
Synthesis of 1,4-benzodiazepinones and 1,4-benzoxazepinones via palladium-catalyzed amino and oxyacetoxylation†‡
A. D.
Manick
,
G.
Duret
,
D. N.
Tran
,
F.
Berhal
* and
G.
Prestat
*
Laboratoire de Chimie et Biochimie Pharmacologiques et Toxicologiques, UMR CNRS 8601, Université Paris Descartes, 45 rue des Saints Pères, 75006 Paris, France. E-mail: farouk.berhal@parisdescartes.fr; guillaume.prestat@parisdescartes.fr
First published on 20th August 2014
Abstract
Palladium-catalyzed amino and oxyacetoxylation have been developed to furnish 1,4-benzodiazepinones and 1,4-benzoxazepinones through the diheterofunctionalization of alkenes. This study demonstrates the ability of this new methodology to allow 7-exo ring-closure.
Metal-catalyzed vicinal dioxygenation1 and oxyamination2 of alkenes is a powerful tool in organic synthesis. In 2005, Sorensen reported a palladium-catalyzed aminoacetoxylation of unactivated olefins in the presence of (diacetoxyiodo)benzene as an oxidant.3 This new synthetic methodology has emerged as an interesting alternative to the classical osmium-based Sharpless dihydroxylation4 and aminohydroxylation.5 The reaction proceeds through an initial nucleopalladation6 followed by oxidation of the organopalladium(II) intermediate thus generating an organopalladium(IV) complex allowing the formation of the second hetero-carbon bond with concomitant release of the catalyst. The flexibility of such a process allows the oxyamination3,7 dioxygenation,8 diamination,9 as well as aminofluorination10 of alkenes.11 Although this strategy appears highly attractive from a synthetic point of view, it has been the subject of a very limited number of reports dealing with the synthesis of biological relevant compounds. Moreover, cyclizing processes have been studied for the formation of five- and six-membered rings only.
Following our efforts towards palladium-catalyzed synthesis of heterocycles,12 we were intrigued to know if such a methodology would allow the formation of the seven-membered heterocyclic core found in benzodiazepine that is considered as a privileged structure.13 Palladium-catalyzed access to 1,4-benzodiazepines has been previously reported via alkene aminopalladation,14 alkene carboamination,15 and an allene carbopalladation/amination domino sequence.16 We report in this communication the successful synthesis of 1,4-benzodiazepin-5-ones via a palladium-catalyzed aminoacetoxylation of alkenes. Moreover, we demonstrate that a slight modification of the reaction conditions can give access to 1,4-benzoxazepin-5-ones through the corresponding oxyacetoxylation.
Compound 1a was chosen as a model substrate to study the aminoacetoxylation reaction (Scheme 1).
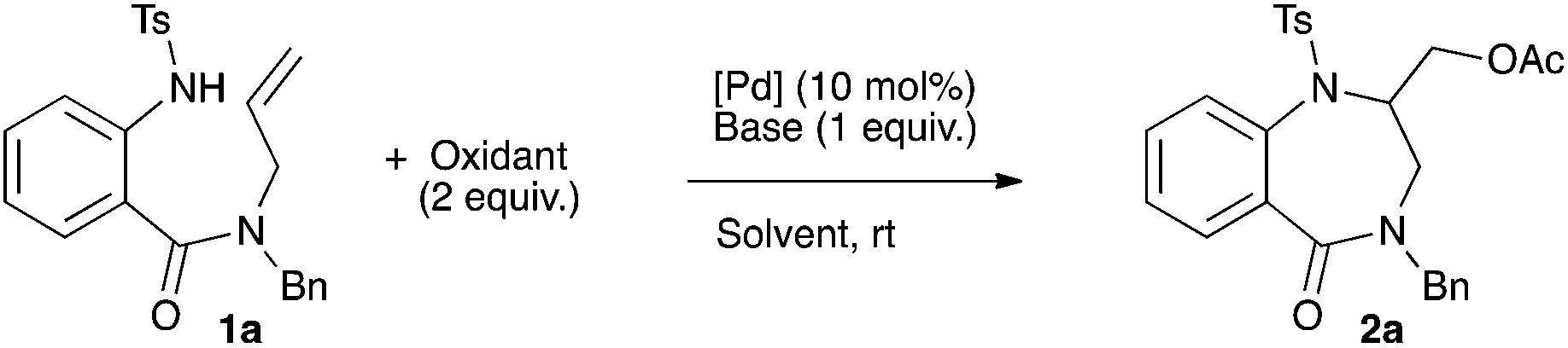 | ||
| Scheme 1 Synthesis of 1,4-benzodiazepinone 2avia palladium-catalyzed aminoacetoxylation. | ||
In the first set of experiments, the reaction was run in the presence of Pd(OAc)2 (10 mol%) and PhI(OAc)2 (2 equiv.) in DCM at rt and the influence of the base was studied. The use of Bu4NOAc allowed the formation of the desired product 2a in 56% yield (entry 1, Table 1). Sodium, potassium and silver acetates led to a large decrease of the yield (10, 25 and 15% respectively, entries 2–4, Table 1). A poor 7% yield was obtained with the use of both potassium and cesium carbonates (entries 5 and 6, Table 1). Organic bases were next tested. The use of pyridine completely inhibited the reaction while DABCO, DBU and proton sponge were found to be poorly effective (entries 7–10, Table 1). Triethylamine allowed a moderate 44% yield (entry 11, Table 1) while diisopropylethyl amine (DiPEA) afforded the desired benzodiazepinone 2a in 71% yield (entry 12, Table 1). The palladium source was next tested. Replacing Pd(OAc)2 with PdCl2(CH3CN)2 or PdCl2(PhCN)2 in the presence of DiPEA gave less satisfactory results (54% and 55% respectively, entries 13 and 14, Table 1). In the absence of any palladium source no product formation was observed (entry 15, Table 1). Moreover, keeping DiPEA as the base and decreasing the palladium concentration to 5 mol% induced a lowering of the yield to 40% (entry 16, Table 1). Finally, raising the amount of oxidant PhI(OAc)2 (2.5 equiv.) in the presence of Pd(OAc)2 (10 mol%) led to a 74% yield (entry 17, Table 1).
| Entry | [Pd] | Base | Yieldb of 2a (%) |
|---|---|---|---|
| a Reaction conditions: 1a (1 equiv.), PhI(OAc)2 (2 equiv.), [Pd] (10 mol%), base (1 equiv.), DCM, rt, 2 h. b Yields refer to isolated products. c Pd(OAc)2 (5 mol%). d PhI(OAc)2 (2.5 equiv.). | |||
| 1 | Pd(OAc)2 | Bu4NOAc | 56 |
| 2 | Pd(OAc)2 | NaOAc | 10 |
| 3 | Pd(OAc)2 | KOAc | 25 |
| 4 | Pd(OAc)2 | AgOAc | 15 |
| 5 | Pd(OAc)2 | K2CO3 | 7 |
| 6 | Pd(OAc)2 | Cs2CO3 | 7 |
| 7 | Pd(OAc)2 | Pyridine | 0 |
| 8 | Pd(OAc)2 | DABCO | 5 |
| 9 | Pd(OAc)2 | DBU | 21 |
| 10 | Pd(OAc)2 | Proton sponge | 13 |
| 11 | Pd(OAc)2 | NEt3 | 44 |
| 12 | Pd(OAc)2 | DiPEA | 71 |
| 13 | PdCl2(CH3CN)2 | DiPEA | 54 |
| 14 | PdCl2(PhCN)2 | DiPEA | 55 |
| 15 | — | DiPEA | 0 |
| 16 | Pd(OAc)2 | DiPEA | 40c |
| 17 | Pd(OAc)2 | DiPEA | 74d |
The influence of the oxidant was next evaluated using Pd(OAc)2 (10 mol%) and DiPEA (1 equiv.), in DCM (Scheme 1 and Table 2). Replacing PhI(OAc)2 with PhI(OTFA)2 or Cu(OAc)2 completely inhibited the formation of any benzodiazepinone (entries 2 and 3 vs. 1, Table 2). On the other hand, the use 3-NO2, 4-MeO-(diacetoxyiodo)benzene and (diacetoxyiodo)benzene gave the same result (entries 4 and 5 and 1, Table 2).
A solvent screening was next undertaken using our optimized conditions (Scheme 1 and Table 3). DCM proved to be the solvent of choice for this reaction leading to the highest yield and shortest reaction time. A correlation between the efficiency of the solvent and its physical characteristics (dipole moment, dielectric constant) could not be established (entries 1–9, Table 3).
| Entry | Solvent | Reaction time (h) | Yieldb of 2a (%) |
|---|---|---|---|
| a Reaction conditions: 1a (1 equiv.), PhI(OAc)2 (2.5 equiv.), Pd(OAc)2 (10 mol%), DiPEA (1 equiv.), DCM, rt. b Yields refer to isolated products. | |||
| 1 | DCM | 2 | 74 |
| 2 | Toluene | 24 | 68 |
| 3 | Cyclohexane | 24 | 44 |
| 4 | THF | 24 | 24 |
| 5 | Dioxane | 24 | 46 |
| 6 | AcOEt | 24 | 47 |
| 7 | DMF | 2 | 27 |
| 8 | Acetonitrile | 3 | 59 |
| 9 | DMSO | 2 | 27 |
Finally the nature of the nitrogen atom protecting group was examined. The replacement of the tosyl group with a nosyl group induced a slight decrease in yield (70% vs. 74%, entries 1 and 2, Table 4) and a longer reaction time to reach complete conversion. After 24 h of reaction, the starting material was completely recovered with either Boc or Ac derivatives 4 and 5 (entries 3 and 4, Table 4). On the other hand, decomposition of the starting material was observed with the free aniline derivative 6 (entry 5, Table 4).
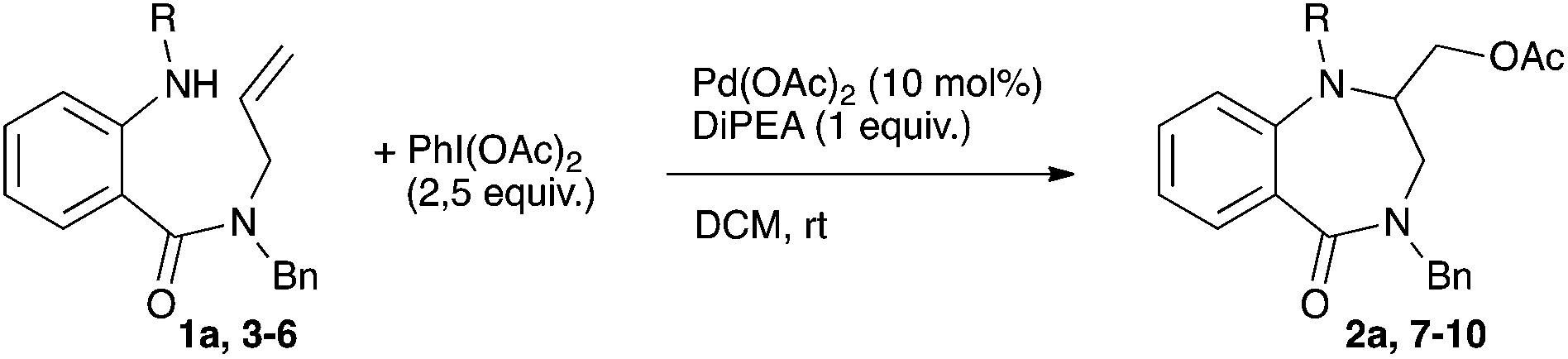
Having in hand the fully optimized conditions the scope of the reaction was then studied (Scheme 2). The reaction proved to easily tolerate electron-donating as well as electron-withdrawing substituents on the aryl ring. The desired amino-acetoxylated compounds were isolated in yields ranging from 58 to 82%. Unsurprisingly, the use of 1,2-disubstituted alkene 1k completely inhibited the aminoacetoxylation process. This is a known limitation of the palladium-catalyzed difunctionalization of alkenes that has been overcome for styrene derivatives only.3,8a,9b However, using our conditions, a complete lack of reactivity was observed starting with the styryl derivative 1l.
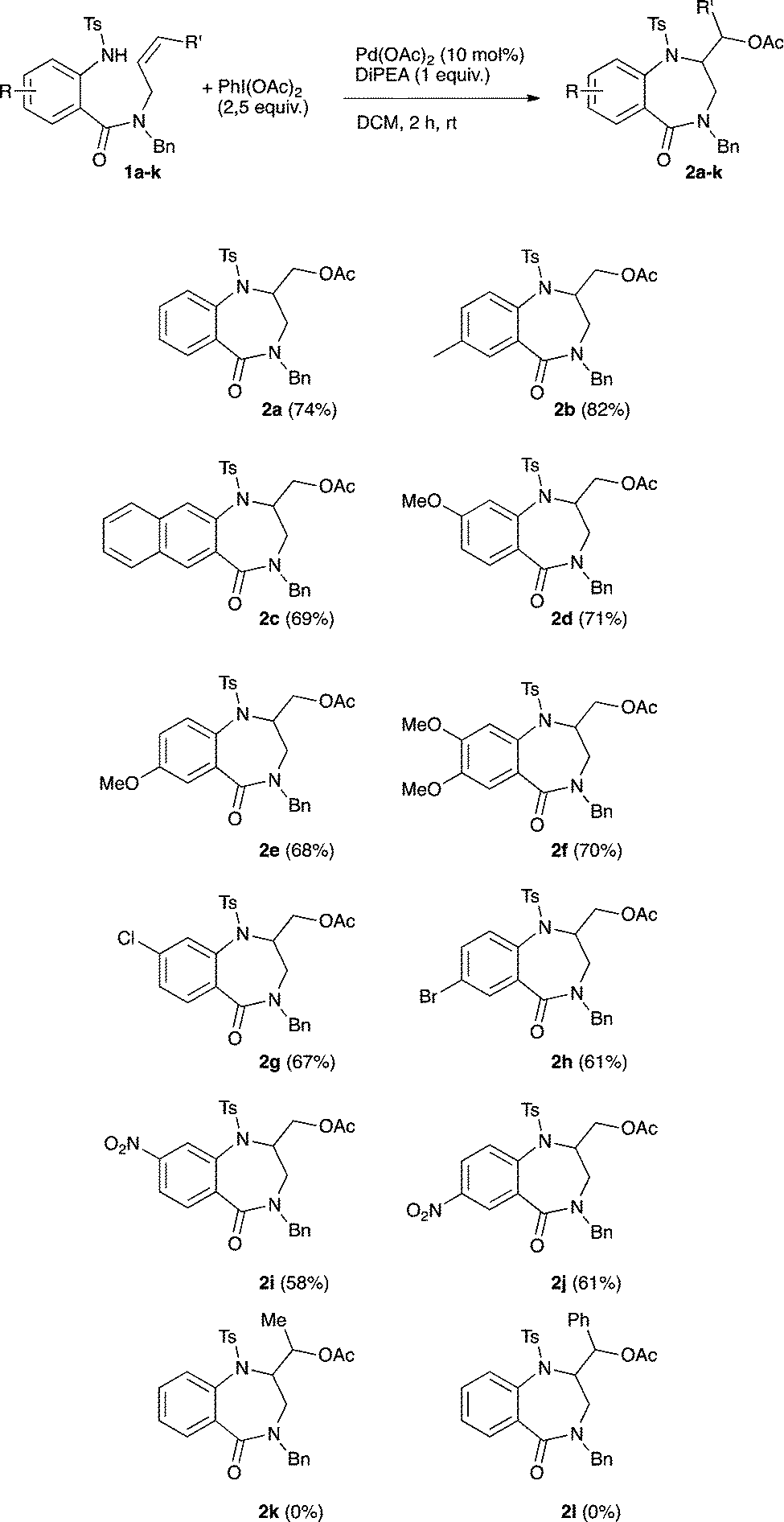 | ||
| Scheme 2 Scope of the palladium-catalyzed aminoacetoxylation. | ||
In order to validate our synthetic strategy towards 1,4-benzodiazepin-5-ones, the removal of the tosyl group was undertaken using 2a as a model substrate. The treatment of 2a by magnesium turnings in methanol led to the removal of both the tosyl and acetyl group. The 1,4-benzodiazepinone 11 was isolated in 75% yield (Scheme 3).
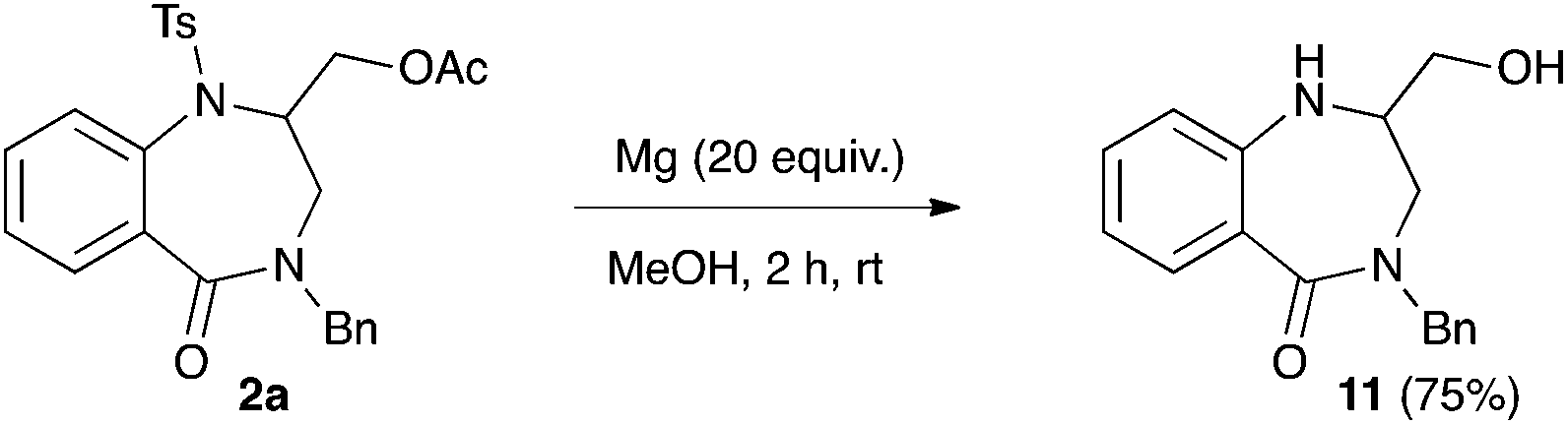 | ||
| Scheme 3 Removal of the tosyl group from 2a. | ||
We next envisioned extending the scope of the reaction by replacing the nucleophilic nitrogen atom with an oxygen atom. By turning from aminoacetoxylation to oxyacetoxylation and starting with the phenol derivative 12a, the reaction should open a new route to 1,4-benzoxazepin-5-one such as 13a. Using our optimized conditions for aniline substrates, the desired benzoxazepinone 13a was isolated in a limited 30% yield (entry 1, Table 5). A short optimization of the reaction conditions was thus undertaken. With DCM as the solvent, the use of t-BuOK or AcOK led to the almost complete inhibition of the reaction (3 and 0% respectively, entries 2 and 3, Table 5). When AcOK was used in the presence of 18-C-6 (1 equiv.) the desired benzoxazepinone 13a was isolated in 27% yield (entry 4, Table 5). Conversely, Bu4NOAc allowed an increase in yield up to an encouraging 52% while Bu4NBr completely failed to promote the cyclization (entries 5 and 6, Table 5). Keeping Bu4NOAc as the base, the solvent effects were next studied. When the reaction was run in toluene or DMSO, a slight decrease in yield was observed (48 and 46% yield respectively, entries 7 and 8, Table 5). Finally, a satisfying 71% yield was reached upon running the reaction in DMF (entry 9, Table 5). In the absence of a palladium source no cyclization product was observed (entry 10, Table 5).
|
|
||||
|---|---|---|---|---|
| Entry | Additive | Solvent | Reaction time (h) | Yieldb of 13a (%) |
| a Reaction conditions: 12a (1 equiv.), PhI(OAc)2 (2.5 equiv.), Pd(OAc)2 (10 mol%), additive (1 equiv.), solvent, rt. b Yields refer to isolated products. c Reaction was run with AcOK (1 equiv.), 18-C-6 (1 equiv.). d Without Pd(OAc)2. | ||||
| 1 | DiPEA | DCM | 2 | 30 |
| 2 | t-BuOK | DCM | 2 | 3 |
| 3 | AcOK | DCM | 2 | 0 |
| 4c | AcOK/18-C-6 | DCM | 2 | 27 |
| 5 | Bu4NOAc | DCM | 1 | 52 |
| 6 | Bu4NBr | DCM | 2 | 9 |
| 7 | Bu4NOAc | Toluene | 1 | 48 |
| 8 | Bu4NOAc | DMSO | 1 | 46 |
| 9 | Bu4NOAc | DMF | 1 | 71 |
| 10d | Bu4NOAc | DMF | 1 | 0 |
The scope of the oxyacetoxylation leading to benzoxazepinone was next examined (Scheme 4). The yields typically ranged between 60 and 71% using substrates bearing various substituents on the phenyl ring. The methoxy-substituted benzoxazepinone was obtained in a limited 35% yield.
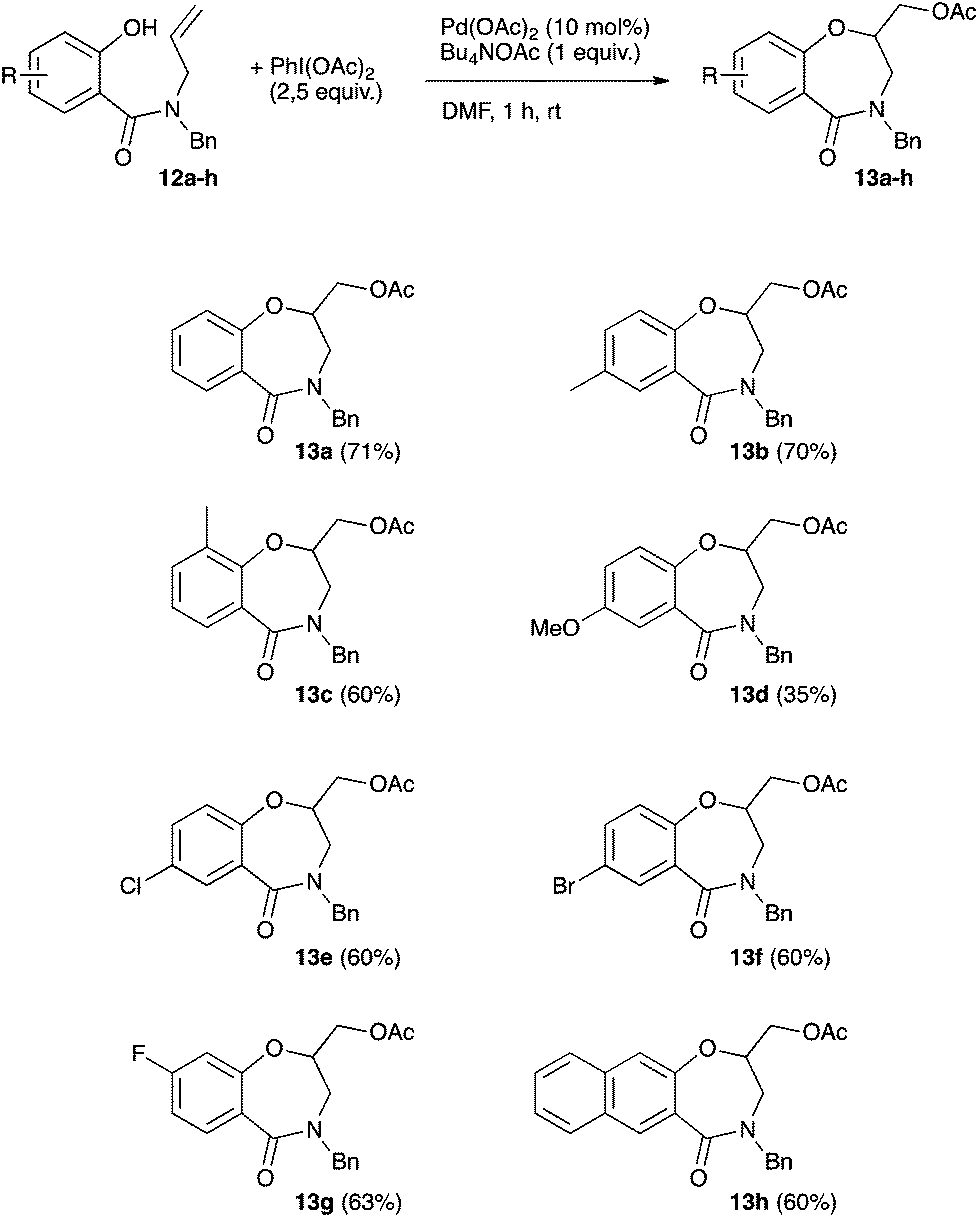 | ||
| Scheme 4 Scope of the palladium-catalyzed oxyacetoxylation. | ||
The mechanism of palladium-catalyzed alkene difunctionalization in the presence of hypervalent iodine has been previously studied by Stahl,7a,d Sanford,7b and Liu and Muñiz.7d Aminopalladation of the alkene is followed by a Pd(II) to Pd(IV) oxidation mediated by PhI(OAc)2. The organopalladium(IV) complex evolves via reductive elimination to form the C–OAc bond and regenerates the Pd(II) catalyst (Scheme 5). Cis- or trans-aminopalladation has been proposed, while reductive elimination might occur with retention or inversion of configuration. The stereoselectivity of these elementary steps seemed highly substrate- and condition-dependent and are still under debate.7d
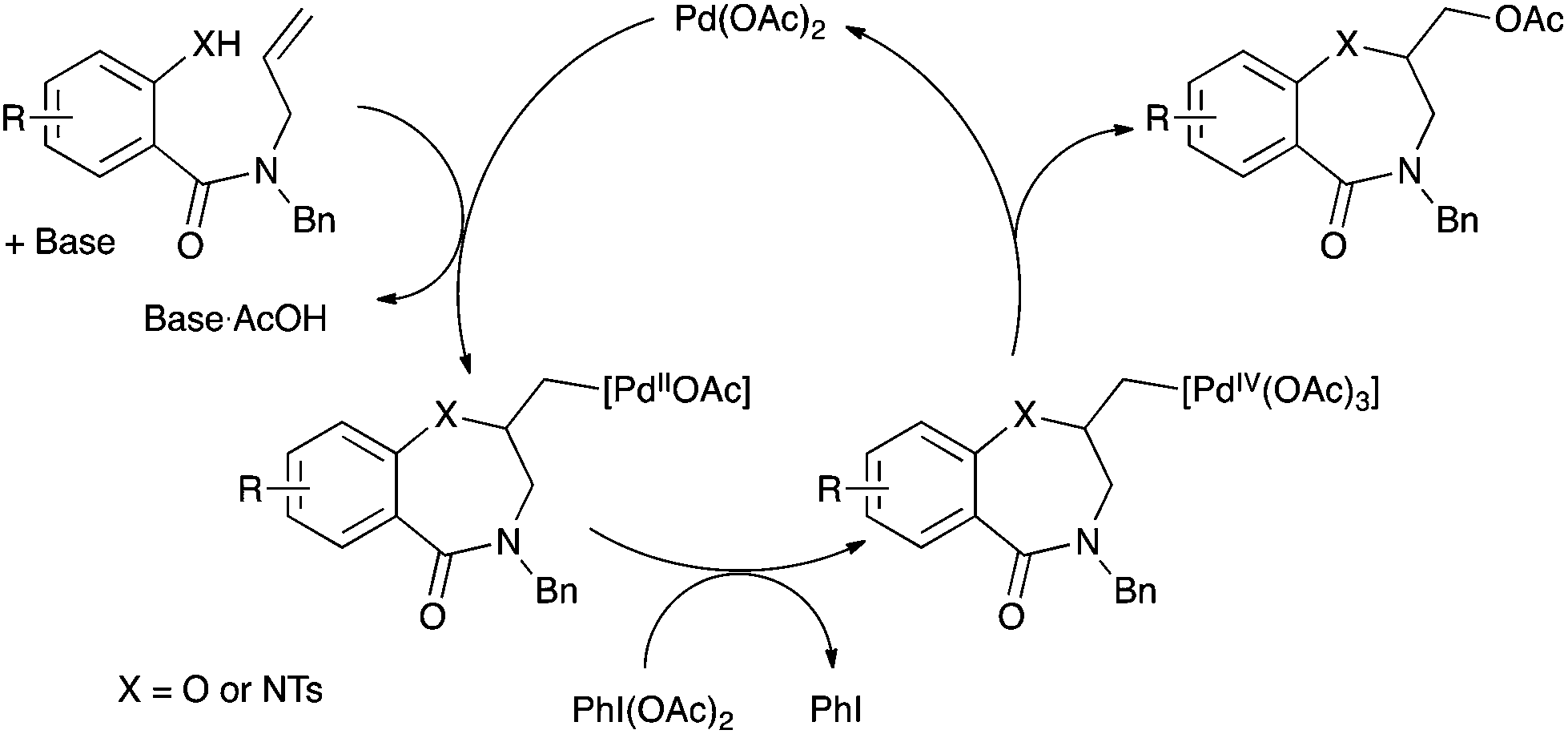 | ||
| Scheme 5 Putative mechanism. | ||
In summary, we have developed a new route to 1,4-benzodiazepin-5-ones based on a palladium-catalyzed aminoacetoxylation process. Slight modifications of the reaction conditions opened up access to 1,4-benzoxazepinones via the corresponding oxyacetoxylation. This study describes for the first time a seven-membered ring-closure diheterofunctionalization of alkenes.
Acknowledgements
We thank the CNRS and Université Paris Descartes for financial support and the Ministère de l'Education Nationale de l'Enseignement Supérieur et de la Recherche for a grant (A. D. M.). Dr Patricia Busca is warmly acknowledged for fruitful discussions and careful proofreading.Notes and references
- A. B. Zaitsev and H. Adolfsson, Synthesis, 2006, 1725–1756 CAS.
- T. J. Donohoe, C. K. A. Callens, A. Flores, A. R. Lacy and A. H. Rathi, Chem. – Eur. J., 2011, 17, 58–76 CrossRef CAS PubMed.
- E. J. Alexanian, C. Lee and E. J. Sorensen, J. Am. Chem. Soc., 2005, 127, 7690–7691 CrossRef CAS PubMed.
- H. C. Kolb, M. S. VanNieuwenhze and K. B. Sharpless, Chem. Rev., 1994, 94, 2483–2547 CrossRef CAS.
- J. A. Bodkin and M. D. McLeod, J. Chem. Soc., Perkin Trans. 1, 2002, 2733–2746 RSC.
- R. I. McDonald, G. Liu and S. S. Stahl, Chem. Rev., 2011, 111, 2981–3019 CrossRef CAS PubMed.
- (a) G. Liu and S. S. Stahl, J. Am. Chem. Soc., 2006, 128, 7179–7181 CrossRef CAS PubMed; (b) L. V. Desai and M. S. Sanford, Angew. Chem., Int. Ed., 2007, 46, 5737–5740 CrossRef CAS PubMed; (c) S. Cui, L. Wojtas and J. C. Antilla, Angew. Chem., Int. Ed., 2011, 50, 8927–8930 CrossRef CAS PubMed; (d) C. Martínez, Y. Wu, A. B. Weinstein, S. S. Stahl, G. Liu and K. Muñiz, J. Org. Chem., 2013, 78, 6309–6315 CrossRef PubMed.
- (a) Y. Li, D. Song and V. M. Dong, J. Am. Chem. Soc., 2008, 130, 2962–2964 CrossRef CAS PubMed; (b) S. R. Neufeldt and M. S. Sanford, Org. Lett., 2013, 15, 46–49 CrossRef CAS PubMed; (c) W. Wang, F. Wang and M. Shi, Organometallics, 2010, 29, 928–933 CrossRef CAS.
- (a) J. Streuff, C. H. Hövelmann, M. Nieger and K. Muñiz, J. Am. Chem. Soc., 2005, 127, 14586–14587 CrossRef CAS PubMed; (b) C. Martínez and K. Muñiz, Angew. Chem., Int. Ed., 2012, 51, 7031–7034 CrossRef PubMed.
- T. Wu, G. Yin and G. Liu, J. Am. Chem. Soc., 2009, 131, 16354–16355 CrossRef CAS PubMed.
- For diheterofunctionalization of alkenes catalyzed by other metals than Pd see: (a) T. P. Zabawa, D. Kasi and S. R. Chemler, J. Am. Chem. Soc., 2005, 127, 11250–11251 CrossRef CAS PubMed; (b) F. C. Sequeira and S. R. Chemler, Org. Lett., 2012, 14, 4482–4485 CrossRef CAS PubMed; (c) M. Nakanishi, C. Minard, P. Retailleau, K. Cariou and R. H. Dodd, Org. Lett., 2011, 13, 5792–5795 CrossRef CAS PubMed; (d) T. De Haro and C. Nevado, Angew. Chem., Int. Ed., 2011, 50, 906–910 CrossRef CAS PubMed; (e) G.-S. Liu, Y.-Q. Zhang, Y.-A. Yuan and H. Xu, J. Am. Chem. Soc., 2013, 135, 3343–3346 CrossRef CAS PubMed; (f) K. S. Williamson and T. P. Yoon, J. Am. Chem. Soc., 2012, 134, 12370–12373 CrossRef CAS PubMed; (g) D.-F. Lu, G.-S. Liu, C.-L. Zhu, B. Yuan and H. Xu, Org. Lett., 2014, 16, 2912–2915 CrossRef CAS PubMed.
- (a) M. Vitale, G. Prestat, D. Lopes, D. Madec, C. Kammerer, G. Poli and L. Girnita, J. Org. Chem., 2008, 73, 5795–5805 CrossRef CAS PubMed; (b) C. Kammerer, G. Prestat, D. Madec and G. Poli, Chem. – Eur. J., 2009, 15, 4224–4227 CrossRef CAS PubMed; (c) X. Bantreil, G. Prestat, A. Moreno, D. Madec, P. Fristrup, P.-O. Norrby, P. S. Pregosin and G. Poli, Chem. – Eur. J., 2011, 17, 2885–2896 CrossRef CAS PubMed; (d) A. Boutier, C. Kammerer-Pentier, N. Krause, G. Prestat and G. Poli, Chem. – Eur. J., 2012, 18, 3840–3844 CrossRef CAS PubMed.
- L. Constantino and D. Barlocco, Curr. Med. Chem., 2006, 13, 65–85 CrossRef.
- E. M. Beccalli, G. Broggini, G. Paladino, A. Penoni and C. Zoni, J. Org. Chem., 2004, 69, 5627–5630 CrossRef CAS PubMed.
- (a) J. D. Neukom, A. S. Aquino and J. P. Wolfe, Org. Lett., 2011, 13, 2196–2199 CrossRef CAS PubMed; (b) C. F. Rosewall, P. A. Sibbald, D. V. Liskin and F. E. Michael, J. Am. Chem. Soc., 2009, 131, 9488–9489 CrossRef CAS PubMed.
- M. Rigamonti, G. Prestat, G. Broggini and G. Poli, J. Organomet. Chem., 2014, 760, 149–155 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Professor Max Malacria on the occasion of his 65th birthday. |
| ‡ Electronic supplementary information (ESI) available: Experimental details, compound characterization and NMR spectra. See DOI: 10.1039/c4qo00179f |
| This journal is © the Partner Organisations 2014 |