and substituted pyridines from ketones and ammonium acetate.16 We repeated our reaction on benzamide (1) by using NH4I instead of APS, however there was no reaction at all, thus suggesting a different mode of activation in our process.
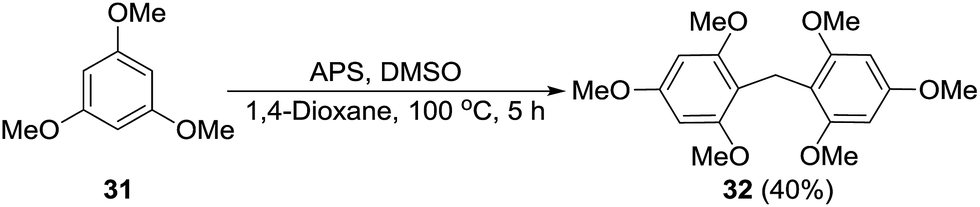
Formation of a formaldehyde equivalent intermediate during the reaction was confirmed by performing the standard reaction on 1,3,5-trimethoxybenzene (31) to obtain methylene inserted product 32. Diarylmethanes of this type are important because of their presence in supramolecular architectures, pharmaceuticals, and biologically active compounds (Scheme 6).17
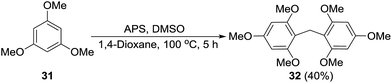 |
| | Scheme 6 Demonstration of DMSO as a formaldehyde equivalent. | |
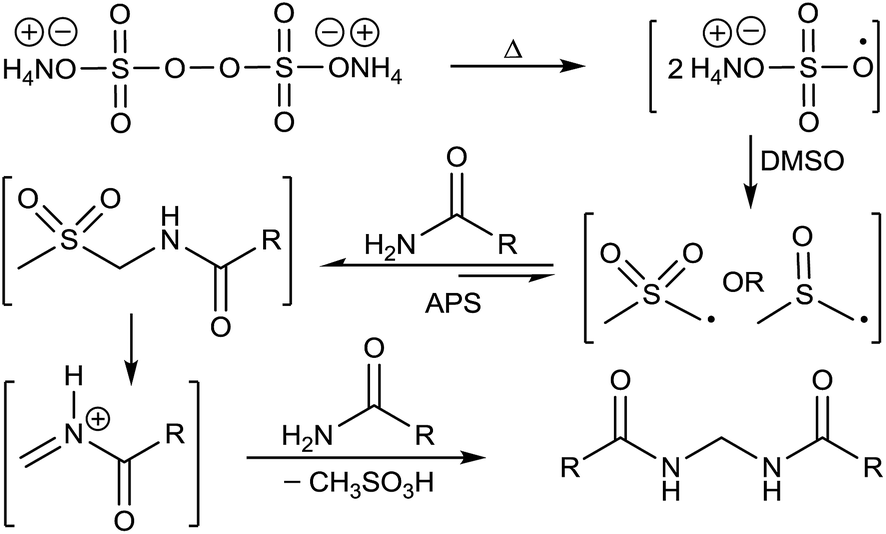
We have previously proved that APS oxidizes DMSO to form dimethyl sulfone (DMS) in 1,4-dioxane.15 Taking that into consideration we envisaged formation of the sulfonyl compound 34 as a probable intermediate formed by the reaction of amide with DMSO/DMS radical species. Reaction of benzamide (1) using DMS instead of DMSO under the standard protocol also furnished the desired bisamide 2 (Scheme 7).
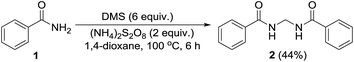 |
| | Scheme 7 Reaction using dimethyl sulfone instead of DMSO. | |
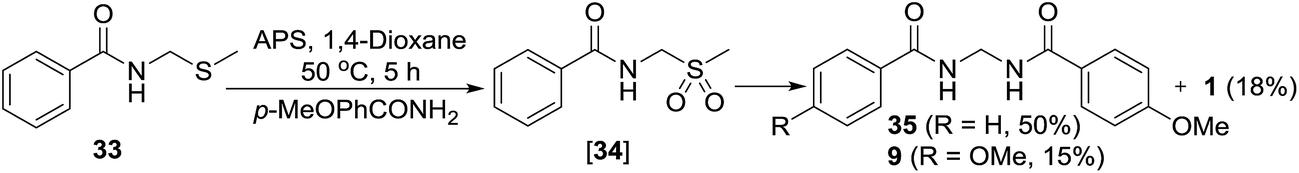
For indirect evidence, the thio compound 33 was prepared as per the literature procedure18 and reacted with p-methoxybenzamide in the presence of APS. The expected unsymmetrical methylenebisamide 35 was obtained as a major product, which confirms our both hypothesis; oxidation of thio compound 33 to form sulfonyl intermediate 34 and its further elimination and reaction with another amide to furnish unsymmetrical bisamide 35. Formation of symmetrical bisamide 9 and benzamide (1) as minor products were also observed, which probably suggests that the formation of compound 34 and its conversion to DMS radical and benzamide (1) is a reversible reaction (Scheme 8). Bisamide 2 was not observed under this condition probably because of low temperature of the reaction and less reactivity of benzamide (1) as compared to p-methoxybenzamide.
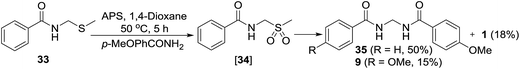 |
| | Scheme 8 Intermediate 34 is a plausible intermediate in the key reaction. | |
A plausible mechanism has been depicted in Fig. 7 based on all the above observations and literature precedence.17b,19 Formation of methane sulfonic acid is also known.19a
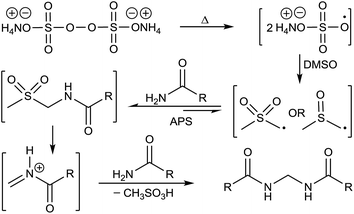 |
| | Fig. 7 Plausible mechanism. | |
In summary, we have developed an efficient protocol for the conversion of primary amides to the corresponding symmetrical methylenebisamides by using environmentally benign DMSO as the source of the methylene unit. The activation of DMSO was achieved by using APS, a commonly used oxidant. The scope of the developed protocol is wide and it avoids the use of formaldehyde, strong acids and transition-metal catalysts. Application of the protocol to a three component Mannich reaction was demonstrated. It is also applicable in the synthesis of thiadiazoles and bis(phenyl)methane. A plausible mechanism of the protocol has been proposed and supported by mechanistic studies. Currently, we are exploring the other synthetic applications of this interesting reagent combination.
Experimental section
All reagents and solvents were used as received from commercial sources unless otherwise noted. All amides were procured from commercial sources and used as it is. All experiments were carried out under air atmosphere. Pre-coated plates (silica gel 60 PF254, 0.25 mm or 0.5 mm) were utilized for thin layer chromatography (TLC). Column chromatographic purifications were carried out on flash silica-gel (240–400 mesh) using petroleum ether and ethyl acetate as eluents. The 1H, 13C NMR spectra were recorded on 200/400/500 MHz, and 50/100/125 MHz NMR spectrometers, respectively in CDCl3/DMSO-d6. Chemical shifts were reported as δ values from standard peaks. Melting points are uncorrected. Mass spectra were taken on LC-MS (ESI) mass or GC-MS mass spectrometer. HRMS were scanned on Quadrupole-Orbitrap Mass Spectrometer available at our institutional facility.
General procedure for the synthesis of methylenebisamides
A solution of amide (50 mg, 1 equiv.), ammonium persulfate [(NH4)2S2O8] (2 equiv.) and DMSO (6 equiv.) in 1,4-dioxane (2 mL) was heated at 100 °C in a round bottom flask, equipped with a stirring bar and water condenser, until the reaction was complete as indicated by thin layer chromatography. After completion, the reaction mixture was filtered through a cotton plug and 1,4-dioxane was removed under vacuum. The residue was then dissolved in ethyl acetate (10 mL) and washed with warm water (4 mL) and brine (3 mL × 2). The organic layer was dried over anhydrous sodium sulfate and the crude product was purified by flash column chromatography to furnish corresponding methylenebisamides in good to excellent yields.
Acknowledgements
P. S. M. thanks CSIR-New Delhi for the research fellowship. S. B. M. gratefully acknowledges generous financial support from DST, CSIR-ORIGIN, CSIR-OSDD New Delhi as well as CSIR-NCL (start-up grant).
Notes and references
-
(a) T. V. Nguyen and M. Hall, Tetrahedron Lett., 2014, 55, 6895 CrossRef CAS;
(b) J. R. McConnell, J. E. Hitt, E. D. Daugs and T. A. Rey, Org. Process Res. Dev., 2008, 12, 940 CrossRef CAS;
(c) L. de Luca, G. Giacomelli and A. Porcheddu, J. Org. Chem., 2001, 66, 7907 CrossRef CAS PubMed;
(d) A. J. Mancuso and D. Swern, Synthesis, 1981, 29377 Search PubMed.
-
(a) S. Mohammed, R. A. Vishwakarma and S. B. Bharate, J. Org. Chem., 2015, 80, 6915 CrossRef CAS PubMed;
(b) S. Song, X. Sun, X. Li, Y. Yuan and N. Jiao, Org. Lett., 2015, 17, 2886 CrossRef CAS PubMed;
(c) X. Gao, X. Pan, J. Gao, H. Jiang, G. Yuan and Y. Li, Org. Lett., 2015, 17, 1038 CrossRef CAS PubMed;
(d) Y.-F. Liang, K. Wu, S. Song, X. Li, X. Huang and N. Jiao, Org. Lett., 2015, 17, 876 CrossRef CAS PubMed;
(e) K. Sun, X. Wang, Y. Jiang, Y. Lv, L. Zhang, B. Xiao, D. Li, Z. Zhu and L. Liu, Chem.–Asian J., 2015, 10, 536 CrossRef CAS PubMed;
(f) X. Jiang, C. Wang, Y. Wei, D. Xue, Z. Liu and J. Xiao, Chem.–Eur. J., 2014, 20, 58 CrossRef CAS PubMed;
(g) F.-L. Liu, J.-R. Chen, Y.-Q. Zou, Q. Wei and W.-J. Xiao, Org. Lett., 2014, 16, 3768 CrossRef CAS PubMed;
(h) Y. Lv, Y. Li, T. Xiong, W. Pu, H. Zhang, K. Sun, Q. Liu and Q. Zhang, Chem. Commun., 2013, 49, 6439 RSC.
-
(a) J. Mottweiler, T. Rinesch, C. Besson, J. Buendia and C. Bolm, Green Chem., 2015, 17, 5001 RSC;
(b) W. Ghezali, K. D. O. Vigier, R. Kessas and F. Jérôme, Green Chem., 2015, 17, 4459 RSC;
(c) S. K. R. Parumala and R. K. Peddinti, Green Chem., 2015, 17, 4068 RSC;
(d) S. Song, X. Li, X. Sun, Y. Yuan and N. Jiao, Green Chem., 2015, 17, 3285 RSC;
(e) S. Song, X. Huang, Y.-F. Liang, C. Tang, X. Li and N. Jiao, Green Chem., 2015, 17, 2727 RSC;
(f) S. N. Gockel and K. L. Hull, Org. Lett., 2015, 17, 3236 CrossRef CAS PubMed;
(g) X. Wu, Y. Zhao and H. Ge, J. Am. Chem. Soc., 2015, 137, 4924 CrossRef CAS PubMed;
(h) J. Mao, Faming Zhuanli Shenqing CN 104447391 A 20150325, 2015 Search PubMed;
(i) A. Borah, L. Goswami, K. Neog and P. Gogoi, J. Org. Chem., 2015, 80, 4722 CrossRef CAS PubMed;
(j) C. Laugel, B. Estrine, J. le Bras, N. Hoffmann, S. Marinkovic and J. Muzart, ChemCatChem, 2014, 6, 1195 CAS;
(k) Y. Li, D. Xue, W. Lu, C. Wang, Z.-T. Liu and J. Xiao, Org. Lett., 2014, 16, 66 CrossRef CAS PubMed.
- C. Alemh and J. Puiggali, J. Org. Chem., 1995, 60, 910 CrossRef.
-
(a) K. H. Kim, S. U. Choi and K. R. Lee, Tetrahedron Lett., 2012, 53, 1490 CrossRef CAS;
(b) T. N. Duong, R. A. Edrada, R. Ebel, V. Wray, W. Frank, A. T. Duong, W. H. Lin and P. Proksch, J. Nat. Prod., 2007, 70, 1640 CrossRef CAS PubMed.
- M. Szostak and J. Aube, Org. Biomol. Chem., 2011, 9, 27 CAS.
-
(a) N. Mameda, M. R. Marri, S. Peraka, A. K. Macharla, S. Kodumuri, D. Chevella, G. Naresh and N. Nama, Catal. Commun., 2015, 61, 41 CrossRef CAS;
(b) B. F. Mirjalili and M. A. Mirhoseini, J. Chem. Sci., 2013, 125, 1481 CrossRef CAS;
(c) M. R. M. Shafiee, J. Saudi Chem. Soc., 2014, 18, 115 CrossRef;
(d) A. H. Fernadez, R. M. Alvarez and T. M. Abajo, Synthesis, 1996, 1299 CrossRef.
-
(a) T. Yamazaki, K.-I. Nunami and M. Goodman, Biopolymers, 1991, 31, 1513 CrossRef CAS PubMed;
(b) M. Rodriguez, P. Dubreuil, J.-P. Bali and J. Martinez, J. Med. Chem., 1987, 30, 758 CrossRef CAS PubMed;
(c) P. V. Pallai, R. S. Struthers and M. Goodman, Biochemistry, 1985, 24, 1933 CrossRef CAS PubMed.
-
(a) L. Pan, L. Huang and C. Xie, Lett. Org. Chem., 2013, 10, 770 CrossRef CAS;
(b) M. Tajbakhsh, R. Hosseinzadeh, H. Alinezhad and P. Rezaee, Synth. Commun., 2013, 43, 2370 CrossRef CAS;
(c) G. Hrichandran, S. D. Amalraj and P. Shanmugam, Indian J. Chem., 2011, 50B, 77 Search PubMed;
(d) E. E. Gilbert, Synthesis, 1972, 30 CrossRef CAS;
(e) E. E. Magat, B. F. Faris, J. E. Reith and L. F. Salisbury, J. Am. Chem. Soc., 1951, 73, 1028 CrossRef CAS;
(f) R. C. Brian and A. H. Lamberton, J. Chem. Soc., 1949, 1633 RSC.
- C. W. Sauer and R. J. Bruni, J. Am. Chem. Soc., 1955, 77, 2559 CrossRef CAS.
-
(a) N. N. Bochkareva and E. P. Trub, J. Gen. Chem., 1984, 54, 619 Search PubMed;
(b) T. E. Varkey, G. F. Whitfield and D. Swern, J. Org. Chem., 1974, 39, 3365 CrossRef CAS.
- Q. Wang, L. Sun, Y. Jiang and C. Li, Beilstein J. Org. Chem., 2008, 4, 51 Search PubMed.
- A. R. Katritzky, W.-Q. Fan, M. Black and J. Pernak, J. Org. Chem., 1992, 57, 541 Search PubMed.
-
(a) K. Yajima, K. Yamaguchi and N. Mizuno, Chem. Commun., 2014, 50, 6748 RSC;
(b) A. Yoshimura, A. D. Todora, B. J. Kastern, S. R. Koski and V. V. Zhdankin, Eur. J. Org. Chem., 2014, 5149 CrossRef CAS;
(c) L. Forlani and C. Boga, J. Chem. Soc., Perkin Trans. 2, 2002, 768 RSC;
(d) L. Forlani, A. Lugli, C. Boga, A. B. Corradi and P. Sgarabotto, J. Heterocycl. Chem., 2000, 37, 63 CrossRef CAS;
(e) C. Boga, L. Forlani, C. Silvestroni, A. B. Corradi and P. Sgarabotto, J. Chem. Soc., Perkin Trans. 1, 1999, 1363 RSC;
(f) T. Isobe, J. Org. Chem., 1999, 64, 6989 CrossRef CAS.
- D. N. Garad, S. D. Tanpure and S. B. Mhaske, Beilstein J. Org. Chem., 2015, 11, 1008 CrossRef CAS PubMed.
- X. Pan, Q. Liu, L. Chang and G. Yuan, RSC Adv., 2015, 5, 51183 RSC.
-
(a) Q. Chen, K. Gao, C. Peng, H. Xie, Z. K. Zhao and M. Bao, Green Chem., 2015, 17, 4546 RSC;
(b) Y. Xu, T. Cong, P. Liu and P. Sun, Org. Biomol. Chem., 2015, 13, 9742 RSC;
(c) D. Chen, C. Xu, J. Deng, C. Jiang, X. Wen, L. Kong, J. Zhang and H. Sun, Tetrahedron, 2014, 70, 1975 CrossRef CAS;
(d) X. Li, Y. Feng, L. Lin and G. Zou, J. Org. Chem., 2012, 77, 10991 CrossRef CAS PubMed.
- L. Bernardi, R. DeCastiglione and U. Scarponl, J. Chem. Soc., Chem. Commun., 1975, 320 RSC.
-
(a) R. Herscu-Kluska, A. Masarwa, M. Saphier, H. Cohen and D. Meyerstein, Chem.–Eur. J., 2008, 14, 5880 CrossRef CAS PubMed;
(b) X. Qiao, S. Chen, L. Tan, H. Zheng, Y. Ding and Z. Ping, Magn. Reson. Chem., 2001, 39, 207 CrossRef CAS;
(c) M. K. Eberhardt and R. Colina, J. Org. Chem., 1988, 53, 1071 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra21801b |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*