
Enantioselective Pd-catalyzed tandem allylic alkylation reaction using monodentate phosphoramidite ligands for the formal total synthesis of huperzine A†‡
Chi-Feng
Lin
,
Chih-Wei
Chien
and
Iwao
Ojima
*
Department of Chemistry, Stony Brook University, Stony Brook, New York 11794-3400, USA. E-mail: iwao.ojima@stonybrook.edu; Fax: +1-631-632-7942
First published on 20th August 2014
Abstract
A small library of fine-tunable monodentate phosphoramidite (MPN) ligands was found useful in selecting a highly efficient chiral ligand for the enantioselective Pd-catalyzed tandem allylic alkylation reaction. The reaction gave a critical tricyclic key intermediate with high enantiopurity for the formal total synthesis of (−)-huperzine A, a Lycopodium alkaloid derived from the club moss Huperzia serrata that possesses potent inhibitory activity against acetylcholine esterase (ACHe).
Introduction
Metal-catalyzed enantioselective reactions serve as one of the most powerful reactions in the modern organic synthesis arsenal, particularly for the synthesis of biologically active compounds.1–4 Among those reactions, palladium-catalyzed asymmetric allylic substitution reactions provide unique and powerful methods for the regio- and stereo-controlled formation of carbon–carbon bonds as well as carbon–heteroatom bonds.5–7In order to promote this powerful catalytic process, extensive investigations have been made for the development of efficient chiral ligands.8 “Modular” diphosphine ligands developed by Trost et al.8,9 and a series of P–N ligands developed by Pfaltz, Helmchen, and Williams10,11 have been among the most successful ligands and widely used. However, these chiral ligands do not always achieve high enantioselectivity and catalyst efficiency in different reaction systems.5,12 Accordingly, continued efforts are necessary for the development of new and efficacious chiral ligands in this process.
We have designed and synthesized a series of new chiral biphenols bearing various substituents at the 3,3′-positions,13 and used them to create libraries of novel monodentate phosphite (MPO) and phosphoramidite (MPN) ligands with fine-tuning capabilities.13–18 These novel monodentate phosphorus ligands have demonstrated excellent efficiency in various transition metal-catalyzed asymmetric transformations.13–18 We have also developed novel diphosphonite ligands (BOPs) with wide bite angles. BOP ligands exhibited excellent efficiency in the intermolecular asymmetric allylic amination reactions.19–21
In 1989, Kozikowski applied Pd-catalyzed allylic alkylation as the key step in the total synthesis of racemic huperzine A (Fig. 1).22 This sesquiterpene alkaloid, derived from the club moss Huperzia serrata (Thunb. Lycopodium serratum), gained prominence in the 1980s when (−)-huperzine A was identified as a selective and potent inhibitor of acetylcholine esterase (AChe).23 As a selective inhibitor of AChe, huperzine A has been studied as a clinical agent in the treatment of Alzheimer's disease (AD). Currently, naturally occurring huperzine A is in clinical use as a treatment mode for AD in China and also advanced to phase III clinical trials in the US.22,23 In efficacy studies only the (−)-enantiomer was shown to be active against AD.24 In 1998, Terashima reported the first catalytic asymmetric variant of Kozikowski's proposed route using chiral ferrocenylphosphine ligands,25 which were able to achieve rather modest enantioselectivity for the formation of a key intermediate to (−)-huperzine A (66% ee). In 2003, Bai achieved 90% ee in the same reaction, using a slightly modified ferrocenylphosphine ligand.26
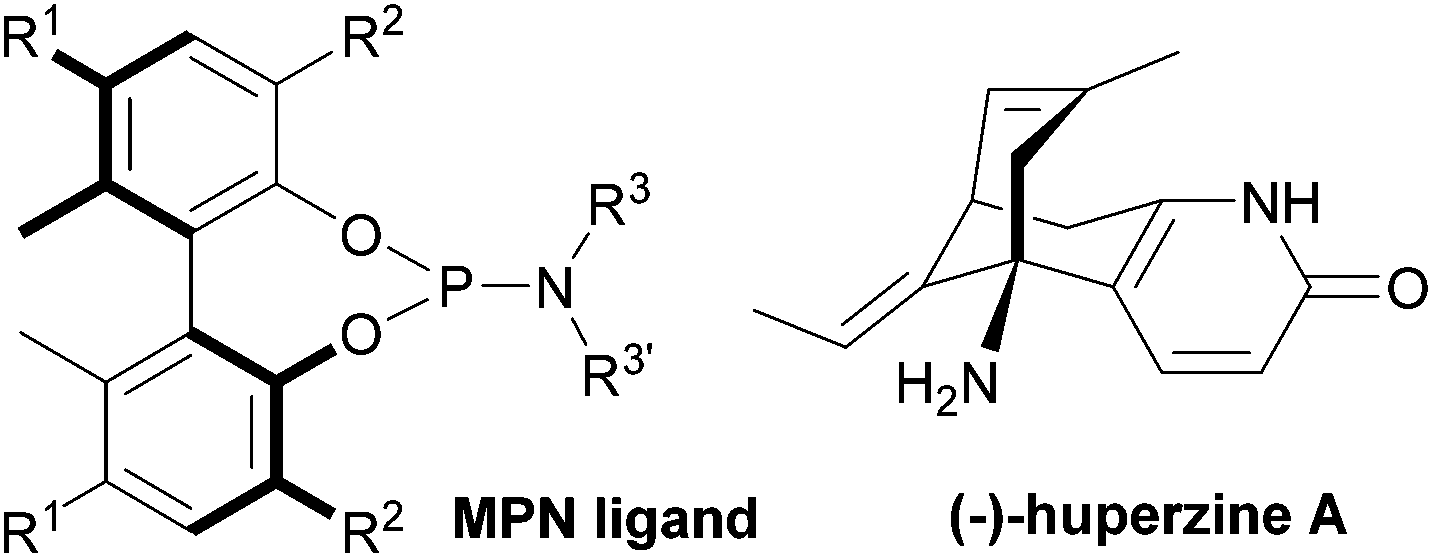 | ||
| Fig. 1 MPN ligand and (−)-huperzine A. | ||
Although the phase III clinical trials did not provide strong enough evidence for FDA approval as a drug for treatment of AD,27 (−)-huperzine A has been attracting considerable interest as a neuroprotective agent to counteract organophosphate chemical weapons such as sarin and VX.28–31 Thus, several reports on the practical synthesis of (−)-huperzine A have recently been published, starting from affordable enantiopure 4-methylcyclohexenones.32–34 Nevertheless, no new process has been reported on the use of catalytic asymmetric synthesis using transition metal catalysts since Bai's work in 2001 and a practical process development by Azadi-Ardakani's team in 2012 using chiral ferrocenyl-phosphines,26,35 although an excellent result was recently reported using an organocatalyst.36
Accordingly, we revisited Kozikowski's process, using our monodentate phosphoramidite (MPN) ligands (Fig. 1), which achieved excellent enantioselectivity (>99% ee) in the short total synthesis of (+)-γ-lycorane using Pd-catalyzed asymmetric allylic alkylation in the key step.16
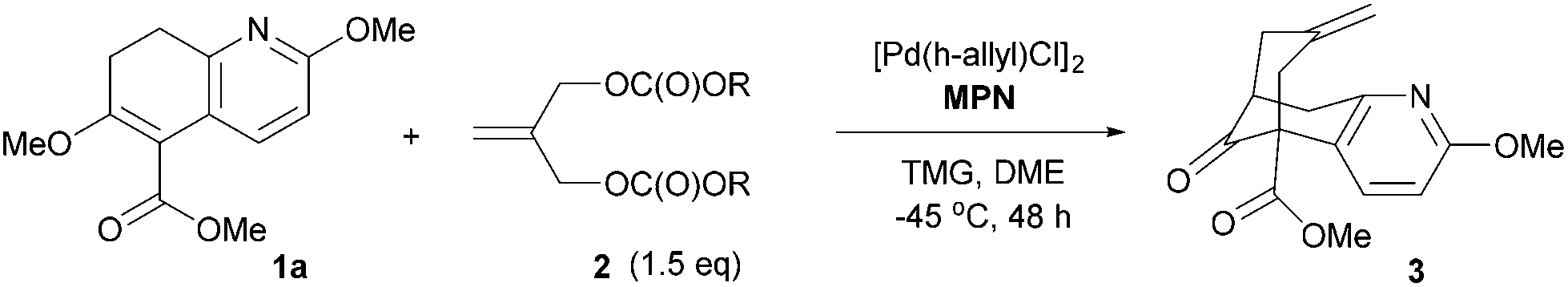
We describe here a successful application of our MPN ligands in the Pd-catalyzed asymmetric tandem allylic alkylation process in the formal total synthesis of huperzine A (Scheme 1).
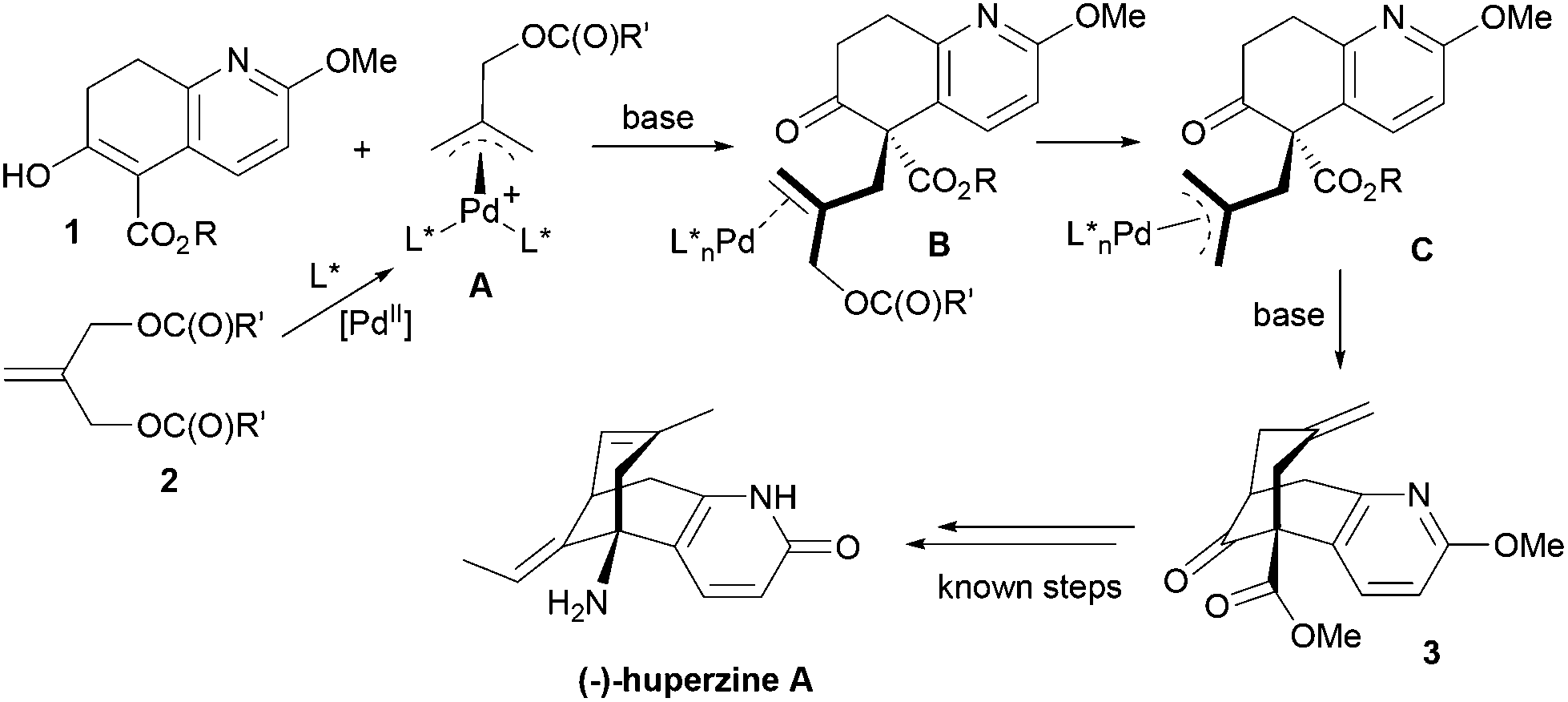 | ||
| Scheme 1 Asymmetric tandem allylic alkylation of 1 with 2. | ||
Results and discussion
Kozikowski's key nucleophile 1a was prepared from commercially available mono-protected 1,4-cyclohexanedione (4), following the literature method22 with modifications, as shown in Scheme 2. This preparation features a modified Stork-enamine synthesis with methyl propiolate in the presence of 7 M NH3 in MeOH to afford compound 5.37 The subsequent O-methylation and deprotection gave 2-methoxytetrahydroquinolin-6-one (7), which was converted to 1avia carbomethoxylation. It is worth noting that the enol-tautomer,38 as shown, was isolated as an exclusive stable species. In a similar manner, four more nucleophiles 1b–e were prepared by reacting chloroformates with 7 or ester exchange reaction with 1a, as shown in Scheme 2.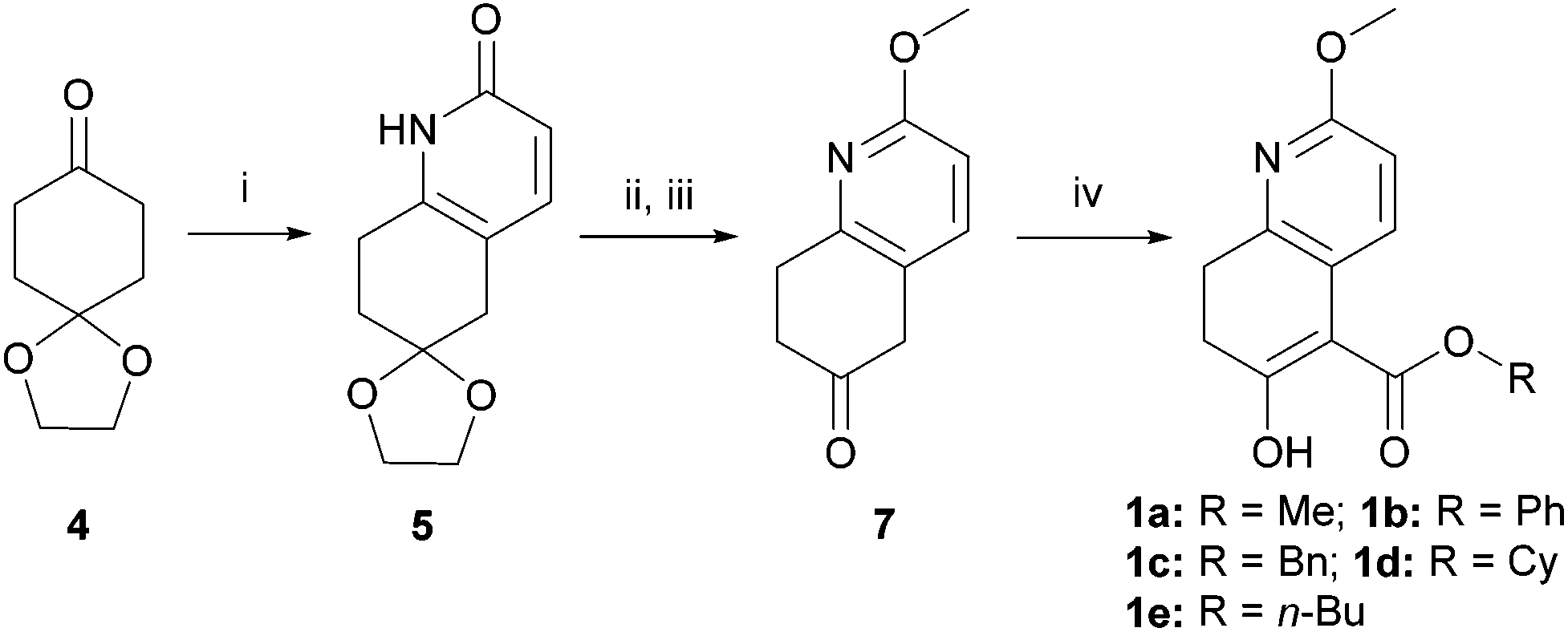 | ||
Scheme 2 Preparation of nucleophile 1. (i) Methyl propiolate (2 equiv.), 7 M NH3, MeOH, 100 °C, 0.69 MPa, 15 h, 51–72%; (ii) Ag2CO3 (2 equiv.), MeI (10 equiv.), CHCl3, reflux, 3 h, 84%; (iii) 5% aq. HCl–acetone (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), reflux, 18 h; (iv) NaH (4 equiv.), (a) CO(OMe)2, reflux, 3 h, 77% for 2 steps, (b) ClCO2Ph, r.t., overnight, 67% for 2 steps, (c) ClCO2Bn, r.t., overnight, 72% for 2 steps, (d) 1a, Cy-OH, p-TsOH, benzene, reflux, 48 h, 82%, (e) 1a, n-BuOH, p-TsOH, benzene, reflux, 48 h, 75%. :1), reflux, 18 h; (iv) NaH (4 equiv.), (a) CO(OMe)2, reflux, 3 h, 77% for 2 steps, (b) ClCO2Ph, r.t., overnight, 67% for 2 steps, (c) ClCO2Bn, r.t., overnight, 72% for 2 steps, (d) 1a, Cy-OH, p-TsOH, benzene, reflux, 48 h, 82%, (e) 1a, n-BuOH, p-TsOH, benzene, reflux, 48 h, 75%. | ||
A series of allylic substrates 2 for tandem dialkylation was prepared from commercially available 2-methylene-1,3-propane-diol (8) and the corresponding acid anhydride or chloroformate. The reactions were carried out under conventional or microwave conditions to afford the corresponding allylic diacetate 2a38 and allylic dicarbonates 2b–i39 in good to excellent yields (Scheme 3).
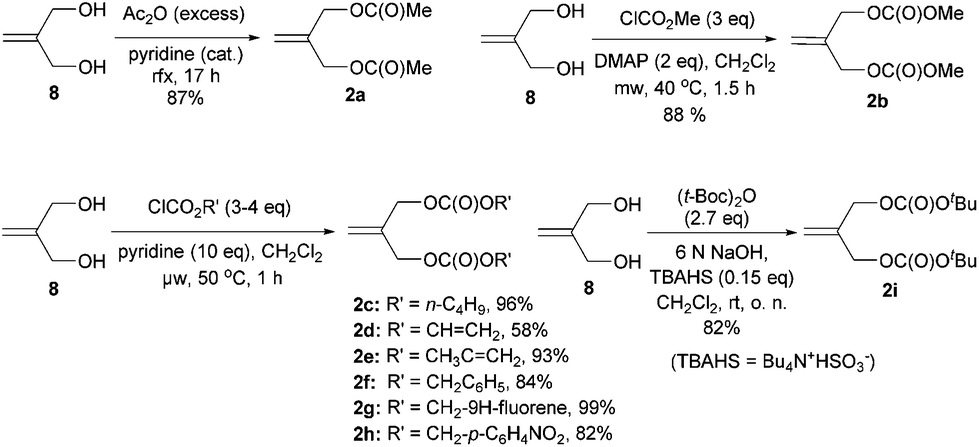 | ||
| Scheme 3 Preparation of allylic substrates 2a–i. | ||
The chiral MPN ligands that were employed in this study are shown in Fig. 2. This series of ligands exhibited high to excellent enantioselectivity in the Pd-catalyzed intermolecular asymmetric allylic alkylation reaction in the key step of (+)-lycorane total synthesis.16
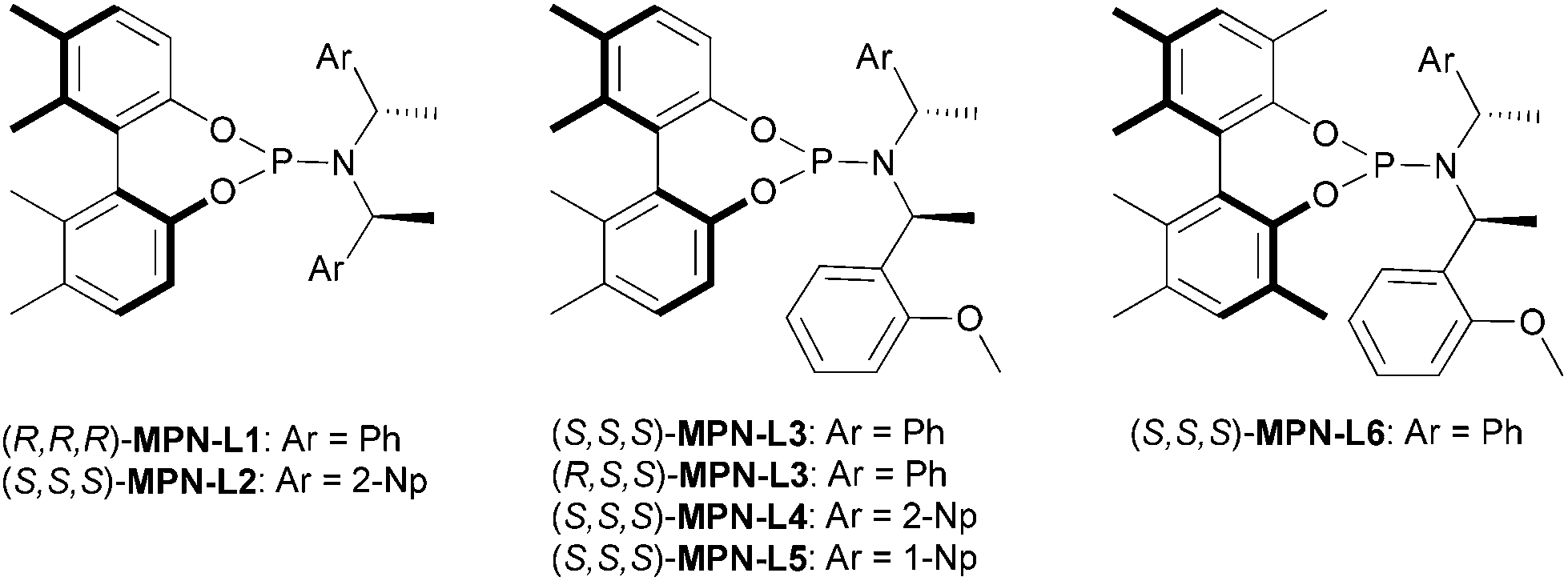 | ||
| Fig. 2 A small library of chiral biphenol-based MPN ligands. | ||
First, we performed a screening of allylic substrates 2 and chiral MPN ligands, using [Pd(η3-allyl)Cl]2 as the catalyst precursor, 1,1,3,3-tetramethylguanidine (TMG) as the base in dimethoxyethane (DME) at −45 °C for 48 h under nitrogen. Results are summarized in Table 1.
|
|
||||
|---|---|---|---|---|
| Entrya | Allylic substrate | Ligand (L*) | Conv.b (%) | % eec |
| a All reactions were run using [Pd(η3-C3H3)Cl]2 (2.5 mol%) with a MPN ligand (15 mol%) and TMG (2.5 eq.) in DME [0.025] at −45 °C for 48 h under N2. b Determined by GC-MS. c Determined by HPLC using Chiralcel OD-H normal phase column with an eluent of 3% isopropanol in hexanes. | ||||
| 1 | 2a | (R,R,R)-MPN-L1 | 100 | 33 (−) |
| 2 | 2b | (R,R,R)-MPN-L1 | 100 | 38 (−) |
| 3 | 2c | (R,R,R)-MPN-L1 | 100 | 36 (−) |
| 4 | 2i | (R,R,R)-MPN-L1 | 100 | 40 (−) |
| 5 | 2i | (S,S,S)-MPN-L2 | 100 | 2 (+) |
| 6 | 2i | (S,S,S)-MPN-L3 | 100 | 49 (−) |
| 7 | 2i | (R,S,S)-MPN-L3 | <5 | n.d. |
| 8 | 2i | (S,S,S)-MPN-L4 | 100 | 61 (−) |
| 9 | 2i | (S,S,S)-MPN-L5 | 100 | 76 (−) |
| 10 | 2i | (S,S,S)-MPN-L6 | <5 | n.d. |
| 11 | 2d | (S,S,S)-MPN-L5 | 100 | 55 (−) |
| 12 | 2e | (S,S,S)-MPN-L5 | 100 | 54 (−) |
| 13 | 2f | (S,S,S)-MPN-L5 | 100 | 87 (−) |
For the allylic substrate screening, the (R,R,R)-MPN-L1 ligand was used and 2a–c and 2i were examined (entries 1–4). Although only moderate enantioselectivity was observed in those substrates, 2i gave the best result among them. Thus, we fixed the substrate to 2i and screened MPN ligands (entries 5–10). As the results clearly indicate, the slight change in the MPN ligand structure exerts dramatic differences in the direction of asymmetric induction, catalytic activity and enantioselectivity. Introduction of 2-Np instead of Ph to the C2 symmetrical MPN ligand was detrimental to enantioselectivity (entry 5). Also, a mismatched pair in the chiral amine moiety and axial chirality, i.e., (R,S,S)-MPN-L3, led to the loss of catalytic activity as well as enantioselectivity (entry 7). Introduction of methyl groups at the 3 and 3′ positions, i.e., (S,S,S)-MPN-L6, was detrimental to the catalytic activity and enantioselectivity (entry 10). Similar to the case of (+)-lycorane synthesis, MPN ligands with an asymmetric chiral amine moiety gave substantially better results than those with a symmetrical moiety (entries 6, 8 and 9). Among the MPN ligands examined, (S,S,S)-MPN-L5 gave the best result (76% ee, entry 9). Thus, this MPN ligand was selected as the ligand of choice for optimization.
Then, we examined the effect of the allylic substrate structure further with (S,S,S)-MPN-L5. Thus, the reactions with allylic substrates, 2d, 2e and 2f, were carried out (entries 11–13). It was found that the allylic dicarbonate bearing benzyl groups, 2f, gave a substantially better result than those bearing ethenyl (2d) and 2-propenyl (2e) groups, as well as tert-butyl groups (2i) (87% ee, entry 13).
Next, we examined the effect of the ester moiety of nucleophile 1 on the reactivity and enantioselectivity. To our surprise, the attempted reactions with phenyl ester 1b and benzyl ester 1c did not afford any desirable product 3b and 3c, respectively. It was found that 1b and 1c existed as the keto ester form in sharp contrast with 1a, 1d and 1e, which existed as the enol ester form as shown in Scheme 2. This difference in properties appears to be a significant factor for the marked contrast in reactivity. The reactions of 1d and 1e using the (S,S,S)-MPN-L5 ligand under the same conditions as those shown in Table 1 gave the corresponding products 3d and 3e with 59% ee (with 2a) and 82% ee (with 2f; 67% ee with 2a), respectively. Thus, 1a was selected as the best nucleophile among those examined.
We also examined the effects of solvent and reaction temperature on the enantioselectivity and reaction rate in the tandem allylic alkylation of 1 with 2f using the MPN-L5 ligand. Results are summarized in Table 2. Among the five common solvents employed, DME, dichloromethane and DMF gave comparable results, i.e., 87–89% ee (entries 1–3) and dichloromethane was so far the best (89% ee, entry 2). Toluene gave lower enantioselectivity (entry 4) and ether was even worse (entry 5).
| Entrya | Solvent | Temp. (°C) | Time (h) | Conv.b (%) | % eec |
|---|---|---|---|---|---|
| a All reactions were run using [Pd(η3-C3H3)Cl]2 (2.5 mol%) with MPN-L5 (15 mol%) and TMG (2.5 eq.) in a solvent under N2. b , c See the footnote of Table 1. | |||||
| 1 | DME | −45 | 48 | 100 | 87 (−) |
| 2 | CH2Cl2 | −45 | 48 | 100 | 89 (−) |
| 3 | DMF | −45 | 48 | 100 | 88 (−) |
| 4 | Toluene | −45 | 48 | 100 | 81 (−) |
| 5 | Et2O | −45 | 48 | 100 | 70 (−) |
| 6 | CH2Cl2 | −25 | 24 | 100 | 85 (−) |
| 7 | CH2Cl2 | −55 | 48 | 100 | 86 (−) |
| 8 | CH2Cl2 | −78 | 96 | 100 | 68 (−) |
The effect of temperature on enantioselectivity was found to be rather unexpected, although the reaction rate was temperature dependent as anticipated. As Table 2 shows (entries 2, 6–8), the enantioselectivity appears to be optimal at or around −45 °C, and it goes down at either higher or lower temperatures. The result implies that the mechanism is not entropy dependent and is thus rather sophisticated.
Since dibenzyl dicarbonate 2f was the best allylic substrates thus far, additional allylic dicarbonates, 2g (9H-fluorenylmethyl) and 2h (4-nitrobenzyl), were prepared and compared with 2f in dichloromethane at −45 °C. Results are shown in Table 3.
Allylic dicarbonate 2g gave virtually the same results (89.0% ee, entry 2) as 2f (89.2% ee, entry 1). Thus, the table shows the enantioselectivity with one decimal. Introduction of a 4-nitro group to the benzyl ester moiety slightly lowered enantioselectivity (87.4% ee, entry 3). However, it is not clear if this is an electronic effect or steric effect at present.
Since the mechanism of asymmetric induction in this reaction is rather unique, i.e., π-allyl-Pd complex A (Scheme 1) generated as the key intermediate possesses a carbobenzoxymethyl group at the C2 carbon of the π-allyl system as shown in Fig. 3. Then, the first carbon–carbon bond formation takes place between the C5 (*) of nucleophile 1 and either C1 or C3 of the allyl moiety, generating the corresponding π-olefin-Pd complex B (Scheme 1). The second carbon–carbon bond formation occurs intramolecularly at the C7 (**) of the second π-allyl-Pd complex C (Scheme 1). Although the second allylic alkylation reaction creates a chiral carbon center, the overall stereochemistry is already determined in the first reaction. Thus, the absolute configuration of the product is solely determined by the enantioface differentiation of nucleophile 1 in the first allylic alkylation reaction. As Fig. 3 illustrates, there are four possible scenarios for this process, i.e., Pro-S or Pro-R face of 1 reacts with either the C1 or C3 carbon of the π-allyl-Pd complex A, generating S or R product.
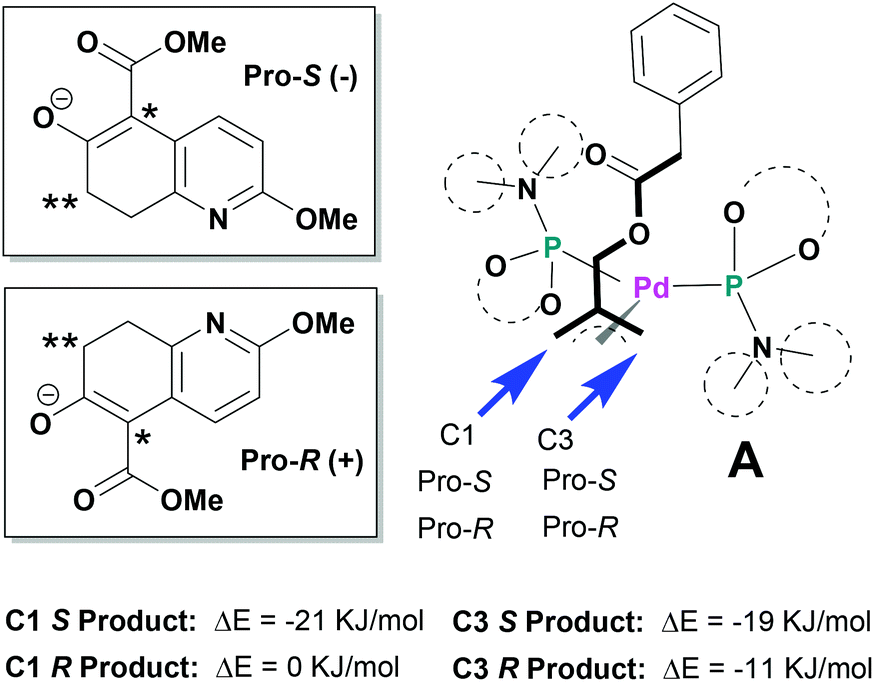 | ||
| Fig. 3 Mechanism of asymmetric induction. | ||
As we have successfully analysed the mode of enantioselection in the intramolecular and intermolecular asymmetric allylic substitution reactions,18,19 we have performed a molecular modeling study using the Spartan program (PM3). From our previous studies,14,15,18,19 it is reasonable to assume that the active chiral catalyst species bears two MPN ligands on the Pd metal, forming a pseudo-C2-symmetrical π-allyl-Pd complex. When this Pd-complex reacts with an allylic dicarbonate, it generates the putative π-allyl-Pd complex A (Fig. 3). We have assumed that this allylic alkylation reaction goes through a late transition state, which is product-like. Then, we calculated the relative energy for four possible products, i.e., π-olefin-Pd complexes B. The results are shown in Fig. 3. The calculations suggest that nucleophile 1 reacts with both C1 and C3, but the S product is substantially more favorable over the R product in both cases, and the C1 site has higher selectivity. It is worth noting that the molecular modeling study has shed light on an interesting aspect of the mechanism of asymmetric induction in this reaction.
Conclusions
(S,S,S)-MPN-L5, selected from fine-tunable MPN ligands, was found to be an excellent chiral ligand for the Pd-catalyzed tandem asymmetric allylic alkylation reaction to afford 3 with high enantiopurity (89.2% ee), which is a critical key intermediate for the formal total synthesis (−)-huperzine. Logical optimization of reaction variables was carried out together with the screening of MPN ligands and allylic substrates 2. The mechanism of asymmetric induction was investigated and discussed based on the molecular modeling of key Pd-complex species involved in the catalyst cycle using the Spartan program. The molecular modeling analysis correctly indicated that the S enantiomer should be the predominant product. Further studies on the applications of fine-tunable biphenol-based MPN and BOP ligands are actively underway in our laboratory.Experimental
A typical procedure for asymmetric tandem allylic alkylations: the chiral ligand (S,S,S)-MPN-L5 (17.2 mg, 15 mol%), a catalyst [Pd(η3-C3H3)Cl]2 (1.8 mg, 2.5 mol%), 2-methylenepropane-1,3-diyl dibenzyl dicarbonate (2f) (107 mg, 0.30 mmol, 1.5 eq.) and 1 (47 mg, 0.20 mmol) were dissolved in 8 mL of dry CH2Cl2 in a 35 mL Schlenk tube. The mixture was then stirred at room temperature for 30 min and cooled to −45 °C for 30 min. Then, tetramethylguanidine (TMG) (57.6 mg, 0.50 mmol) was slowly added to this solution and the reaction mixture was stirred at −45 °C for 48 h. The solvent was evaporated and the resulting yellow oil was submitted to a normal phase chiral HPLC column (Chiralcel OD-H) with an eluent of 3% isopropanol in hexanes (tR = 19.0 and 24.2 min) to determine the enantiopurity. The crude product was submitted to flash column chromatography on silica gel (hexanes–EtOAc = 20:1→10:1) to give 3 as a light yellow oil (26 mg, 70% yield): 1H NMR (CDCl3, 300 MHz) δ 2.53 (2H, m), 2.75 (1H, m), 2.92 (m, 1H), 3.06 (2H, m), 3.40 (1H, dd, J = 18.4, J = 6.8), 3.78 (3H, s), 3.86 (3H, s), 4.47 (1H, s), 4.80 (1H, s), 6.54 (1H, d, J = 8.4 Hz), 6.94 (1H, d, J = 8.4 Hz); 13C NMR (CDCl3, 100 MHz) δ 40.4, 43.9, 45.6, 47.8, 52.6, 53.4, 62.0, 109.5, 116.3, 124.7, 137.5, 138.9, 151.3, 162.9, 171.3, 208.3. All data are consistent with the literature values.25
Acknowledgements
This work was supported by a grant from the National Science Foundation. Generous support from Mitsubishi Chemical Corporation is also gratefully acknowledged. The authors thank Dr. Stephen Chaterpaul and Dr. Hojae Choi for technical assistance.References
- B. C. G. Söderberg, Coord. Chem. Rev., 2004, 248, 1085–1158 CrossRef PubMed.
- J. Terao and N. Kambe, Bull. Chem. Soc. Jpn., 2006, 79, 663–672 CrossRef CAS.
- Y. J. Park, J.-W. Park and C.-H. Jun, Acc. Chem. Res., 2008, 41, 222–234 CrossRef CAS PubMed.
- K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442–4489 CrossRef CAS PubMed.
- B. M. Trost and C. Lee, in Catalytic Asymmetric Synthesis, ed. I. Ojima, Wiley-VCH, New York, 2nd edn, 2000, pp. 593–649 Search PubMed.
- B. M. Trost, J. Org. Chem., 2004, 69, 5813–5837 CrossRef CAS PubMed.
- B. M. Trost and G. Dong, J. Am. Chem. Soc., 2006, 128, 6054–6055 CrossRef CAS PubMed.
- B. M. Trost and M. L. Crawley, Chem. Rev., 2003, 103, 2921–2943 CrossRef CAS PubMed.
- B. M. Trost, Chem. Pharm. Bull., 2002, 50, 1–14 CrossRef CAS.
- G. Helmchen and A. Pfaltz, Acc. Chem. Res., 2000, 33, 336–345 CrossRef CAS PubMed.
- J. M. J. Williams, Synlett, 1996, 705 CrossRef CAS PubMed.
- G. Helmchen, U. Kazmater and S. Förster, in Catalytic Asymmetric Synthesis, ed. I. Ojima, Wiley, Hoboken, 3rd edn, 2010, pp. 497–641 Search PubMed.
- Z. Hua, V. C. Vassar and I. Ojima, Org. Lett., 2003, 5, 3831–3834 CrossRef CAS PubMed.
- Z. Hua, V. C. Vassar, H. Choi and I. Ojima, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5411–5416 CrossRef CAS PubMed.
- B. D. Chapsal, Z. Hua and I. Ojima, Tetrahedron: Asymmetry, 2006, 17, 642–657 CrossRef CAS PubMed.
- B. D. Chapsal and I. Ojima, Org. Lett., 2006, 8, 1395–1398 CrossRef CAS PubMed.
- C. Shi and I. Ojima, Tetrahedron, 2007, 63, 8563–8570 CrossRef CAS PubMed.
- C.-W. Chien, C. Shi, C.-F. Lin and I. Ojima, Tetrahedron, 2011, 67, 6513–6523 CrossRef CAS PubMed.
- C. Shi, C.-W. Chien and I. Ojima, Chem. – Asian J., 2011, 6, 674–680 CrossRef CAS PubMed.
- C.-F. Lin and I. Ojima, J. Org. Chem., 2011, 76, 6240–6249 CrossRef CAS PubMed.
- Y. Zang and I. Ojima, J. Org. Chem., 2013, 78, 4013–4018 CrossRef CAS PubMed.
- Y. Xia and A. P. Kozikowski, J. Am. Chem. Soc., 1989, 111, 4116–4117 CrossRef CAS.
- D. L. Bai, X. C. Tang and X. C. He, Curr. Med. Chem., 2000, 7, 355–374 CrossRef CAS.
- X. C. Tang, G. H. Kindel, A. P. Kozikowski and I. Hanin, J. Enthnopharmacol., 1994, 44, 147–155 CrossRef CAS.
- S. Kaneko, T. Yoshino, T. Katoh and S. Terashima, Tetrahedron: Asymmetry, 1997, 8, 829–832 CrossRef CAS.
- X.-C. He, B. Wang, G. Yu and D. Bai, Tetrahedron: Asymmetry, 2001, 12, 3213–3216 CrossRef CAS.
- G. Yang, Y. Wang, J. Tian and J. Liu, PLoS One, 2013, 8, e74916 CAS.
- J. Haigh, S. Johnston, A. Peppernay, P. Mattern, G. Garcia, B. Doctor, R. Gordon and P. Aisen, Chem.-Biol. Interact., 2008, 175, 380–386 CrossRef CAS PubMed.
- J. Z. Karasova, J. Bajgar, L. Novotny and K. Kuca, Lett. Drug Des. Discovery, 2009, 6, 563–567 CrossRef CAS and references therein.
- T. Koshiba, S. Yokoshima and T. Fukuyama, Org. Lett., 2009, 11, 5354–5356 CrossRef CAS PubMed.
- J. White, Y. Li, J. Kim and M. Terinek, Org. Lett., 2013, 15, 882–885 CrossRef CAS PubMed.
- M. K. M. Tun, D. Wüstmann and S. B. Herzon, Chem. Sci., 2011, 2, 2251–2253 RSC.
- R. Ding, B.-F. Sun and G.-Q. Lin, Org. Lett., 2012, 14, 4446–4449 CrossRef CAS PubMed.
- R. Ding, J.-G. Fu, G.-Q. Xu, B.-F. Sun and G.-Q. Lin, J. Org. Chem., 2014, 79, 240–250 CrossRef CAS PubMed.
- S. R. Tudhope, J. A. Bellamy, A. Ball, D. Rajasekar, M. Azadi-Ardakani, H. S. Meera, J. M. Gnanadeepam, R. Saiganesh, F. Gibson, L. He, C. H. Behrens, G. Underiner, J. Marfurt and N. Favre, Org. Process Res. Dev., 2012, 16, 635–642 CrossRef CAS.
- X. Ding, X. Li, D. Liu, W. Cui, X. Ju, S. Wang and Z. Yao, Tetrahedron, 2012, 68, 6240–6248 CrossRef CAS PubMed.
- A. P. Kozikowski, E. R. Reddy and C. P. Miller, J. Chem. Soc., Perkin Trans. 1, 1990, 195–197 RSC.
- G. Campiani, L. Q. Sun, A. P. Kozikowski, P. Aagaard and M. McKinney, J. Org. Chem., 1993, 58, 7660–7669 CrossRef CAS.
- S. G. Salamone and G. B. Dudley, Org. Lett., 2005, 7, 4443–4445 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Professor Max Malacria on the occasion of his 65th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/C4QO00180J |
| This journal is © the Partner Organisations 2014 |