
A facile and efficient method for the synthesis of alkynone by carbonylative Sonogashira coupling using CHCl3 as the CO source†
Guanglong
Sun
,
Min
Lei
* and
Lihong
Hu
*
State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, P. R. China. E-mail: mlei@simm.ac.cn; lhhu@simm.ac.cn
First published on 15th March 2016
Abstract
A facile and efficient method for the synthesis of alkynones by a Pd-catalyzed carbonylative Sonogashira coupling reaction starting from aryl iodide, terminal alkyne and chloroform (CHCl3) as the CO source is described. This procedure proves that CHCl3 is a cheap and efficient CO source in the presence of CsOH·H2O as the base. Furthermore, it is applied successfully for the modification of natural products, such as vindoline and tabersonin, to obtain the corresponding products in good yields.
Alkynones are important intermediates in various organic syntheses of heterocyclic derivatives due to their multifunctional nature, such as pyrimidines,1,2 quinolones,3 furans,4,5 pyrazoles,6–9 pyrroles,10,11 flavones,12 and benzodiazepines.13 Furthermore, they are also used for the synthesis of many natural products (e.g., azaspiracid,14 calystegine B2,15 anthrapyran metabolite16). In addition, alkynones have been reported to possess diverse pharmacological properties such as antiinflammatory and analgesic activities,17 PKC inhibitors,18 CCR2 antagonists,19 and so on.20–23 Due to the unique structural feature and also to the biological activities of alkynones,14–23 the synthesis of alkynones has received a considerable attention.
Recently, many methods have been reported for the synthesis of alkynones: (1) oxidation of propargylic alcohols;24–27 (2) reaction of acyl chlorides with terminal alkynes;28–34 (3) reaction of nitriles with terminal alkynes;35 (4) reaction of aldehydes with terminal alkynes;36 (5) carbonylative Sonogashira coupling reaction;37–41 (6) other methods.42–46 Among all of the methods mentioned above, carbonylative Sonogashira coupling reaction of aryl iodide, terminal alkyne and CO plays a key role due to its good yields, mild reaction conditions, and excellent functional group tolerance. However, a high CO pressure is always required to achieve efficient carbonylative coupling for the preparation of alkynones. High toxicity and pressure of CO severely limits the using of this method in organic synthesis. In recent years, to avoid using CO directly, several precursors of CO have been reported to replace the CO in organic synthesis.47–51 As shown in Scheme 1, methyldiphenylsilanecarboxylic acid,47,48 phenyl formate49 and 9-methyl-9H-fluorene-9-carbonyl chloride50,51 are applied successfully in organic synthesis as CO precursors. However, the use of these precursors has been limited due to their high prices, especially on a large scale organic synthesis. Therefore, there is a need to search a better precursor of CO for the application in organic synthesis in terms of operational simplicity and economic viability.
 | ||
| Scheme 1 Reported CO precursors. | ||
During the course of our studies, a Pd(II)-mediated carbonylative Sonogashira coupling reaction for the synthesis of alkynones using CHCl3 as the CO precursor in the presence of CsOH·H2O was also discovered.52–55 Herein, we describe a Pd(II)-catalyzed carbonylative Sonogashira coupling reaction of aryl iodide, terminal alkyne and CHCl3 to synthesis of alkynones.
Initially, to exam whether the CHCl3 could be used as CO precursor, we carried out the reaction of iodobenzene 1a, ethynylbenzene 2a, and CHCl3 using Pd(OAc)2 as catalyst, BINAP as ligand and CsOH·H2O as base in toluene. After stirred for 8 h at 80 °C, the desired product 3aa was obtained in 70% yield. This result clearly indicated that CHCl3 could be as CO precursor to synthesis of alkynone. To further improve the efficiency of this reaction, a series of optimization experiments were performed, and the results are summarized in Table 1.
|
|
|||
|---|---|---|---|
| Entry | Ligand | Solvent | Yieldb (%) |
| a Reaction conditions: 1a (0.5 mmol), 2a (0.6 mmol), CHCl3 (1.5 mmol), Pd(OAc)2 (2.5 mol%), ligand (10 mol%), CsOH·H2O (5 mmol), solvent (3 mL), 80 °C, 8 h. b Yields were determined by LC-MS; the number in parentheses refers to the yield of isolated 3aa. | |||
| 1 | — | Toluene | 0 |
| 2 | BINAP | Toluene | 70 |
| 3 | dppf | Toluene | 80 |
| 4 | dppp | Toluene | 76 |
| 5 | PPh3 | Toluene | 96 (91) |
| 6 | DPEphos | Toluene | 85 |
| 7 | CEMTPP | Toluene | 91 |
| 8 | (2-Furyl)3P | Toluene | 50 |
| 9 | o-Phenanthroline | Toluene | 15 |
| 10 | PPh3 | 1,4-Dioxane | 81 |
| 11 | PPh3 | DMF | 0 |
| 12 | PPh3 | MeCN | 0 |
| 13 | PPh3 | EtOH | 5 |
| 14 | PPh3 | THF | 65 |
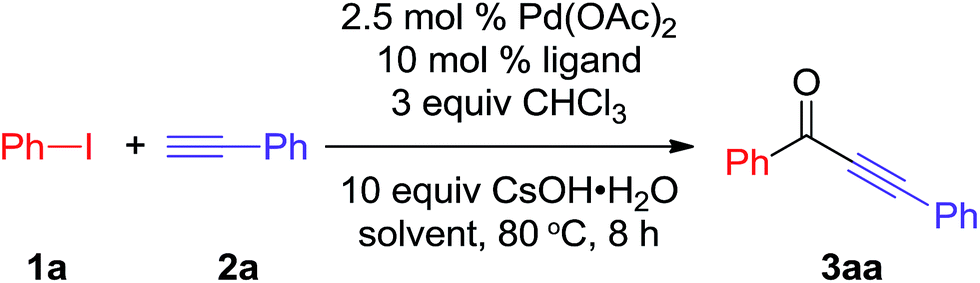
Preliminary experiments suggested that the ligands had a significant impact on the yields of 3aa. Hence, various ligands such as BINAP, dppf, dppp, PPh3, DPEphos, CEMTPP, (2-furyl)3P, and o-phenanthroline were applied to promote this coupling reaction. As shown in Table 1, the reaction could not proceed smoothly to obtain the corresponding product 3aa under ligand-free conditions (Table 1, entry 1). All the ligands studied on this reaction showed good ligand effect in terms of the yields of 3aa (50–96%) but o-phenanthroline (15%). By screening of ligands, PPh3 was found to be the superior one than others to obtained 3aa in 96% yield determined by LC-MS and in 91% isolated yield, respectively. Therefore, PPh3 was chosen as the ligand for all further reactions.
Furthermore, to examine the solvent effect, various solvents such as toluene, 1,4-dioxane, DMF, MeCN, EtOH, and THF were applied to this coupling reaction. As shown in Table 1, the reaction could not proceed smoothly in DMF and MeCN and only 5% yield of 3aa was obtained in EtOH (Table 1, entries 11–13). Moderate to excellent yields were obtained when using toluene, 1,4-dioxane, and THF as the solvent (65–96%) (Table 1, entries 5, 81 and 14), and toluene displayed the best solvent effect.
In addition, the other factors, such as catalyst, base and temperature, were also investigated and these contents were provided in ESI.† Having established the optimized reaction conditions, we then carried out the reaction under the similar conditions using aromatic compounds with different leaving groups, such as Br-, Cl-, TsO-, and TfO-, as substrates to replace iodobenzene. In these cases, the results showed that only 1,4-diphenylbuta-1,3-diyne was obtained in 75% yield, and the desired 3aa was not formed (see ESI†).
To explore the generality and scope of the carbonylative Sonogashira coupling reaction, the reaction of various aryl iodides and phenylacetylene was carried out under the optimized conditions (Table 2). As shown in Scheme 2, ortho, meta and para substituted substrates were chosen to exam the steric hindrance on this reaction. The desired products 3aa–na were obtained in 78–91% yields. These results clearly indicated that the steric hindrance of aryl iodides without significant effect on this carbonylative Sonogashira coupling reaction. Moreover, both electron-rich and electron-deficient aryl iodides could afford the corresponding alkynones 3aa–na in good yields (78–91%).
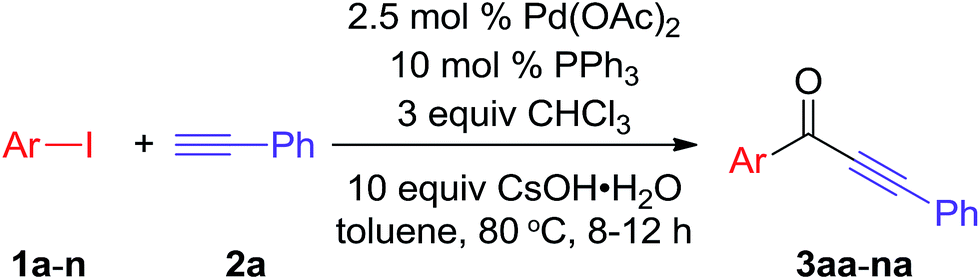

 | ||
| Scheme 2 Reaction of iodobenzene with substituted amino-ethynylbenzene. | ||
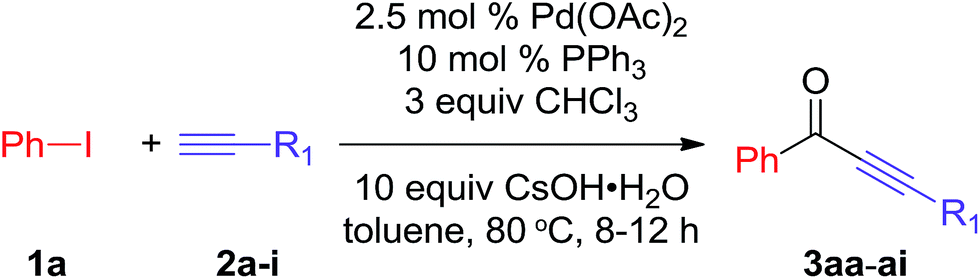
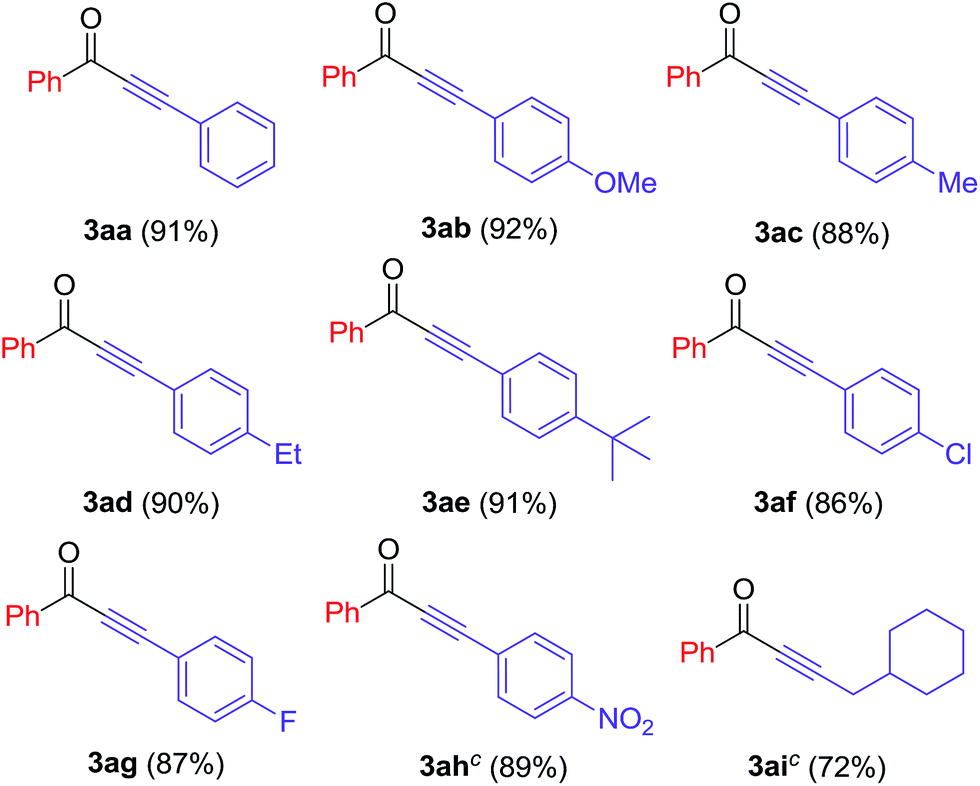
We further investigated the reactions of iodobenzene with various terminal alkynes (Table 3). All of the aryl alkynes were coupled with iodobenzene to afford the alkynones 3aa–ah in good yields (86–92%). The reaction of 3-cyclohexyl-1-propyne with iodobenzene proceeded to yield 72% of the desired product 3ai.
In addition, several substituted amino-ethynylbenzenes (2j, 2k and 2l) were selected for this reaction, and some interesting results were obtained (Scheme 2). Normal product 3aj was formed when using 3-(Boc-amino)-ethynylbenzene 2j as starting material. However, only detrifluoroacetyl product 3ak was obtained under similar conditions when using 3-(trifluoroacetyl-amino)-ethynylbenzene 2j. If the amino without any protection group 2l, both 4aa and 3ak were formed in 80% and 10% yields, respectively. These results provided an alternative approach to synthesis corresponding products as needed.
Furthermore, this carbonylative Sonogashira coupling reaction was also applied in natural products (Scheme 3). At first, vindoline and tabersonin were chosen as starting materials, which were iodinated by NIS to form 15-iodo-vindoline 5 and 10-iodo-tabersonin 6.56,57 Then, the reaction of 5 or 6 with phenylacetylene was carried out under standard conditions for 12 h. The corresponding products 15-alkynone-vindoline 7 and 10-alkynone-tabersonin 8 were obtained in 79% and 75% yields, respectively.
 | ||
| Scheme 3 Reaction of ethynylbenzene with natural products. | ||
Conclusions
In conclusion, we have developed a convenient and efficient procedure for the carbonylative Sonogashira coupling of aryl iodides with terminal alkynes using CHCl3 as the CO precursor. It is noteworthy that this protocol is also suitable for the modification of natural products. The mild reaction conditions, short reaction time, high yields of the products, and compatibility with various functional groups, will make the present method a useful and important addition to the present methodologies for the synthesis of alkynones.Acknowledgements
This work was supported by the National Natural Science Foundation of China (grants 81273397 and 81561148011), the Chinese National Science & Technology Major Project “Key New Drug Creation and Manufacturing Program” (grant 2013ZX09508104).Notes and references
- A. S. Karpov and T. J. J. Müller, Org. Lett., 2003, 5, 3451 CrossRef CAS
.
- D. M. D'Souza and T. J. J. Muller, Nat. Protoc., 2008, 3, 1660 CrossRef
- G. Abbiati, A. Arcadi, F. Marinelli, E. Rossi and M. Verdecchia, Eur. J. Org. Chem., 2009, 7, 1027 CrossRef
- J. Kiji, T. Okano, H. Kimura and K. Saiki, J. Mol. Catal. A: Chem., 1998, 130, 95 CrossRef CAS
- A. V. Kel'in and V. Gevorgyan, J. Org. Chem., 2002, 67, 95 CrossRef
- J. D. Kirkham, S. J. Edeson, S. Stokes and J. P. A. Harrity, Org. Lett., 2012, 14, 5354 CrossRef CAS
- D. B. Grotjahn, S. Van, D. Combs, D. A. Lev and C. Schneider, J. Org. Chem., 2002, 67, 9200 CrossRef CAS
- H. Liu, H. Jiang, M. Zhang, W. Yao, Q. Zhu and Z. Tang, Tetrahedron Lett., 2008, 49, 3805 CrossRef CAS
- B. Willy and T. J. J. Müller, ARKIVOC, 2008, 1, 195 Search PubMed
- J. H. Shen, G. L. Cheng and X. L. Cui, Chem. Commun., 2013, 49, 10641 RSC
- A. V. Kel'in, A. W. Sromek and V. Gevorgyan, J. Am. Chem. Soc., 2001, 123, 2074 CrossRef
- K. Sakamoto, E. Honda, N. Ono and H. Uno, Tetrahedron Lett., 2000, 41, 1819 CrossRef CAS
- B. Willy, T. Dallos, F. Rominger, J. Schönhaber and T. J. Müller, Eur. J. Org. Chem., 2008, 28, 4796 CrossRef
- C. J. Forsyth, J. Xu, S. T. Nguyen, I. A. Samdai, L. R. Briggs, T. Rundberget, M. Sandvik and C. O. Miles, J. Am. Chem. Soc., 2006, 128, 15114 CrossRef CAS
- J. Marco-Contelles and E. Opazo, J. Org. Chem., 2002, 67, 3705 CrossRef CAS
- L. F. Tietze, R. R. Singidi, K. M. Gericke, H. Bockemeier and H. Laatsch, Eur. J. Org. Chem., 2007, 35, 5875 CrossRef
- P. N. P. Rao, Q.-H. Chen and E. E. Knaus, J. Med. Chem., 2006, 49, 1668 CrossRef CAS
- A. Takashima, B. English, Z. H. Chen, J. X. Cao, R. T. Cui, R. M. Williams and D. V. Faller, ACS Chem. Biol., 2014, 9, 1003 CrossRef CAS
- C. Z. Cai, D. F. McComsey, C. F. Hou, J. C. O'Neill, E. Opas, S. McKenney, D. Johnson and Z. H. Sui, Bioorg. Med. Chem. Lett., 2014, 24, 1239 CrossRef CAS
- C. François-Endelmond, T. Carlin, P. Thuery, O. Loreau and F. Taran, Org. Lett., 2010, 12, 40 CrossRef
- N. Wu, A. Messinis, A. S. Batsanov, Z. Yang, A. Whiting and T. B. Marder, Chem. Commun., 2012, 48, 9986 RSC
- A. S. Karpov, E. Merkul, F. Rominger and T. J. Müller, Angew. Chem., Int. Ed., 2005, 44, 6951 CrossRef CAS
- M. Ono, H. Watanabe, R. Watanabe, M. Haratake, M. Nakayama and H. Saji, Bioorg. Med. Chem. Lett., 2011, 21, 117 CrossRef CAS
- K. C. Weerasiri and A. E. V. Gorden, Eur. J. Org. Chem., 2013, 1546 CrossRef CAS
- L. Wang, J. Li, H. Yang, Y. Lv and S. Gao, J. Org. Chem., 2012, 77, 790 CrossRef CAS
- Y. Zhu, B. Zhao and Y. Shi, Org. Lett., 2013, 15, 992 CrossRef CAS
- C. Han, M. Yu, W. Sun and X. Yao, Synlett, 2011, 22, 2363 Search PubMed
- L. Chen and C. Li, Org. Lett., 2004, 6, 3151 CrossRef CAS
- B. Wang, M. Bonin and L. Micouin, J. Org. Chem., 2005, 70, 6126 CrossRef CAS
- K. Y. Lee, M. J. Lee and J. N. Kim, Tetrahedron, 2005, 61, 8705 CrossRef CAS
- R. J. Cox, D. J. Ritson, T. A. Dane, J. Berge, J. P. H. Charmant and A. Kantacha, Chem. Commun., 2005, 1037 RSC
- S. J. Yim, C. H. Kwon and D. K. An, Tetrahedron Lett., 2007, 48, 5393 CrossRef CAS
- M. Navidi, B. Movassagh and S. Rayati, Appl. Catal., A, 2013, 452, 24 CrossRef CAS
- W. J. Sun, Y. Wang, X. Wu and X. Q. Yao, Green Chem., 2013, 15, 2356 RSC
- H. Ding, C. Lu, X. Hu, B. Zhao, B. Wu and Y. Yao, Synlett, 2013, 24, 1269 CrossRef CAS
- J. W. Yuan, J. Wang, G. H. Zhang, C. Liu, X. T. Qi and Y. Lan, Chem. Commun., 2015, 51, 576 RSC
- X.-F. Wu, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 11142 CrossRef CAS
- C. Bai, S. Jian, X. Yao and Y. Li, Catal. Sci. Technol., 2014, 4, 3261 RSC
- T. Tang, X.-D. Fei, Z.-Y. Ge, Z. Chen, Y.-M. Zhu and S.-J. Ji, J. Org. Chem., 2013, 78, 3170 CrossRef CAS
- C. Zhang, J. Liu and C. Xia, Org. Biomol. Chem., 2014, 12, 9702 RSC
- J. Liu, X. Peng, W. Sun, Y. Zhao and C. Xia, Org. Lett., 2008, 10, 3933 CrossRef CAS
- D. K. Friel, M. L. Snapper and A. H. Hoveyda, J. Am. Chem. Soc., 2008, 130, 9942 CrossRef CAS PubMed
- M. M. Jackson, C. Leverett, J. F. Toczko and J. C. Roberts, J. Org. Chem., 2002, 67, 5032 CrossRef CAS
- L. Anastasia and E. Negishi, Org. Lett., 2001, 3, 3111 CrossRef CAS
- W. Kim, K. Park, A. Park, J. Choe and S. Lee, Org. Lett., 2013, 15, 1654 CrossRef CAS
- H. Wang, L. N. Guo, S. Wang and X. Y. Duan, Org. Lett., 2015, 17, 3054 CrossRef CAS
- S. D. Friis, R. H. Taaning, A. T. Lindhardt and T. Skrydstrup, J. Am. Chem. Soc., 2011, 133, 18114 CrossRef CAS
- C. Lescot, D. U. Nielsen, I. S. Makarov, A. T. Lindhardt, K. Daasbjerg and T. Skrydstrup, J. Am. Chem. Soc., 2014, 136, 6142 CrossRef CAS
- S. P. Chavan and B. M. Bhanage, Eur. J. Org. Chem., 2015, 11, 2405 CrossRef
- P. Hermange, A. T. Lindhardt, R. H. Taaning, D. Lupp and T. Skrydstrup, J. Am. Chem. Soc., 2011, 133, 6061 CrossRef CAS
- S. L. Buchwald, T. Skrydstrup and S. D. Friis, Org. Lett., 2014, 16, 4296 CrossRef
- S. N. Gockel and K. L. Hull, Org. Lett., 2015, 17, 3236 CrossRef CAS
- V. V. Grushin and H. Alper, Organometallics, 1993, 12, 3846 CrossRef CAS
- M. Xia and Z. C. Chen, J. Chem. Res., 1999, 5, 328 RSC
- Z. Li and L. Wang, Adv. Synth. Catal., 2015, 357, 3469 CrossRef CAS
- P. D. Johnson, J. H. Sohn and V. H. Rawal, J. Org. Chem., 2006, 71, 7899 CrossRef CAS
- G. Lewin, Y. Rolland and J. Poisson, Heterocycles, 1980, 14, 1915 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, copies of the 1H and 13C NMR spectra. See DOI: 10.1039/c6ra02424f |
| This journal is © The Royal Society of Chemistry 2016 |