
An efficient one-pot synthesis of N,N′-disubstituted ureas and carbamates from N-acylbenzotriazoles†
Anoop S. Singha,
Dhananjay Kumarb,
Nidhi Mishraa and
Vinod K. Tiwari*a
aDepartment of Chemistry, Centre of Advanced Study, Institute of Science, Banaras Hindu University, Varanasi-221005, India. E-mail: Tiwari_chem@yahoo.co.in
bDepartment of Chemistry, University of Delhi, New Delhi-110007, India
First published on 30th August 2016
Abstract
A facile and high-yielding one-pot synthesis of carbamates and N,N′-disubstituted symmetrical ureas from N-acylbenzotriazoles has been devised. It is believed that, the intermediate acyl-azide undergo Curtius rearrangement and in different solvents gives different products i.e. carbamates in alcohols and N,N′-disubstituted symmetrical urea in THF.
Introduction
Functionalized urea and carbamate motifs are essential structural elements of many biologically active compounds.1 Also, their derivatives play a vital role in the research of pharmaceuticals and organic chemistry.2 Generally, synthesis of substituted urea involves the reaction of suitable amines with urea,3 phosgenation4 and reductive or oxidative carbonylation of amines.5 But these well-established protocols include some drawbacks, such as, insufficiency for symmetric ureas, involvement of highly toxic reagents and longer reaction time. During past few years, a number of papers have reported synthesis of urea and carbamate derivatives via metal catalyst.6 Katritzky et al. have synthesized the symmetric ureas7a,b where the benzotriazole moiety acts as a leaving group and is substituted by amines. This methodology has been further explored in a number of ways by many research groups and contributed thousands of pharmaceutical compounds.8 However, a better yielding, non-toxic, mild and practical approach for synthetic, pharmaceutical as well as industrial significance is still under investigation.In this manuscript, we have introduced N-acylbenzotriazole as a suitable reagent for the preparation of ureas and carbamates via Curtius rearrangement.9 Good leaving tendency of benzotriazole moiety has been used in present work to afford ureas and carbamates without adding amine (Scheme 1).
 | ||
| Scheme 1 Comparative illustration of previous and present work. | ||
Certainly, an N-acyl azide intermediate is formed, which on hydrolysis generates amines in situ. Acyl-azide, on heating, undergoes Curtius rearrangement which leads to formation of corresponding isocyanates. These isocyanates, on reaction with variety of amines (in situ generated by hydrolysis of isocyanates) and/or alcohols, give corresponding ureas and carbamates.
Results and discussion
Our strategy began with the synthesis of core compound N-acyl benzotriazoles by the reaction of corresponding aromatic/aliphatic acids with benzotriazole using SOCl2 reagent in dichloromethane, following the sound known process reported in literature.10 The one-pot reaction procedure completed within two hours and the crude mass was easily purified by flash column chromatography. The corresponding N-acylbenzotriazoles 2a–q (Table 1) were characterized by IR, mass and NMR spectroscopic studies.| a Molar ratios: carboxylic acids (1a–q) (1.0 equiv.), SOCl2 (1.2 equiv.), benzotriazole (3.25 equiv.). Yields reported after purification by column chromatography (SiO2). |
|---|
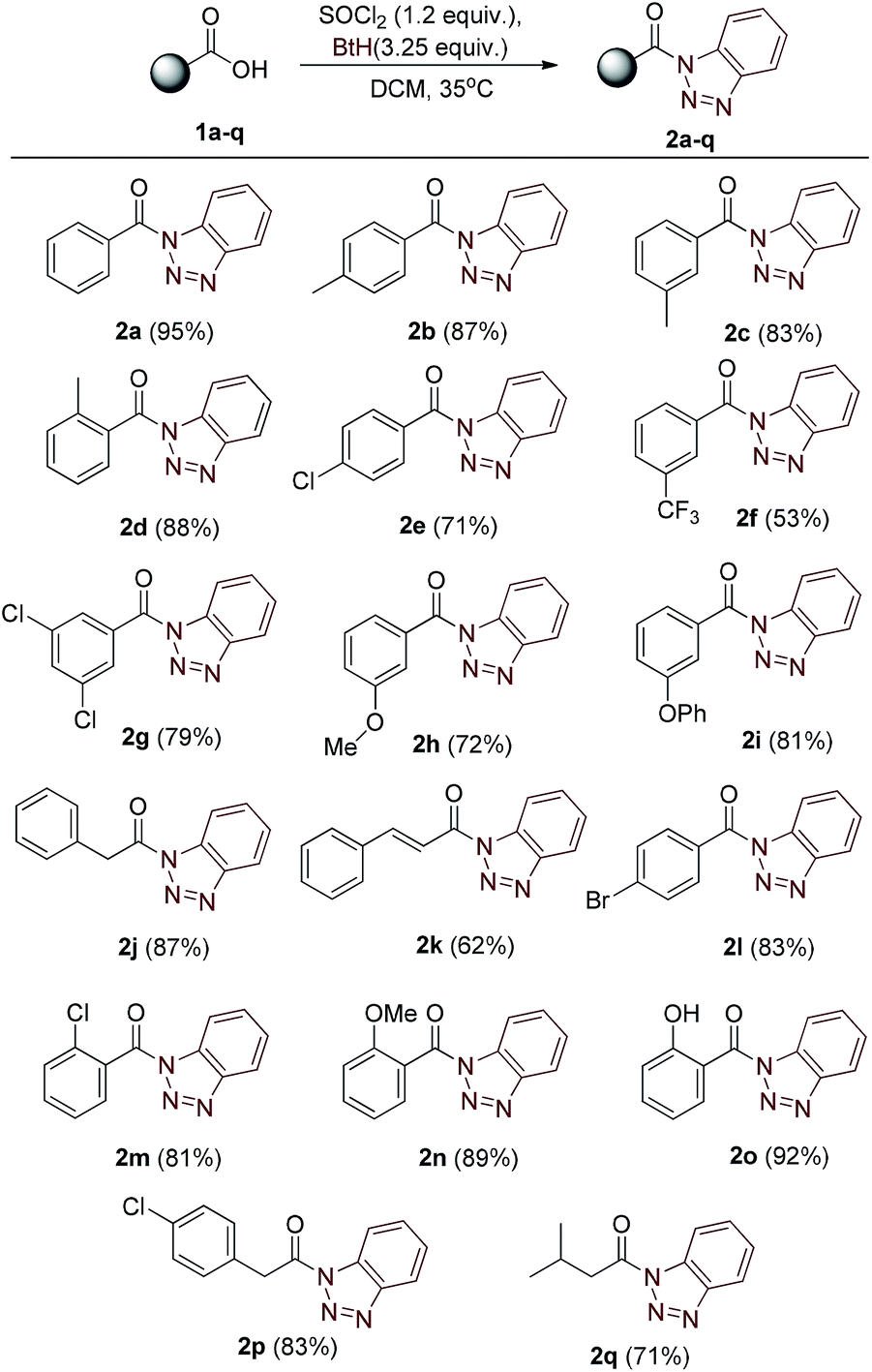 |
The acylated benzotriazole derivatives play a key role in large number of reactions and can be used to synthesize a variety of compounds.11 Formerly, we have also synthesized amide by the help of acylated benzotriazole derivatives in excellent yields.12 Our present work is focussed on further exploration of benzotriazole methodology for Curtius rearrangement, through which synthesis of carbamates and symmetric ureas can be achieved. N-acyl benzotriazole, when refluxed with sodium azide in presence of homogeneous medium of water and protic organic solvent, gives carbamates via isocyanate intermediate, generated in situ when N-acylazides were heated. Similar protocol, when applied in water and organic aprotic solvent system, affords symmetric ureas.
In our proto-type reaction, a mixture of (1H-benzo[d][1,2,3]triazol-1-yl)(phenyl)methanone 2a (2.0 mmol) & sodium azide (3.0 mmol) was refluxed in water–ethanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio mixture) for 3 hours (Scheme 2).
:1 ratio mixture) for 3 hours (Scheme 2).
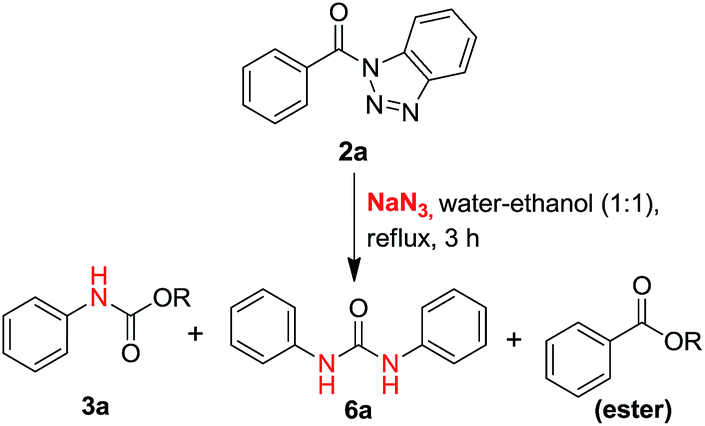 | ||
| Scheme 2 Synthesis of carbamate (3a) and symmetric ureas (6a). | ||
Among the three spots appeared on TLC (10% ethyl acetate/n-hexane) of the reaction mass, first spot (Rf = 0.7) was identified as ester, second spot (Rf = 0.5) was characterized as carbamate and the third spot (Rf = 0.4) was symmetric urea. The ratio of the three products i.e. ester, carbamate and urea, was found to be 1.35:1.61:1 respectively and these compounds have been successfully isolated in pure form after column chromatography (SiO2) using gradient of EtOAc/n-hexane. The formation of carbamate 3a and urea 6a, without externally added amines suggests that Curtius rearrangement must be involved here which gives amines by the rearrangement of acyl azides. Further, in search of appropriate solvent system to obtain optimum yield of our products of interest, i.e. urea and carbamate, we performed the reaction in different type of organic solvents with water in different compositions (Table 2).
|
||||
|---|---|---|---|---|
| Entrya | Solvent systemb | Ratios | Yield of 3ac (%) | Yield of 6ac (%) |
| a Molar ratio: 1H-benzo[d][1,2,3]triazol-1-yl)(phenyl)methanone 2a (2.0 mmol) & sodium azide (6.0 mmol).b Binary solvents.c Yields reported after purification by column chromatography (SiO2). | ||||
| 1 | Ethanol/water | 50/50 | 37 | 23 |
| 2 | Ethanol/water | 85/15 | 64 | 17 |
| 3 | Ethanol/water | 90/10 | 65 | 7 |
| 4 | Ethanol/water | 95/05 | 67 | 5 |
| 5 | Ethanol/water | 100/0 | 20 | 0 |
| 6 | Ethanol/water | 15/85 | 29 | 20 |
| 7 | i-PrOH/water | 50/50 | 41 | 26 |
| 8 | THF/water | 50/50 | 0 | 67 |
| 9 | THF/water | 15/85 | 0 | 18 |
| 10 | THF/water | 70/30 | 0 | 25 |
| 11 | THF/water | 85/15 | 0 | 81 |
| 12 | t-BuOH/water | 50/50 | 0 | 43 |
| 13 | Toluene/water | 50/50 | 0 | 53 |
| 14 | Benzene/water | 50/50 | 0 | 54 |
| 15 | DCM/water | 50/50 | 0 | Trace |
| 16 | Acetone/water | 50/50 | 0 | 57 |
| 17 | Ethyl acetate/water | 50/50 | 0 | 55 |
It was observed that carbamates were formed preferentially when the binary solvent system was a mixture of nucleophilic-protic solvent (generally alcohols) and water (Table 2; entry 1–7). Greater composition of nucleophilic alcohols ensures the capture of intermediate isocyanate leading to formation of carbamate as the major product (Table 2; entry 3 & 4). A 19:1 ratio of alcohol and water was found to be the most reliable solvent system. However, symmetric urea was the major product, when the binary solvent includes a non-nucleophilic-aprotic organic solvent along with water (Table 2; entry 8–17). The carbamate formation was inhibited possibly due to the capture of intermediate isocyanate with aniline in absence of nucleophilic alcohols. The essentiality of water is for the solubility of sodium azide as well as for Curtius rearrangement. Further, to find out optimum ratios of different solvents for the most favourable solvent system to obtain better yields of urea and carbamate separately, the same proto-type reaction was carried out in different solvent ratios as depicted in Table 2 & Fig. 1.
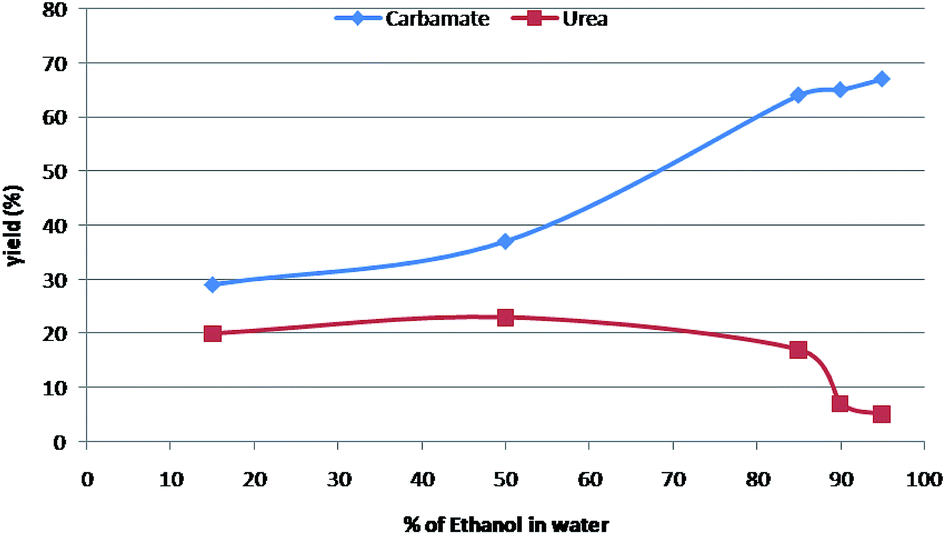 | ||
| Fig. 1 Optimization for carbamate. | ||
After the optimization of solvent system, we investigated the reaction for generalization by varying the alcohol and acylated benzotriazole derivatives. Experimentally, it was found that primary and secondary alcohols (for example, methanol, ethanol, 1-propanol, 1-butanol, 1-pentanol, and isopropyl alcohol) give good yields of carbamates. It was noticed that reaction with tertiary alcohols lead to the formation of symmetric urea as the major product. For example, reaction of compound 2a with sodium azide in presence of t-butanol/water (in 1:1) ratio gave symmetric urea 6a. A library of 20 compounds has been prepared which contains both aliphatic (saturated/unsaturated) and aromatic carbamates in moderate to good yields (Table 3). The yield of carbamate 3l, formed with methanol/water as solvent, is fairly low since methanol undergoes substitution relatively at higher rate than rearrangement. Benzotriazole derivatives of phenyl acetic acid 2j and 2p afforded carbamates 3i, 3s and 3t in good yields. Reaction of bezotriazole derivative of isovaleric acid 2q under similar reaction condition did not produce carbamate product even in trace amount. Compounds have been characterized on the basis of their NMR, mass and IR spectra.
|
|---|
| a Molar ratios: acyl benzotriazoles (1.0 equiv.), NaN3 (3.0 equiv.), alcohols:water (19:1). Yields reported after purification by column chromatography. |
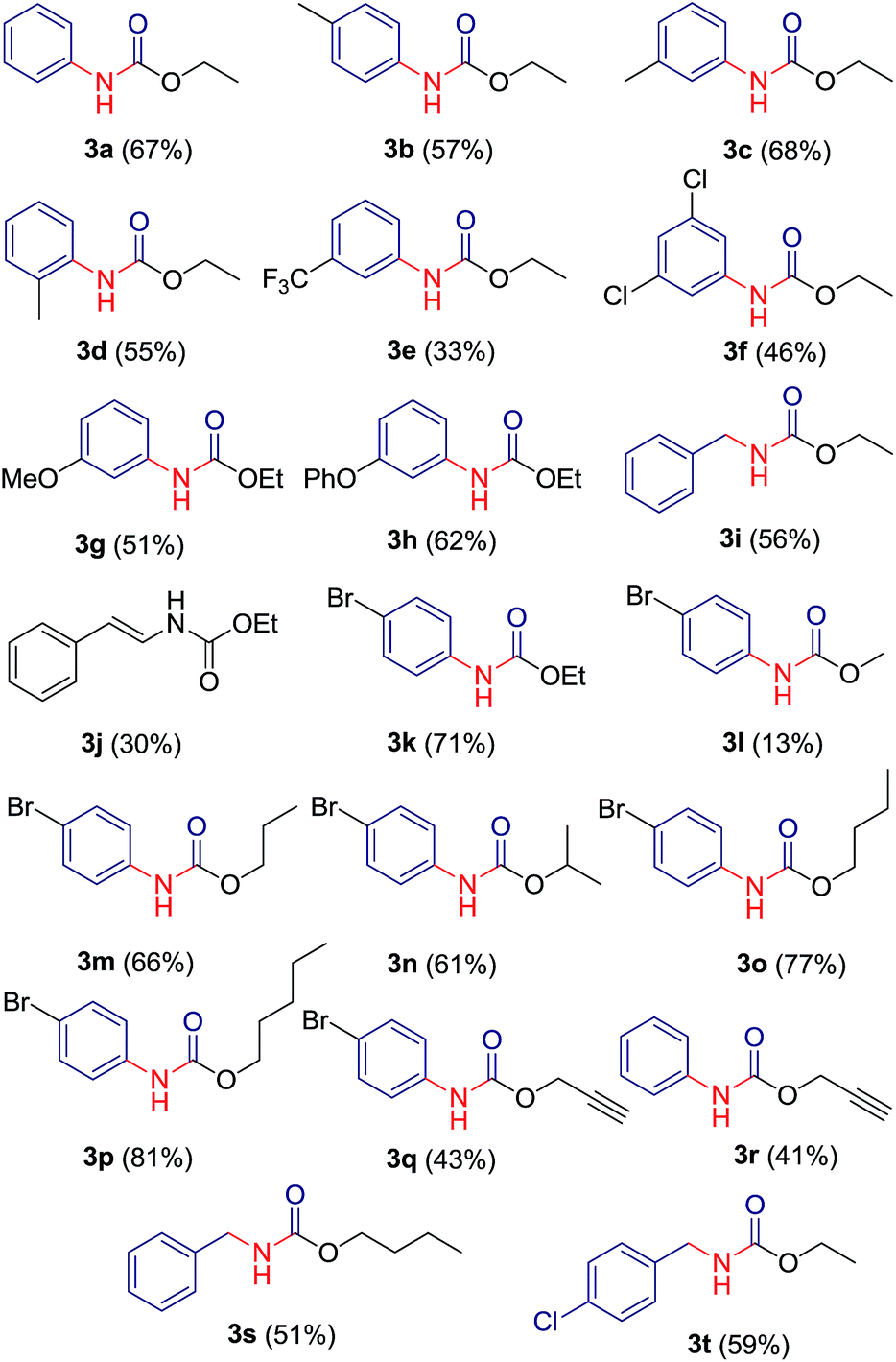 |
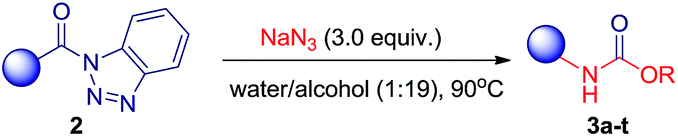
To enhance the generality and utility of this methodology, we further explored the reaction for the synthesis of intramolecular carbamates (Scheme 3). The reaction of compound 2o under above mentioned conditions affords cyclic carbamate 4 via intramolecular cyclization. The intermediate isocyanate formed by Curtius rearrangement must have been captured by hydroxyl at ortho to the carbonyl. The structure of compound 4 has been characterized by NMR, mass and IR spectra. Also, the structure of compound 4 was established by single crystal X-ray analysis. The crystallographic and instrumental details for compound 4 have been summarized in Table 4.
 | ||
| Scheme 3 Preparation of carbamate 4 from corresponding acyl benzotriazole (2o). Molar ratio: acyl benzotriazole (1.0 equiv.), NaN3 (3 equiv.), alcohols/THF:water (8:2). Yields reported after purification by column chromatography (SiO2). | ||
| a For details, see ESI file (CIF) enclosed with manuscript. | |
|---|---|
| Property | 4 |
| Mol. formula | C7H5NO2 |
| Formula weight | 135.12 |
| Crystal system | Orthorhombic |
| Space group | P212121 |
| a (Å) | 4.4436(14) |
| b (Å) | 6.641(2) |
| c (Å) | 20.957(6) |
| α, β, γ (o) | 90, 90, 90 |
| V (Å3) | 618.5(3) |
| Z | 4 |
| Density (calc.) | 1.451 |
| F(000) | 264 |
| μ (mm−1) | 0.108 |
| Crystal size [mm] | 0.14 × 0.15 × 0.22 |
| Temperature (K) | 293 |
| Radiation (MoKα) | 0.71073 |
| θ min–max [o] | 3.22, 24.94 |
| h, k, l | −5:5; −6:89; −16:27 |
| Tot., uniq. data, R(int) | 3269, 1471, 0.0267 |
| Obs. data [I > 2.0 σ(I)] | 1322 |
| Nref, Npar | 1528, 91 |
| R1, wR2, S | 0.0386, 0.0969, 1.061 |
| Min–max resd. dens. [e Å−3] | −0.258, 0.133 |
| CCDC | 1482294 |
During our investigation, it was found that in intramolecular reaction, single and same product is formed with almost 100% conversion, regardless the solvent system we used. However the results are optimum with aprotic polar solvents. Interestingly, when we used thiols instead of alcohol in the reaction, thiocarbamates were obtained in moderate yields (Scheme 4).
 | ||
| Scheme 4 Preparation of thiocarbamates 5a and 5b from corresponding acyl benzotriazole (2a, 2l). Yields reported after purification by column chromatography (SiO2). | ||
Contrary to carbamates, urea was preferably formed in aprotic organic solvent–water system and THF/water medium was found the best in this regards. Further optimization of solvent-system composition and temperature for the reaction revealed that THF/water in 85:15 ratios at 90 °C gave most significant results. We explored this altered reaction path by changing functionality of N-acyl benzotriazole and a series containing seven different symmetric ureas (Table 5) has been developed.
|
|---|
| a Molar ratios: acyl benzotriazoles (1.0 equiv.), NaN3 (3.0 equiv.), THF:water (85:15). Yields reported after purification by column chromatography (SiO2). |
 |

The structures of compounds 6a–g have been characterized by NMR, mass and IR spectra. Also, the structure of compound 6a was established by single crystal X-ray analysis (see ESI CIF file for details†).
Mechanistic consideration
A possible mechanistic way to explicate the product formation and rearrangement is given in mechanistic consideration (Scheme 5). The mechanism certainly involves Curtius rearrangement as the key step of reaction which gives amines by the rearrangement of acyl azides.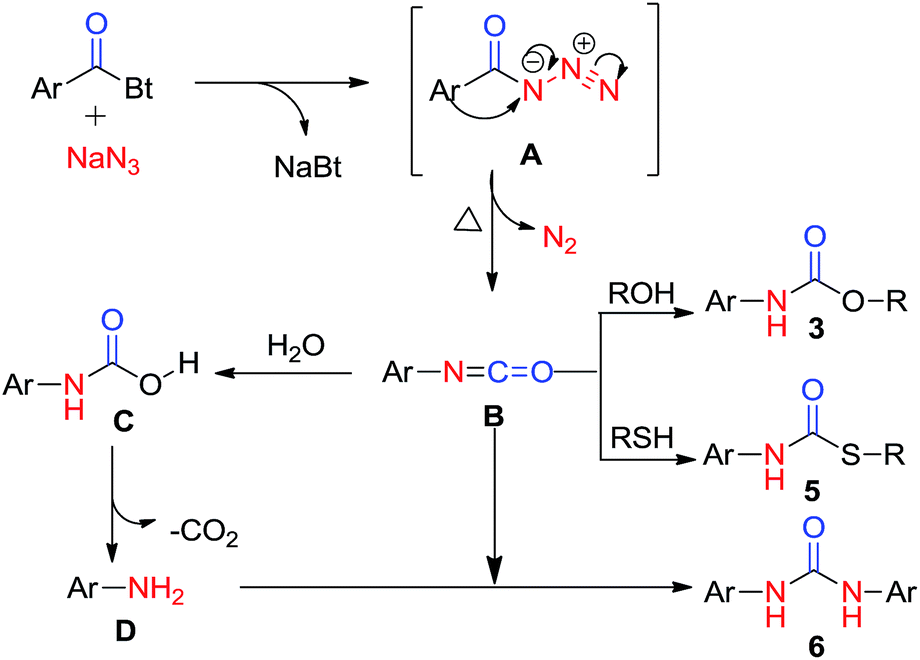 | ||
| Scheme 5 Plausible mechanism involving Curtius rearrangement. | ||
At first, intermediate acyl azide A, formed by attack of azide ion on carbonyl carbon to replace the benzotriazole moiety in acyl benzotriazole, undergoes Curtius rearrangement to form isocyanate intermediate B with the consequent loss of molecular nitrogen (N2). Finally, the intermediate isocyanate is immediately captured by nucleophilic alcoholic solvent to afford carbamate product. Alternatively, isocyanate B generates aniline via carbamic acid, which undergoes decarboxylation to afford anilines. This aniline, on reaction with intermediate isocyanate B, generates symmetric urea.
Combination with click chemistry
With escalating importance of click-chemistry13 as an efficient tool for development of biologically and pharmaceutically relevant compounds, the significance of the methodologies which can easily and effectively afford starting materials for click reaction (i.e. azides and alkynes) has also increased. After successfully employing our methodology in generation of carbamates with a terminal alkyne functionality, we extended this work for cascade synthesis of 1,4-substituted 1,2,3-triazolyl carbohydrate derivatives via click chemistry (Scheme 6). | ||
| Scheme 6 Click-inspired synthesis of novel triazoyl glycoconjugate carbamates (8a–c). Molar ratios: acyl benzotriazoles (1.0 equiv.), NaN3 (6.0 equiv.), tBuOH–water mixture (8:2), sugar azide (1.1 equiv.), CuSO4·H2O (0.02 mol%) and NaAsc (0.2 mol%). Method-I: one-pot sequential reaction; method-II: successive reaction. Yields reported after purification by column chromatography (SiO2). | ||
The reaction involved heating N-acyl benzotriazole with NaN3 in presence of water and propargyl alcohol (as reactant as well as solvent) followed by removal of excess propargyl alcohol by evaporation and addition of sugar azide along with CuSO4 and sodium ascorbate in tBuOH–water mixture (8:2). Though the reaction moved in the way we expected, but the desired triazolyl sugar derivatives were formed with very low yields (8–13%). On the other hand conventional click reaction of developed alkyne-functionalized carbamates (3q and 3r) with sugar azide resulted in excellent yields (88–94%) of triazolyl products. The low yield in cascade approach may be due to some side reactions involved in this process. We are trying to find out the exact causes behind it to improve this reaction for better results.
Conclusions
In summary, we have developed a one-pot, low-toxic and efficient method for synthesis of ureas and carbamates from N-acyl benzotriazole. Through this synthetic route, we have successfully achieved good yields under mild reaction conditions in aqueous medium without using catalyst and with simple purification methods. Furthermore, we have efficiently utilized this methodology for generation of terminal alkyne armed carbamates which were clicked with sugar azides to afford novel triazolyl sugar derivatives. A cascade approach was also attempted for synthesis of triazolyl sugar derivatives by employing this methodology along with click-chemistry, but results were not very pleasing. Work on improvement of cascade approach is ongoing in our laboratory.Experimental
General remarks
All reagents and solvents were of pure analytical grade. Thin-layer chromatography (TLC) was performed on 60 F254 silica gel, pre-coated on aluminium plates and revealed with either a UV lamp (λmax = 254 nm) or a specific colour reagent (Dragendorff reagent or iodine vapour) or by spraying with methanolic H2SO4 solution and subsequent charring by heating at 100 °C. Solvents were evaporated under reduced pressure at temperature <50 °C. Column chromatography was carried out on silica gel (230–400 mesh, E Merck). Distilled n-hexane and ethyl acetate were used for the column chromatography. 1H and 13C NMR were recorded at 300 and 75 MHz, respectively. Chemical shifts are given in ppm downfield from internal TMS; J values in Hz. Mass spectra were recorded using electron spray ionization mass spectrometry (ESI-MS). Elemental analyses were performed on a Perkin-Elmer 2400 C, H, N analyzer and values were found to be within ±0.5% of the calculated values. Infrared spectra recorded as Nujol mulls in KBr plates.Single-crystal X-ray data of compound 6a were collected on Xcalibur Eos (Oxford) CCD-Diffractometer using graphite monochromated MoKα radiation (λ = 0.71073 Å). The data integration and reduction were processed with CrysAlis Pro software.14 Data of compound 4 was collected on Bruker SMART CCD-Diffractometer using graphite monochromated MoKα radiation (λ = 0.71073 Å). The structures were solved by the direct method and then refined on F2 by the full matrix least-squares technique with the SHELX-97 set of software15 using the WinGX (version 1.80.05) program package.16 All non-hydrogen atoms were refined anisotropically and hydrogen atoms were treated as riding atoms using SHELX default parameters. Molecular structures have been drawn using ORTEP software. Further information on the crystal structure (excluding structure factors) has been given in CIF file, Tables S1 & S2 and Fig. S1 & S2 (ESI†) and also deposited in the Cambridge Crystallographic Data Centre as supplementary publication numbers 1482293 (6a) and 1482294 (4).
Procedure for crystallization of compound 6a and 4
For crystallization a mixture of ethyl acetate and hexane (2:8) has been used and kept in dark place at temperature 25 °C. The single crystal appeared after three days was isolated in its initial state of growth.
Typical experimental procedure for synthesis of N-acylbenzotriazoles
Compound 1 (1.0 g, 7.34 mmol) was added to a RB flask containing dichloromethane (15.0 mL) equipped with freshly prepared CaCl2 guard tube and temperature was maintained 0–5 °C. Thionyl chloride (0.6 mL, 8.27 mmol) was added dropwise with vigorous stirring and constant cooling. The reaction mixture was allowed to stir for 15 min then added 1H-1,2,3-benzotriazole (3.06 g, 25.69 mmol) in fraction after complete addition stirred the reaction mixture 2–3 hour at room temperature. After completion of reaction (monitored by TLC), the excess SOCl2 was quenched with ice maintaining the temperature at 0 °C. Extracted with CH2Cl2, washed with 10% Na2CO3, water, and brine solution, the organic layer was dried over anhydrous Na2SO4, concentrated till dry under reduced pressure. Further, purification using flash column chromatography using gradient mixtures of ethyl acetate and n-hexane afforded product 2a–q in pure.Physical data of developed compounds (2a–q)
Typical experimental procedure for the synthesis of carbamates (3a–t)
To a stirring solution of compound 2 (1.0 equiv.) in mixture of alcohol/water (19:1) was added sodium azide (3.0 equiv.) in portions. The reaction was stirred under heating at 90–100 °C for 4 hours. After completion of the reaction (monitored by TLC), the reaction mixture was concentrated in vacuo. The crude reaction mixture was purified directly using flash column chromatography to afford carbamate derivative (3a–t). Purified compounds were recrystalised using CHCl3/n-hexane.
Physical data of developed compounds (3a–t)
Typical experimental procedure for the synthesis of thiocarbamates (5a & 5l)
To a stirring solution of compound 2a & 2l (1.0 equiv.) in mixture of thiol/water (9:1) was added sodium azide (3.0 equiv.) in portions. The reaction was stirred at 100 °C for 4 hours. After completion of the reaction (monitored by TLC), the reaction mixture was concentrated in vacuo. The crude reaction mixture was purified directly using flash column chromatography to afford thiocarbamate derivative (5a & 5b).
Typical experimental procedure for the synthesis of symmetric urea
To a stirring solution of compound 2 (1 equiv.) in mixture of THF/water (85:15) was added sodium azide (3.0 equiv.) in portions. The reaction mixture was stirred at 90–100 °C for 4 hours. After, completion of the reaction (monitored by TLC), the reaction mixture was concentrated in vacuo. The crude reaction mixture was purified directly using flash column chromatography to afford carbamates derivative (6a–g). Purified compounds were recrystallized using ethyl acetate/n-hexane.
Typical experimental procedure for the synthesis of triazolyl sugar derivatives
:1) was added sodium azide (1.2 equiv.) and allow to stir for 10 hours at 90–100 °C. After completion of the reaction (monitored by TLC), then propargyl alcohol was removed and added tBuOH–water (8:2) and allow to stir. Sugar azide (1.1 equiv.) was added in portions followed by addition of CuSO4·H2O (0.02 mol%) and NaAsc (0.2 mol%) and stirred for 4 hours at rt. After completion of reaction the reaction mixture was concentrated in vacuum. The crude reaction mixture was purified directly using flash column chromatography to afford triazolyl derivatives of carbamate (8a–c).:2) was added sugar azide (1.1 equiv.), CuSO4·H2O (0.02 mol%) and NaAsc (0.2 mol%) and stirred for 4 hours at rt. After completion of reaction the reaction mixture was concentrated in vacuum. The crude reaction mixture was purified directly using flash column chromatography to afford triazolyl derivatives of carbamate (8a–c).Acknowledgements
The authors thank Council of Scientific and Industrial Research (CSIR), New Delhi (Grant No. 02(0173)/13/EMR-II) for funding and CISC, Banaras Hindu University for providing basic infrastructure and spectroscopic studies.References
-
(a) T. W. von Geldern, C. Hutchins, J. A. Kester, J. R. Wu-Wong, W. Chiou, D. B. Dixon and T. J. Opgenorth, J. Med. Chem., 1996, 39, 957 CrossRef CAS PubMed
; (b) T. W. von Geldern, J. A. Kester, R. Bal, J. R. Wu-Wong, W. Chiou, D. B. Dixon and T. J. Opgenorth, J. Med. Chem., 1996, 39, 968 CrossRef CAS PubMed
-
(a) H. Irschik, K. Gerth, G. Höfle, W. Kohl and H. Reichenbach, J. Antibiot., 1983, 36, 1651 CrossRef CAS PubMed
- M. A. Pasha and M. B. M. Reddy, Synth. Commun., 2009, 39, 2928 CrossRef CAS
- A. Zhang, Y. Kuwahara, Y. Hotta and A. Tsuda, Asian J. Org. Chem., 2013, 2, 572 CrossRef CAS
-
(a) A. F. Hegarty and L. J. Drennan, in Comprehensive Organic Functional Group Transformations, ed. T. L. Gilchrist, Pergamon Press, Oxford, 1995, vol. 6, p. 499 Search PubMed
-
(a) J. E. McCusker, A. Denise Main, K. S. Johnson, C. A. Grasso and L. McElwee-White, J. Org. Chem., 2000, 65, 5216 CrossRef CAS PubMed
-
(a) A. R. Katritzky, N. Kirichenko and B. V. Rogovoy, ARKIVOC, 2003, viii, 8 Search PubMed
-
(a) A. R. Katritzky, X. Lan, J. Z. Yang and O. V. Denisko, Chem. Rev., 1998, 98, 409 CrossRef CAS PubMed
-
(a) P. A. S. Smith, Org. React., 1946, 3, 337 Search PubMed
- A. R. Katritzky, Y. Zhang and S. K. Singh, Synthesis, 2003, 2795 CrossRef CAS
-
(a) A. R. Katritzky, K. Suzuki and Z. Wang, Synlett, 2005, 1656 CrossRef CAS
- R. R. Kale, V. Prasad and V. K. Tiwari, Lett. Org. Chem., 2010, 7, 136 CrossRef CAS
-
(a) V. K. Tiwari, B. B. Mishra, K. B. Mishtra, N. Mishra, A. S. Singh and X. Chen, Chem. Rev., 2016, 116, 3086 CrossRef CAS PubMed
- CrysAlis Pro, ver. 171.35.21, Agilent Technologies, P. Ltd Search PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837 CrossRef CAS
- A. R. Katritzky, C. Cai and S. K. Singh, J. Org. Chem., 2006, 71, 3375 CrossRef CAS PubMed
- A. R. Katritzky, A. A. Shestopalov and K. Suzuki, Synthesis, 2004, 11, 1806 CrossRef
- A. R. Katritzky, A. V. Vakulenko and R. Jain, ARKIVOC, 2003, xiv, 131 Search PubMed
- X. Wang and Y. Zhang, Tetrahedron, 2003, 59, 4201 CrossRef CAS
- A. R. Katritzky, C. Cai, K. Suzuki and S. K. Singh, J. Org. Chem., 2004, 69, 811 CrossRef CAS PubMed
- A. R. Katritzky, H. Y. He and K. Suzuki, J. Org. Chem., 2000, 65, 8210 CrossRef CAS PubMed
- A. R. Katritzky, S. K. Singh, C. Cai and S. Bobrov, J. Org. Chem., 2006, 71, 3364 CrossRef CAS PubMed
- S. O'Sullivan, E. Doni, T. Tuttle and J. A. Murphy, Angew. Chem., Int. Ed., 2014, 53, 474 CrossRef PubMed
- Z. H. Guan, H. Lei, M. Chen, Z. H. Ren, Y. Bai and Y. Y. Wang, Adv. Synth. Catal., 2012, 354, 489 CrossRef CAS
- Y. Watanabe, Y. Tsuji, R. Takeuchi and N. Suzuki, Bull. Chem. Soc. Jpn., 1983, 56, 3343 CrossRef CAS
- S. Owen, T. Otani, S. Masaoka and T. Ohe, Biosci., Biotechnol., Biochem., 1996, 60, 244 CrossRef CAS PubMed
- A. Chilin, G. Marzaro, S. Zanatta, V. Barbieri and G. Pastorini, Tetrahedron, 2006, 62, 12351 CrossRef CAS
- A. Inesi, V. Mucciante and L. Rossi, J. Org. Chem., 1998, 63, 1337 CrossRef CAS
- C. C. Chen, L. Y. Chen, R. Y. Lin, C. Y. Chu and S. A. Dai, Heterocycles, 2009, 78, 2979 CrossRef CAS
- E. Kianmehr and M. H. Baghersad, Adv. Synth. Catal., 2011, 353, 2599 CrossRef CAS
- N. V. Reddy, K. R. Prasad, P. S. Reddy, M. L. Kantam and K. R. Reddy, Org. Biomol. Chem., 2014, 12, 2172 CAS
- C. Mellin, H. M. Vargas and B. Ringdahl, J. Med. Chem., 1989, 32, 1590 CrossRef CAS PubMed
- C. Stock and R. Brückner, Adv. Synth. Catal., 2012, 354, 2309 CrossRef CAS
- X. Guo, J. Shang, J. Li, L. Wang, Y. Ma, F. Shi and Y. Deng, Synth. Commun., 2011, 41, 1102 CrossRef CAS
- X. Zhang and S. Lu, Synth. Commun., 2007, 37, 3291 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H and 13C NMR for all the developed compounds has been provided. CCDC 1482293 and 1482294. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra14131e |
| This journal is © The Royal Society of Chemistry 2016 |