
Carbon coated manganese monoxide octahedron negative-electrode for lithium-ion batteries with enhanced performance†
Abstract
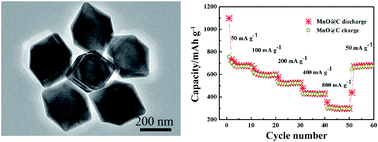
Trimanganese tetraoxide (Mn3O4) octahedra have been successfully synthesized by a one-pot hydrothermal process with the assistance of polyvinyl pyrrolidone (PVP). PVP is observed to be beneficial to control the morphology and agglomeration of octahedra. The growth mechanism has been proposed on the basis of control experiments. The manganese monoxide octahedra with a carbon coating (MnO@C) are generated after carbonizing the pyrrole carbon precursor. The as-prepared MnO@C exhibits a reversible capacity of 503 mA h g−1 after 100 cycles at a current density of 100 mA g−1 and a capacity of 526, 432 and 304 mA h g−1 at a current density of 200, 400 and 800 mA g−1, respectively. The MnO@C also shows much better cycling and rate performance compared with Mn3O4 due to the presence of the carbon coating, which efficiently buffers the volume change during the lithiation/delithiation and improves the electrical conductivity among manganese monoxide (MnO) octahedra.
 Please wait while we load your content...
Please wait while we load your content...

