
Computational prediction and experimental validation of low-affinity target of triptolide and its analogues†
Xiufeng Liua,
Kai Wanga,
Ningjun Duana,
Yan Lana,
Pengcheng Mab,
Heng Zhengd,
Weijuan Zheng*a,
Jiahuang Li*a and
Zi-chun Hua*ac
aThe State Key Laboratory of Pharmaceutical Biotechnology, College of Life Science, Nanjing University, Nanjing 210093, P. R. China. E-mail: zchua@nju.edu.cn; lijiah@nju.edu.cn; wjzheng@nju.edu.cn; Fax: +86-25-83324605; Tel: +86-25-83324605
bInstitute of Dermatology, Peking Union Medical College, Chinese Academy of Medical Sciences, Nanjing, 210042, P. R. China
cChangzhou High-Tech Research Institute of Nanjing University, Jiangsu Target Pharma Laboratories Inc., Changzhou 213164, P. R. China
dSchool of Life Science and Technology, China Pharmaceutical University, Nanjing, 210009, P. R. China
First published on 9th April 2015
Abstract
Triptolide, triptonide and triptriolide are active ingredients of traditional Chinese herbal medicine Tripterygium wilfordii Hook.f. Although these compounds are found to have significant anti-inflammatory, immune-suppressive or anti-tumour effects, the molecular mechanisms of actions, especially their binding to proteins remain unclear. Since the chemical structures of triptolide, triptonide and triptriolide are similar to steroid hormones, we try to identify potential target proteins from the steroid hormone receptors (or “nuclear receptors”). In this study, using the reverse docking strategy, 12 nuclear receptors are reversely docked to triptolide and ranked by the binding energy scores. Based on this, human estrogen receptor alpha (ERα) was selected as a potential interaction protein for triptolide and the binding mode of three compounds to ERα-LBD (ligand binding domain) was further assessed by Docking and Molecular Dynamics (MD) simulation. To further analyze the docking results, Surface Plasmon Resonance (SPR), Isothermal Titration Calorimetry (ITC) and Reporter Gene assays were used to validate the interactions of ERα-LBD with the three compounds. SPR studies together with ITC measurements indicated that the three compounds could bind to ERα-LBD with weak affinity. Triptonide showed the highest affinity and triptriolide exhibited the weakest affinity. Furthermore, the binding of triptonide or triptolide to ERα significantly increased the reporter gene activity in human cervical cancer cell lines HeLa. This study not only further defines the binding proteins of triptolide and its analogues but also provides useful information for application of these compounds.
1. Introduction
Natural products have played a very important role in health care and development of drugs. The extracts of Tripterygium wilfordii Hook.f. (TWHF), also known as Lei Gong Teng or Thunder God Vine, have been used in traditional Chinese medicine for more than two centuries for treatment of inflammation, autoimmune diseases, arthritis and cancers.1–3 Pharmaceutical chemistry study indicated that TWHF contains over 200 kinds of active components.4 They are divided into three categories: alkaloids, diterpenes and triterpenes. Triptolide and its analogues (triptonide and triptriolide) belong to diterpenetriepoxide, one kind of the major biologically active components of the herb. As shown in Fig. 1, triptolide, triptonide and triptriolide have similar chemical structures, containing three epoxide groups next to each other. Numerous studies have shown that these compounds may elicit their bioactivities by modulating multiple cellular targets, however, possible target proteins rarely been reported. Therefore, to screen for the potential molecular targets of these compounds have become an important and challenging task. The chemical structures of the three chemicals are similar with steroid hormones which suggest that the chemicals may target some nuclear receptors (NRs), the receptor of steroid hormones. | ||
| Fig. 1 Chemical structures of triptolide, triptonide and triptriolide. The different groups of three compounds were marked in red. | ||
NRs are a family of transcription factor, and regulate transcription by binding to response elements in the regulatory regions of target genes and thereby affect expression of genes involved in differentiation, growth, lipid homeostasis, inflammation and immunity.5–7 Meanwhile NRs are good drug targets because they have open pockets which can be used to hold a signaling molecule. Drug compounds can also be made to fit in these pockets, switching the nuclear receptor on or off to alter gene expression.8 Over the past two decades, significant advances have been made in understanding the regulation of gene expression by NRs. The knowledge on NRs has delivered novel therapies for lipid control and hormone replacement, and for management of cancer and diabetes.9–13 Therefore, NRs are attractive molecular targets for design of therapy for diabetes, obesity, atherosclerosis, cancer, inflammation, neuro-degeneration and so on.
Potential targets identification of small-molecule is a daunting task. Using proteomic approaches are the major methods for target identification. However, it is laborious and time-consuming.14 To overcome this issue, a series of computational tools have been developed for target identification, such as reverse docking.15 It involves docking the small-molecule in the potential binding targets and has been proved to be an effective method for the analysis of drug toxicity16 or identification of drug targets.17
This study was designed to investigate the potential interactions between NRs and triptolide, triptonide and triptriolide. 12 NRs were first screened through docking calculations to identify the putative molecular targets of triptolide. Then the most likely target ERα-LBD was expressed and purified in vitro. The binding capacities between ERα-LBD and three compounds were determined by Surface Plasmon Resonance (SPR) and Isothermal Titration Calorimetry (ITC) analyses. The results were further validated using reporter gene assays. These data revealed ERα act as a previously unknown binding protein of triptolide and triptonide which may provide valuable information for studying the mechanisms and structure–function relationships of these chemicals in vivo.
2. Materials and methods
2.1 Chemicals, reagents, and plasmids
Triptonide from Shanghai Yuanye Biological Technology Co., Ltd and triptolide and triptriolide from Prof. Pengcheng Ma, Institute of Dermatology, Chinese Academy of Medical Sciences were used in this study. The non-detergent sulfobetaine (NDSB-201) was purchased from TCI Japan Ltd and the Dual-Luciferase Reporter Assay System Kit was purchased from Promega (Madison, WI, USA). The estrogen response element luciferase reporter plasmid ERE-Luc, ERα expression vector ERα/pCI and the control plasmid phRL-tk were kindly provided by Dr Xinru Wang, Department of Toxicology, School of Public Health, Nanjing Medical University.2.2 Molecular docking
Three-dimensional structures for triptolide, triptonide and triptritolide were generated and optimized by Discovery Studio 2.5 (Accelrys Software Inc. USA). Crystal structures of NRs were retrieved from the Brookhaven Protein Data Bank (PDB). Docking studies were performed by AutoDock 4.2 (The Scripps Research Institute, La Jolla, Cal.). Automated docking was used to locate the appropriate binding orientations and conformations of various compounds. Polar hydrogen atoms were added and Kolllman charge, atomic solvation parameters and fragmental volumes were assigned to the protein using AutoDock Tools (ADT). The program AutoGrid was used to generate the grid maps. The grid dimensions were 90 points per dimension separated by 0.180 Å. For all chemicals, random starting positions, random orientations and torsions were used. The translation, quaternion and torsion steps were taken from default values in AutoDock. 100 runs were carried out using the genetic algorithm with a population size of 300 individuals and 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500000 energy evaluations. After docking, the solutions were clustered into groups with RMS deviations lower than 1.0 Å. The clusters were ranked by the lowest energy representative of each cluster. The interactive visualization and analysis of molecular structures and hydrogen bonds between protein and chemicals were performed by Discovery Studio 2.5. To validate the docking reliability of this modelling, the native ligand (estradiol) was re-docked to the binding site of ERα-LBD, and the docked conformation corresponding to the lowest free energies was selected as the most possible binding conformation. The results of Re-docking indicate that the binding conformation of estradiol in ERα-LBD determined by AutoDock matched well with that of the X-ray (Fig. S1†). This result indicate that AutoDock 4.2 process is suitable for the docking study.
500000 energy evaluations. After docking, the solutions were clustered into groups with RMS deviations lower than 1.0 Å. The clusters were ranked by the lowest energy representative of each cluster. The interactive visualization and analysis of molecular structures and hydrogen bonds between protein and chemicals were performed by Discovery Studio 2.5. To validate the docking reliability of this modelling, the native ligand (estradiol) was re-docked to the binding site of ERα-LBD, and the docked conformation corresponding to the lowest free energies was selected as the most possible binding conformation. The results of Re-docking indicate that the binding conformation of estradiol in ERα-LBD determined by AutoDock matched well with that of the X-ray (Fig. S1†). This result indicate that AutoDock 4.2 process is suitable for the docking study.
2.3 Molecular dynamics (MD) simulations
To obtain the stable docking pose of three compounds in ERα-LBD, MD simulations of three docked complexes were performed with AMBER 12. The main procedure of MD simulation was as follows: the ff12SB force field was used for the protein system, whereas the GAFF force field was used for the ligands. Atom charges of the ligand were calculated using the RESP method encoded in the AMBER 12.0 software package at the HF/6-31G* level. Each model was solvated in a truncated octahedron box of TIP3P water molecules that extend 10 Å from the protein atoms, and neutralized by adding Na+ and Cl− counter ions. To remove possible poor contacts between protein atoms and solvent, energy minimization (5000 steps for the water molecules followed by 10000 steps for the whole system) were performed before MD simulation. Langevin dynamics with the collision frequency 2 ps−1 was used to increase the temperature of the solvated system, from 0 K to 298 K in 200 ps. At this stage, the heavy atoms of the protein and the ligand were restrained by a harmonic force of 1.0 kcal (mol−1 Å−2). The SHAKE algorithm was used to constrain bonds involving hydrogen atoms and the non-bonded cutoff was set to be 10.0 Å. Next, each system was subjected to 1 ns equilibration at 298 K under NPT ensemble conditions, with a harmonic force of 1.0 kcal (mol−1 Å−2) restrained on the ligand and the backbone atoms of the protein. Following the equilibration, all the restrains were removed and the system was subjected to MD simulation for 20 ns. The time step was set as 2 fs during all MD simulation stage and a snapshot was saved every 10 ps. The cpptraj analysis program within Amber Tools 13.18 was used for the calculations of the root mean square deviation (RMSD) and MMPBSA.py protocol was used for Molecular Mechanics/Poisson Boltzmann (or Generalized Born) Surface Area (MM/PBSA) calculations.
2.4 Expression and purification of recombinant ERα-LBD
MCF-7 human breast cancer cell lines were obtained from American Type Culture Collection (ATCC) (Manassas, VA, USA). Total RNA was extracted by Trizol reagent (Invitrogen, USA) according to the manufacturer's protocol. Reverse transcription was performed by SuperScript III Reverse Transcriptase (Invitrogen, USA). cDNAs encoding human ERα-LBD were amplified, the primer sequences as follows:Forward: 5′-GGAATTCTCTAAGAAGAACAGCCTG-3′
Reverse: 5′-CCGCTCGAGGCGGTGGGCGTCCAGCAT-3′
hERα-LBD was expressed as an N-terminally His6-tagged fusion protein in BL21 (DE3) plysS cells using pET-28a plasmid (Novagen USA) as described by pET System manual. For purification, the cell pellets from 500 ml cultures were collected by centrifugation and followed by sonication. The soluble fraction containing recombinant proteins was purified by affinity chromatography using Ni–NTA Agarose gel (Qiagen, USA) as previously described.19 Protein purity was analyzed by SDS-PAGE (5% stacking gel and 12% separating gel) and concentration by Bradford assay (BioRad, Hercules, CA, USA).
2.5 Surface plasmon resonance (SPR) assay
SPR assay was performed on a Biacore T200 system using CM5 sensors. First, standard amine coupling was used to immobilize ERα-LBD proteins to the surfaces of preconditioned sensor chips. An immobilization density of 8700 RU was achieved with a coupling buffer of 10 mM sodium acetate at pH 5.0. The proteins were dissolved in a PBS buffer (pH 7.4, 5% DMSO) during immobilization. The compounds were dissolved in a pure DMSO and then the solution was diluted for 4 concentrations (62.5/125/250/500 μM). Injection of the compound was followed by injection of running buffer as a regeneration step to remove any remaining compounds bound to the surfaces at a flow rate of 30 μl min−1. The response data were double-referenced to calculate the KD values.2.6 ITC binding experiments
Micro-ITC200 system with Origin 7.0 was used for graphing functions (GE Healthcare). ERα-LBD proteins were diluted to a concentration of 20 μM in 20 mM sodium phosphate buffer (pH 7.4) and contain 0.5% DMSO. Triptolide, triptonide or triptriolide were prepared in the same buffer at concentration of 300 μM. The experiments were performed at 25 °C. All the solutions were thoroughly degassed by ultrasonic bath. For each experiment, the compounds were placed in the syringe with the stirring rate at 1000 rpm. Protein samples were placed in the sample cell. Control titration of compounds into the same buffer without protein was also performed. The volumes of the sample cell were 200 μl and 40 μl in syringe.2.7 Transfection and ERE-luciferase reporter gene assays
HeLa cell lines without the endogenous ERα were seeded at a density of 5.0 × 103 cells per well in 24-well plates and grown overnight until reaching 70–80% confluence. The cells were co-transfected with 0.5 μg of pERE-Luc, 0.2 μg of ERα-pCI, and 0.1 μg of phRL-tk control plasmid per well. Lipofectamine 2000 (Invitrogen) was used according to the instructions provided by the manufacturer. After 12 h, the transfection medium was replaced with fresh medium containing the tested chemicals at a concentration of 10−9 M and kept for another 18 h, then the cells were harvested for Firefly and Renilla luciferase analysis following the manufacturer's instructions of Dual-Luciferase Reporter Assay System Kit.2.8 Statistical analysis
The data were presented as mean ± SEM. Statistical comparisons were made by Student's t test using GraphPad Prism 5.0 software.3. Results
NRs-LBD are clear historical examples of natural drug binding domains, which respond to small chemicals including intracellular metabolites. Since the scaffold of triptolide is similar to some native ligands (e.g. estradiol, testosterone and cortisol) of NRs, triptolide and its derivatives might target the ligand binding site of NRs and thus mimic their functions. Hence, docking triptolide into the NRs-LBD using molecular docking calculations was attempted.3.1 Docking results for triptolide in NR-LBDs
Twelve potential NRs are listed in Table 1. The free energy of binding to triptolide was used to evaluate the binding affinity. Among them, ERα-LBD has the lowest free energy (−10.7 kcal mol−1) suggesting that ERα is the most possible target for triptolide. Furthermore, in order to gain insight into the energetics of the binding of triptolide and its two analogues to ERα-LBD, three compounds were docked into ERα-LBD. Table 2 shows the free energies of binding (ΔG) and the calculated inhibition constants (Ki) of docked compounds. It indicates that the triptonide had the best binding free energy (−11.2 kcal mol−1) and inhibition constant (6.0 nM), followed by triptolide (ΔG = −10.7 kcal mol−1 and Ki = 14.6 nM), while triptriolide showed the weakest affinity with ERα-LBD (ΔG = −8.8 kcal mol−1 and Ki = 381.5 nM).| PDB_ID | Symbol | Full name | FEB (kcal mol−1) | Poses |
|---|---|---|---|---|
| a Abbreviations: FEB, free energy of binding; “Poses”, refers to number of poses in highest scoring cluster out of total 100 poses. | ||||
| 2YJA | ER-α | Estrogen receptor-α | −10.7 | 99 |
| 2J7Y | ER-b | Estrogen receptor-β | −8.5 | 63 |
| 1KV6 | ERR-3 | Strogen-related receptor-3 | −6.9 | 100 |
| 1E3G | AR | Androgen receptor | −5.5 | 81 |
| 1R1K | HER | Ecdysone hormone receptor | −6.4 | 98 |
| 1PQC | OR | Oxysterols receptor | −5.8 | 56 |
| 1ZH7 | NR5A2 | Orphan nuclear receptor | −4.5 | 77 |
| 2ZY0 | RAR | Retinoic acid receptor | −4.0 | 63 |
| 1N46 | TR | Thyroid hormone receptor | −4.3 | 52 |
| 1HG4 | UP | Ultraspiracle protein | −4.9 | 75 |
| 1P93 | GR | Glucocorticoid receptor | −4.9 | 63 |
| 1ZDT | SF-1 | Steroidogenic factor 1 | −5.8 | 26 |
| Compounds | Inhibition constant Ki (10−9 M) | Free energy of binding ΔG (kcal mol−1) |
|---|---|---|
| Triptolide | 14.6 | −10.7 |
| Triptonide | 6.0 | −11.2 |
| Triptriolide | 381.5 | −8.8 |
3.2 Binding mode between ERα-LBD and three compounds
Considering the binding mode based on the docking study is not sufficient criterions. Therefore, an additional MD simulation was conducted. The starting structures of protein–ligand were obtained from molecular docking, and 20 ns MD simulations were conducted. RMSDs of backbone atoms were calculated against the starting structures (Fig. S2 and S3†). The stabilized RMSD values of the final 10 ns ensured the reliability and suitability of these MD trajectories for further analysis. Based on backbone-atom RMSDs, the last 5 ns of each MD trajectory was clustered and the central structure (the member with the smallest average RMSD values compared to all other members of the major cluster) of the major cluster was extracted to serve as the representative structure of each MD simulation. Based on backbone-atom RMSDs, the last 5 ns of each MD trajectory was extracted to calculate the binding energy. As shown in Table 4, the binding free energies of triptolide or triptonide to ERα-LBD obtained by MD simulation were similar to molecular docking study. The binding free energy value of triptriolide to ERα-LBD by MD simulation (−4.7 kcal mol−1) is almost twice molecular docking results (−8.8 kcal mol−1). One possible reason for such differences is that 12, 13 epoxide ring of triptriolide is opened and changed to hydroxyl groups. Therefore, triptriolide becomes more hydrophilic compared with triptolide and triptonide. During the MD simulation process water molecules were added to the simulation system for better to simulate the “wet”-lab experiments. The existence of water leads to the reduced binding force of triptriolide-receptor. So, the free energy of binding obtained by MD simulation (wet scenario) is much higher compared with docking method (dry scenario). Meanwhile, there are more rotatable bonds in triptriolide compared with triptolide and triptonide when epoxide ring is opened. Triptriolide with rotatable bonds could lead to the conformation change of protein receptor during the MD simulation. However, the protein conformation used in AutoDock is fixed and cannot be affected by the triptriolide. This could be another reason for the difference in free energy of binding for triptriolide, obtained by two methods.Fig. 2A shows the superstition of docking models of estrogen, triptolide, triptonide and triptriolide complexed with ERα-LBD. ERα-LBD is comprised of twelve alpha helices and a beta sheet, forming a ligand-binding pocket. Three compounds were docked deeply into the binding pocket region of ERα-LBD. Fig. 2B–D shows the MD simulated binding model of three compounds along with the ERα-LBD. Triptolide and triptonide adopted the similar conformation and orientation with minor differences in ligand binding site. Met343, Leu346, Ala350, Leu387, Met388, Leu391, Phe404, Met421, Ile424, and Leu525 fixed the heterocycle of triptolide or triptonide by hydrophobic and van der Waals interactions. Triptriolide has a different structure from the triptolide in the hydrolysis of the 12, 13 epoxide. The diterpene planum of triptriolide was twisted and lactone located tricyclic plane rotated about 90° corresponding triptolide as binding in ERα-LBD. This structure torsion may be unfavorable for triptriolide binding.
 | ||
| Fig. 2 Micro environment of the bind site of ERα-LBD and triptolide/triptonide/triptriolide. (A) Superposition of docking models of original ligand estrogen (black in stick), triptolide (blue in stick), triptonide (orange in stick) and triptriolide (violet in stick) complex with ERα-LBD. Active site of ERα-LBD complexed with triptolide (B), triptonide (C) and triptriolide (D), obtained by MD simulation binding model. Triptolide (blue) and triptonide (orange) displayed the similar conformation in binding site. However, diterpene planum of triptriolide twisted and lactone located tricyclic plane rotated about 90° in ERα-LBD binding site. | ||
3.3 Protein expression and purification
Human ERα-LBD (hERα-LBD) proteins were expressed and purified to confirm the above predicted binding mode in vitro. The expression plasmid pET-28a, containing the cDNA encoding ERα-LBD was transfected into BL21 (DE3) competent cells. The proteins were purified by Ni2+ affinity chromatography column. The purified proteins were then analyzed by SDS-PAGE and stained with Coomassie Blue. As shown in Fig. 3, hERα-LBD migrated at 28 kDa as expected and no degradation was observed. | ||
| Fig. 3 Recombinant ERα-LBD protein purification and identification. (A) Expressed and purified hERα-LBD proteins were separated by SDS-PAGE, MW: molecular weight markers. (B) Western blot analysis of hERα-LBD proteins using anti-His antibody. | ||
3.4 Surface plasmon resonance analysis
The interactions of hERα-LBD with the triptolide, triptonide and triptriolide were detected by the Biacore SPR system, and hERα-LBD proteins were immobilized on the SPR sensor chip by aminecoupling method. For the ideal coupling, effective factors such as flow rate, contact time, pH of running buffer and its composition were optimized. We found that pH of protein dilution buffer was the most important factor for coupling. The immobilized amount of hERα-LBD protein under different pH conditions of sodium acetate buffer was determined from the average SPR response. Maximum immobilization of proteins (8700 U; data not show) occurred at pH 5.0 of 10 mM sodium acetate buffer. The flow rate of 30 μl min−1 with contact time 300 s was used to detect the interaction with small ligands. The interactions of hERα-LBD proteins with the triptolide, triptonide or triptriolide were detected using the optimized SPR biosensor system. The SPR analytical conditions, molecular characteristics for the three compounds, and results of the hERα-LBD interaction with the three compounds were summarized in Table 3. The results from the SPR signals responding to the different compounds concentrations clearly indicated the existence of the interactions between ERα-LBD and the three compounds (Fig. 4). Although the binding affinities were different among compounds, a similar trend was found with the computational docking study (Table 4).| Chemical | Molecular characteristics | Analytical conditions | Affinity values KD (10−6 M) | |||
|---|---|---|---|---|---|---|
| MW (g mol−1) | Molecular formula | Concentration (μM) | Flow rate (μl min−1) | Contact time (s) | ||
| Triptolide | 360.4 | C20H24O6 | 0, 62.5, 125, 250, 500 | 30 | 90 | 607.3 |
| Triptonide | 358.4 | C20H22O6 | 0, 62.5, 125, 250, 500 | 30 | 180 | 98.8 |
| Triptriolide | 396.0 | C20H26O7 | 0, 62.5, 125, 250, 500 | 30 | 90 | 1530 |
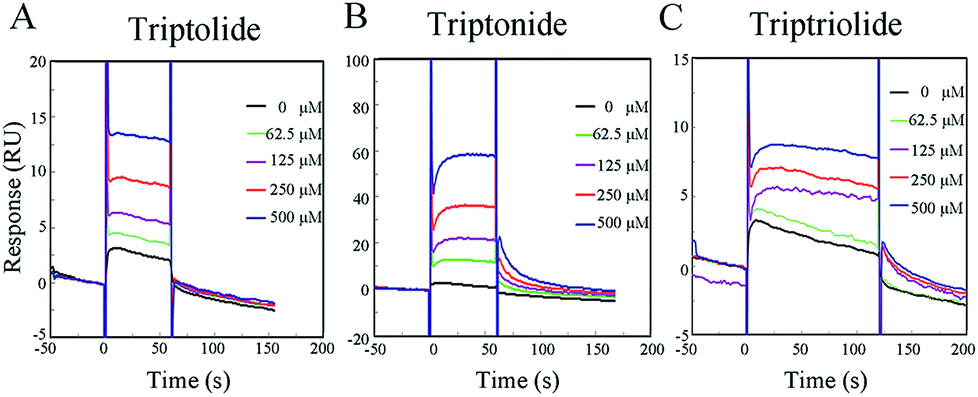 | ||
| Fig. 4 SPR sensorgrams illustrate triptolide (A), triptonide (B) and triptriolide (C) binding to hERα-LBD. | ||
| Chemical | ITC | SPR | MD | AutoDock | ||||
|---|---|---|---|---|---|---|---|---|
| KD (10−6 M) | Rank | KD (10−6 M) | Rank | FEB (kcal mol−1) | Rank | FEB (kcal mol−1) | Rank | |
| a Abbreviations: FEB, free energy of binding. | ||||||||
| Triptolide | 233 | 2 | 607.3 | 2 | −9.7 | 2 | −10.7 | 2 |
| Triptonide | 34.4 | 1 | 98.8 | 1 | −12.1 | 1 | −11.2 | 1 |
| Triptriolide | 604 | 3 | 1530 | 3 | −4.7 | 3 | −8.8 | 3 |
The binding affinity for triptonide to hERα-LBD (98.8 × 10−6 M) was much higher than that for triptolide and triptriolide. In addition, as shown in SPR sensorgrams (Fig. 4), a very quick association and dissociation of ERα-LBD/triptolide and hERα-LBD/triptriolide were observed, indicating that the formed complexes were not as stable as hERα-LBD/triptonide.
3.5 Isothermal titration calorimetry analysis
Because SPR measures the mass of material binding to the sensor surface, small analytes give very small responses. Although the recent improvements in signal to noise ratio have made it possible to measure binding of such small analytes under optimal conditions, it is better to further confirm the results by other methods. ITC analysis was used to measure the binding affinity of the three compounds to ERα-LBD. Fig. 5 shows the ITC curve of the binding of chemicals to protein and the plot of the heat evolved per mole chemicals added against the molar ratio of chemicals to the protein. The binding affinities of triptolide, triptonide and triptriolide to hERα-LBD obtained by microCAL origin 7.0 software were 233 × 10−6 M, 34.4 × 10−6 M, 604 × 10−6 M, respectively. The rankings of binding affinities were consistent with the results from SPR study, following the trend triptonide > triptolide > triptriolide. However, the values of KD obtained in the SPR study were about three times lower than ITC results. This difference could be due to the protein immobilization effects on the conformational and rotational, and therefore, on the chemicals binding in SPR experiment.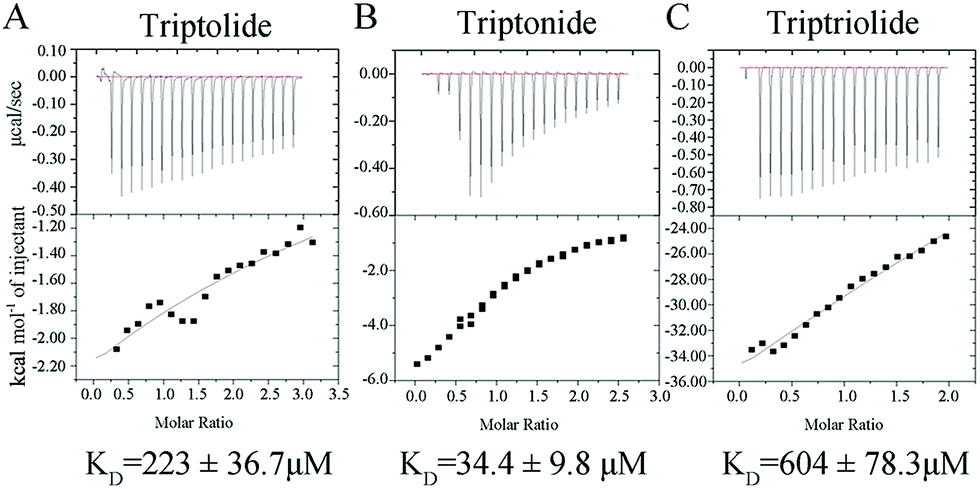 | ||
| Fig. 5 Isothermal titration calorimetry profiles obtained for triptolide (A), triptonide (B) and triptriolide (C) with hERα-LBD. | ||
3.6 Activation of ERE-Luc transcription by triptolide and its analogues
The above findings using the two in vitro methods confirmed the computer docking results: ERα acted as a potential target of triptolide and its analogues. Furthermore, ERE-luciferase reporter gene assays in ER negative cell lines HeLa were conducted to evaluate biological effects of the binding on cells. The cells were co-transfected with pERE-Luc, phRL-tk and ER expression vector, ERα/pCI for 12 h. Then, the cells were treated with triptolide, triptonide and triptriolide at a concentration of 10−9 M for 18 h. As shown in Fig. 6, the results showed that triptonide treatment induced 2-fold of luciferase gene expression compared to the control and triptolide induced gene expression by 1.5-fold, which was weaker than that of triptonide. Triptriolide did not induce higher ERE-Luc activity compared to the vehicle control in our test concentrations. Collectively, these results indicated again that triptolide and triptonide could target ERα. These results were consistent with the findings of in vitro tests, i.e. triptonide and ERα-LBD had the strongest affinity than the other two compounds. | ||
| Fig. 6 Effects of triptonide, triptolide and triptriolide on ERE-Luc activities. ERE-Luc reporter gene assay. Cells were transiently transfected with the ERE-Luc reporter, phRL-tk and ERα expression plasmids ERα/pCI and then treated with, triptonide, triptolide or triptriolide at the concentration of 10−9 M. Each bar represents a mean ± SEM (n = 5). Significant differences from the control values are: *P < 0.05, **P < 0.01. | ||
4. Discussion
The concept of one gene, one drug, and one disease has been significantly challenged. Over the past decade, the binding of one drug to multiple target proteins, with clinical effects being mediated through the modulation of the set of protein target, is gaining favor.20–22 The research work on triptolide and its analogues is attracting more and more attentions because of their outstanding clinical properties. However, the mechanism of its activities remains elusive. In order to extend the applications of these drugs, it is necessary to find the potential targets as many as possible for clinical safety or toxicity. One of the most useful approaches to determine target protein of a drug is to use isotope labeling technique. However, isotopic labeling has some disadvantages which have been discussed in detail in several reviews.23 Docking methods have become increasingly powerful in recent years, as witnessed by their performances in blinding predictions, which can provide valuable insight into drug applications.24 However, their performances remain flexible as there are multiple criteria and approaches.25 Here, blinding docking method was used to predict the possible targets for triptolide and its analogues. Several possible NR–triptolide complexes were selected and ranked due to their free energies. In order to validate the predicted results, the interactions of ERα-LBD with triptolide, triptonide and triptriolide were studied in vitro. Two kinds of label free methods SPR and ITC analysis were used. The results in vitro were close to the docking studies. In combination with in vitro experimental techniques such as SPR and ITC, docking calculation can be a useful tool not only for drug design but also to provide potential mechanisms of natural product activity.In the present work, two methods SPR and ITC were used to quantitative assessment of binding strengths of hERα-LBD proteins with three small molecule ligands. A major advantage of SPR and ITC is that it does not require the labeling or modification of the samples and keeping their properties unchanged. Previous studies show good correlations between the data obtained by SPR and ITC.26,27 However, both of SPR and ITC methods have limitations. During the SPR experiment, hERα-LBD proteins were immobilized onto the surface of chip and then a solution containing the tested compounds were flown over the surface of the sensor. There are two disadvantages with SPR measurements: (1) the protein was immobilized on the surface of sensor that may perturb the conformation and biological activity of the protein.28 (2) The tested compounds flow faster over the surface of sensor, which would be bad for compound diffusion in the activated chip surface.29 For these reasons, the affinity values KD determined using SPR are slightly lower than the values obtained by ITC (Fig. 5 and Table 3). Compared to SPR, ITC experiment can be conducted in a solution phase. But the disadvantage of ITC method is that it needs a higher concentration of compounds (10–15 fold of protein concentration) to injected into protein samples, and sometimes problem of solubility is a major challenge to meet the experimental conditions.30 Triptolide, triptonide and triptriolide are poorly soluble in water environment, so 0.5% (vol/vol) DMSO was added to overcome solubility problems in our study. Also the same amount of DMSO was added to the protein samples to prevent from buffer mismatch induced nonspecific heat effects. Commonly, the concentration should be kept as low as possible and the final concentration should be no more than 5% if DMSO is required.31
In this study, the prediction and determination of ERα-binding affinities with three structural analogues under the same condition is also beneficial to further understand the relationship between structure and affinity, thus providing theoretical helps for drug designs. For example, triptolide, triptonide and triptriolide have the same ring structure. However, triptonide has a higher binding affinity. So we inferred that 14-hydroxyl groups on the ring are not involved in ERα binding, although the effect of hydroxyl groups in the other sites need to be proved. The binding abilities to ERα of many chemicals with various affinities (nM–μM range) have been reported.32 In our test system, the ERα-binding affinities to the three chemicals are belonging to weak-affinity ligands. Ligand binding to ER initiates a series of events, including conformational change, dimerization, binding to a specific estrogen response element in the promoter of estrogen-regulated genes, activation of transcription and recruitment of co-activator complexes.33 So far, dozens of genes have been reported to be regulated by triptolide. Using the SABiosciences' proprietary database (http://www.sabiosciences.com/chipqpcrsearch.php?app=TFBS), the binding sites for ERα were identified. As shown in Table 5, the promoters of 14 triptolide regulated genes contain ERα binding sites, many of them play an important role in cell cycle control and apoptosis, e.g., P53, P21, BCL-2, BAX and TNF-α. These results suggested that triptolide could regulate the transcription of these genes via binding to ERα.
| Gene symbol | Gene name | Numbers EBSs | Fold change | Ref. |
|---|---|---|---|---|
| Genes associated with cell cycle pathways | ||||
| P53 | Tumor protein p53 | 2 | 1.8–3.9 | 34 |
| P21 | Cyclin-dependent kinase inhibitor 1 | 1 | 2.3–7.1 | 35 |
|
||||
| Genes associated with apoptosis pathways | ||||
| Bcl-2 | B-cell CLL/lymphoma 2 | 1 | 1.6–2.4 | 35 |
| Bax | BCL2-associated X protein | 1 | 1.6–2.6 | 36 |
| HSP70 | Heat shock 70 kDa protein | 2 | 2.9–3.8 | 37 |
| TNF-α | Tumor necrosis factor alpha | 2 | 7.8–12.1 | 38 |
|
||||
| Genes associated with both cell cycle and apoptosis pathway | ||||
| NGF | Nerve growth factor | 2 | 1.7–3.9 | 39 |
| MMP19 | Matrix metalloproteinase 19 | 1 | 1.1–3.2 | 40 |
| MDM2 | E3 ubiquitin protein ligase | 2 | 1.3–1.4 | 41 |
| VEGF | Vascular endothelial growth factor | 4 | 1.6–3.0 | 42 |
| MMP9 | Matrix metallopeptidase 9 | 1 | 2 | 43 |
|
||||
| Others | ||||
| C3 | Component 3 | 2 | 1.4–2.3 | 44 |
| SOCS-3 | Suppressor of cytokine signaling-3 | 2 | 2.5 | 45 |
| ALOX5 | 5-Lipoxygenase | 1 | 1–1.3 | 46 |
5. Conclusion
In summary, this study using reverse docking approach discovered ERα as a new binding protein for triptolide and its two structure analogues. Two label-free methods, SPR and ITC, have been used to quantitative determination of the binding strengths of ERα-LBD and three compounds. The results show that there is a weak binding between ERα-LBD and tested compounds. Meanwhile, the ranking of binding affinities is the same as the reverse docking result. These results could make contribution to understand the multiple regulatory actions against various diseases by triptolide and its analogues.Acknowledgements
We sincerely thank Dr Qi Zhao (University of Dundee, UK) and Dr Yao Wang (University of New South Wales, Sydney) for discussions and preparation the manuscript. This study was supported in part by grants from the Chinese National Natural Sciences Foundation (81121062 and 81072712), the Ministry of Science and Technology (2012AA020304 and 2012ZX09401012), the Jiangsu Provincial Nature Science Foundation (BK2011573, BE2013630 and BZ2012050), the Bureau of Science and Technology of Changzhou, Jiangsu, China (CZ20120004, CZ20130011, CE20135013 and CM20122003).Notes and references
- J. Ma, M. Dey, H. Yang, A. Poulev, R. Pouleva, R. Dorn, P. E. Lipsky, E. J. Kennelly and I. Raskin, Phytochemistry, 2007, 68, 1172–1178 CrossRef CAS PubMed.
- Q. Liu, Int. Immunopharmacol., 2011, 11, 377–383 CrossRef CAS PubMed.
- B. J. Chen, Leuk. Lymphoma, 2001, 42, 253–265 CAS.
- R. Xu, J. M. Fidler and J. H. Musser, Stud. Nat. Prod. Chem., 2005, 32, 773–801 CAS.
- D. M. Lonard, R. B. Lanz and B. W. O'Malley, Endocr. Rev., 2007, 28, 575–587 CrossRef CAS PubMed.
- X. Hu and M. A. Lazar, Trends Endocrinol. Metab., 2000, 11, 6–10 CrossRef CAS.
- E. Y. Hsia, M. L. Goodson, J. X. Zou, M. L. Privalsky and H. W. Chen, Adv. Drug Delivery Rev., 2010, 62, 1227–1237 CrossRef CAS PubMed.
- T. Chen, Curr. Opin. Chem. Biol., 2008, 12, 418–426 CrossRef CAS PubMed.
- J. S. Lee, K. I. Kim and S. H. Baek, Cancer Lett., 2008, 267, 189–196 CrossRef CAS PubMed.
- I. G. Schulman, Adv. Drug Delivery Rev., 2010, 62, 1307–1315 CrossRef CAS PubMed.
- M. M. Aagaard, R. Siersbæk and S. Mandrup, Biochim. Biophys. Acta, 2011, 1812, 824–835 CrossRef CAS PubMed.
- R. A. Nemenoff and R. A. Winn, Eur. J. Cancer, 2005, 41, 2561–2568 CrossRef CAS PubMed.
- J. M. Renoir, V. Marsaud and G. Lazennec, Biochem. Pharmacol., 2013, 77, 1249–1261 Search PubMed.
- P. J. Hajduk, J. R. Huth and C. Tse, Drug Discovery Today, 2005, 10, 1675–1682 CrossRef CAS.
- Q. T. Do, I. Renimel, P. Andre, C. Lugnier, C. D. Muller and P. Bernard, Curr. Drug Discovery Technol., 2005, 2, 161–167 CrossRef CAS.
- Q. Liu, T. Chen, H. Chen, M. Zhang, N. Li, Z. Lu, P. Ma and X. Cao, Biochem. Biophys. Res. Commun., 2004, 319, 980–986 CrossRef CAS PubMed.
- B. Shu, W. Duan, J. Yao, J. Huang, Z. Jiang and L. Zhang, Toxicol. In Vitro, 2009, 23, 598–602 CrossRef CAS PubMed.
- J. Chen, H. Chen, T. Zhu, D. Zhou, F. Zhang, X. Lao and H. Zheng, Phys. Chem. Chem. Phys., 2014, 16, 6709–6716 RSC.
- D. Ma, Y. Chen, L. Fang, G. Jin, B. Zhou, L. Cao, J. Ye and Z. Hua, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2007, 857, 231–239 CrossRef CAS PubMed.
- J. P. Overington, B. Al-Lazikani and A. L. Hopkins, Nat. Rev. Drug Discovery, 2006, 5, 993–996 CrossRef CAS PubMed.
- F. Yang, B. R. Zhou, P. Zhang, Y. F. Zhao, J. Chen and Y. Liang, Chem.-Biol. Interact., 2007, 170, 231–243 CrossRef CAS PubMed.
- L. Skarydova, J. Hofman, J. Chlebek, J. Havrankova, K. Kosanova, A. Skarka, A. Hostalkova, T. Plucha, L. Cahlikova and V. Wsol, J. Steroid Biochem. Mol. Biol., 2014, 143C, 250–258 CrossRef PubMed.
- W. J. Qian, J. M. Jacobs, T. Liu, D. G. Camp and R. D. Smith, Mol. Cell. Proteomics, 2006, 5, 1727–1744 CAS.
- H. A. Jung, M. N. Islam, C. M. Lee, S. H. Oh, S. Lee, J. H. Jung and J. S. Choi, Chem.-Biol. Interact., 2013, 206, 55–62 CrossRef CAS PubMed.
- M. Totrov and R. Abagyan, Curr. Opin. Struct. Biol., 2008, 18, 178–184 CrossRef CAS PubMed.
- I. Navratilova, G. A. Papalia, R. L. Rich, D. Bedinger, S. Brophy, B. Condon, T. Deng, A. W. Emerick, H. W. Guan, T. Hayden, T. Heutmekers, B. Hoorelbeke, M. C. McCroskey, M. M. Murphy, T. Nakagawa, F. Parmeggiani, X. Qin, S. Rebe, N. Tomasevic, T. Tsang, M. B. Waddell, F. F. Zhang, S. Leavitt and D. G. Myszka, Anal. Biochem., 2007, 364, 67–77 CrossRef CAS PubMed.
- J. Newman, O. Dolezal, V. Fazio, T. Caradoc-Davies and T. S. Peat, J. Comput.-Aided Mol. Des., 2012, 26, 497–503 CrossRef CAS PubMed.
- J. A. Maynard, N. C. Lindquist, J. N. Sutherland, A. Lesuffleur, A. E. Warrington, M. Rodriguez and S. H. Oh, Biotechnol. J., 2009, 4, 1542–1558 CrossRef CAS PubMed.
- D. G. Myszka, Curr. Opin. Biotechnol., 1997, 8, 50–57 CrossRef CAS.
- K. Bouchemal and S. Mazzaferro, Drug Discovery Today, 2012, 17, 623–629 CrossRef CAS PubMed.
- R. Zhang, T. Mayhood, P. Lipari, Y. Wang, J. Durkin, R. Syto, J. Gesell, C. McNemar and W. Windsor, Anal. Biochem., 2004, 331, 138–146 CrossRef CAS.
- R. M. Blair, H. Fang, W. S. Branham, B. S. Hass, S. L. Dial, C. L. Moland, W. Tong, L. Shi, R. Perkins and D. M. Sheehan, Toxicol. Sci., 2000, 54, 138–153 CrossRef CAS PubMed.
- M. Brown and P. A. Sharp, J. Biol. Chem., 1990, 265, 11238–11243 CAS.
- H. Xiaowen and S. Yi, Mol. Biol. Rep., 2012, 39, 8763–8770 CrossRef PubMed.
- J. Liu, M. Shen, Z. Yue, Z. Yang, M. Wang, C. Li, C. Xin, Y. Wang, Q. Mei and Z. Wang, Phytomedicine, 2012, 19, 756–762 CrossRef CAS PubMed.
- X. F. Wang, Y. B. Zhao, Q. Wu, Z. H. Sun and H. J. Li, Mol. Med. Rep., 2014, 9, 39–44 CAS.
- P. Kizelsztein, S. Komarnytsky and I. J. Raskin, Neuroimmunology, 2009, 217, 28–37 CrossRef CAS PubMed.
- S. M. Johnson, X. Wang and B. M. Evers, J. Surg. Res., 2011, 168, 197–205 CrossRef CAS PubMed.
- B. Xue, J. Jiao, L. Zhang, K. R. Li, Y. T. Gong, J. X. Xie and X. M. Wang, Neurochem. Res., 2007, 32, 1113–1119 CrossRef CAS PubMed.
- H. Zhao, Z. Yang, X. Wang, X. Zhang, M. Wang, Y. Wang, Q. Mei and Z. Wang, Exp. Mol. Med., 2012, 44, 633–641 CrossRef CAS PubMed.
- M. Huang, H. Zhang, T. Liu, D. Tian, L. Gu and M. Zhou, Mol. Cancer Ther., 2013, 12, 184–194 CrossRef CAS PubMed.
- W. Wang, W. Lin, B. Hong, X. Li, M. Zhang, L. Zhang and G. Lv, J. Int. Med. Res., 2012, 40, 2284–2294 CrossRef CAS PubMed.
- S. Yang, C. Gu, G. Zhang, J. Kang, H. Wen, Q. Lu and J. Huang, Asian Pac. J. Trop. Med., 2011, 4, 482–485 CrossRef CAS.
- Y. Hong, W. Zhou, K. Li and S. H. Sacks, Kidney Int., 2002, 62, 1291–1300 CrossRef CAS PubMed.
- Y. Li, Y. Tian, W. Zhu, J. Gong, W. Zhang, C. Yu, L. Gu, N. Li and J. Li, Int. Immunopharmacol., 2013, 16, 268–274 CrossRef CAS PubMed.
- G. X. Zhou, X. L. Ding, J. F. Huang, H. Zhang and S. B. Wu, Biochim. Biophys. Acta, 2007, 1770, 1021–1027 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra17009a |
| This journal is © The Royal Society of Chemistry 2015 |