
Palladium nanoparticles immobilized on an amine-functionalized MIL-101(Cr) as a highly active catalyst for oxidative amination of aldehydes†
Mrinal Saikia ab and
Lakshi Saikia*a
ab and
Lakshi Saikia*a
aMaterials Science Division, CSIR-North East Institute of Science and Technology, Jorhat–785006, Assam, India. E-mail: l.saikia@gmail.com; Fax: +91 376 2370 011; Tel: +91 376 2370 081
bAcademy of Scientific and Innovative Research, Chennai, India
First published on 28th January 2016
Abstract
The present work reports the synthesis of well dispersed palladium nanoparticles immobilized on an amino functionalized nanoscale metal–organic framework MIL-101(Cr), by using a facile polyol method. The resulting Pd/NH2-MIL-101(Cr) nanocomposite is established to be an active heterogeneous catalyst for oxidative amination of aldehydes under solvent free conditions using hydrogen peroxide as an oxidant. The synthesized Pd/NH2-MIL-101(Cr) is characterized by powder X-ray diffraction, X-ray photoelectron spectroscopy, transmission electron microscopy, field emission scanning electron microscopy, Brunauer–Emmett–Teller (BET) surface area analysis, Fourier transform infrared spectroscopy (FT-IR), thermogravimetric analysis (TGA) and atomic absorption spectroscopy (AAS). The catalyst could easily be recovered and recycled without any significant loss of its catalytic activity.
Introduction
Palladium nanoparticles (Pd Nps) have been attractive in recent years owing to their unique properties of demonstrating exceptional performance in hydrogen sensing and hydrogen storage,1,2 Suzuki and Stille coupling reactions,3–5 CO oxidation,6 electron oxidation of formic acid,7 glucose oxidation,8 hydrogenation of unsaturated alcohols,9,10 alkene hydrogenation11 etc. The catalytic activities of Pd Nps are highly dependent on their size and shape. Therefore, the synthetic protocol of this nanoparticle always has an effect on its size and shape and thus its catalytic activity. Among different methods, the ‘polyol method’ is one of the preferred methods of synthesis of Pd Nps where the synthesis of nanoparticles has been carried out at elevated temperature in polyol solvents. By virtue of its reductive character at elevated temperature, polyol can instantaneously reduce dissolved metal cations to readily form metal nanoparticles with sufficient stabilization and surface functionalization. Here, polyol serves as both the solvent and the reducing reagent and that make this method as an energy efficient and environmentally benign process. In fact, the term ‘polyol synthesis’ was first coined by Fievet, Lagier and Figlarz12 in 1989 for the use of polyols for the synthesis of small particles. It was first started with the metals like Co, Ni, Cu, and Pt particles1 and was further extended to other metals as well as intermetallic and alloys.Amides are found to be the important chemical compounds, particularly in organic chemistry as these are the key building blocks of natural products, polymers both natural as well as synthetic, pharmaceuticals,13 many important drugs, agrochemical products, synthetic intermediates for fine chemical industry etc. Moreover these amides exhibit a wide array of important biological activities.14 The basic route of amide formation involves the acylation of amines with carboxylic acid derivatives has the drawbacks of producing a stoichiometric amount of waste product and of using highly hazardous reagents.15 In order to circumvent these problems, great efforts have been committed to develop methods for amide synthesis include Staudinger reaction,16 the Schmidt reaction,17 the Beckmann rearrangement,18 aminocarbonylation of haloarenes,19 iodonium-promoted α-halo nitroalkane amine coupling,20 direct amide synthesis from alcohols with amines or nitroarenes,21 hydroamination of alkynes,22 amidation of thioacids with azides,23 and trans-amidation of primary amides.24 However, the way of formation of amides from the oxidative amidation of aldehydes with amines is always a preferred protocol as the process is more atom economic that exhibits the use of cheap and abundant starting materials. Although reports are available for transition metal catalyzed oxidative amination of aldehydes,25–31 yet a few reports are accessible for palladium metal in particular,32–34 and most of the protocols have some drawbacks like low product yield, using of excess amount of catalyst, harsh reaction conditions, employing of expensive reagent etc. Furthermore, most of the oxidative amination reactions are performed in homogeneous catalytic system which again suffers from catalyst separation from the reaction mixture, multistep preparation, as well as less cost effective. Conversely, heterogeneous catalytic system has the advantage of facile recovery, regeneration and reuse of the catalyst which make the process significant from the environmental perspective.
Metal–organic frameworks (MOFs) have been appearing as very promising functional materials for separation, gas storage, heterogeneous catalysis, drug delivery, and sensing by virtue of their high surface area, porosity and tuneable chemical properties.35–38 These distinctive features make MOFs very fascinating in the field of heterogeneous catalysis. Particularly, metal–organic framework of MIL-101(Cr) type is found to be a good candidate as heterogeneous catalyst for various important organic transformations. It plays its role as catalytic support39 owing to its high surface area, hydrothermal stability and chemical robustness. Several available preparative methods for the synthesis of palladium nanoparticles on MIL-101(Cr) have been reported like anion exchange,40 incipient wetness impregnation,41–44 impregnation,45–48 colloidal deposition,27,49 infiltration,50 chemical vapour deposition51 etc. and the resulting Pd@MIL-101(Cr) has been employed in various important organic transformation.52–65 Apart from MIL-101(Cr), other metal–organic framework like UiO-66 type,66–69 MIL-53 (ref. 63 and 70) can also be act as host matrix for encapsulating palladium nanoparticles. Keep that in mind we have developed a facile method of synthesis of palladium nanoparticles on nanoscale NH2-MIL-101(Cr) by a polyol approach where a multivalent alcohol is used to dissolve the metal precursor followed by reduction of the metal at reflux condition.71
Reports are also available for polyol synthesized palladium nanoparticles.72–74 Recently, Wen-Bin Cai75 and his co-workers have synthesized Pd–Cu/C electrocatalyst by one-pot polyol reduction method and study its catalytic activity towards formic acid oxidation. R. K. Sharma et al. developed a silica nanospheres-supported palladium nano-catalyst for oxidative amination of aldehydes.76 In this context, we have synthesized a heterogeneous palladium catalyst by immobilizing palladium nanoparticles on an amine functionalized metal–organic framework (MOF), i.e., NH2-MIL-101(Cr) and study its catalytic activity towards the oxidative amination of aldehydes under solvent free condition. Here MOF act as a catalytic support that stabilizes the metal nanoparticles with adjustable sizes owing to its high surface area and well defined pore structure. To the best of our knowledge, up to now, no reports have been available employing NH2-MIL-101(Cr) as support for Pd NPs in oxidative amination of aldehydes.
Therefore, we have adopted the polyol method of synthesis of Pd Nps on NH2-MIL-101(Cr) and characterizing the resulting Pd/NH2-MIL-101(Cr) thorough different analytical techniques like PXRD, FE-SEM, TEM, N2 physisorption, XPS, FT-IR and TGA. We have employed hydrogen peroxide (H2O2) as an oxidant for the aforementioned chemical transformation as it is an inexpensive oxidant that gives only water as the sole by product. Hence, facile synthetic route, recovery and reusability of the catalyst, mild reaction condition, using of hydrogen peroxide (H2O2) as oxidant, solvent free condition and high product yields are the prime advantages of our protocol.
Experimental section
Materials and methods
All chemicals are analytical grade and used as purchased without further purification. Cr(NO3)3·9H2O and 2-aminoterephthalic acid, are purchased from Sigma-Aldrich, USA. Palladium(II) acetate is purchased from Himedia. Ethylene glycol, C2H5OH, H2O2 and DMF are purchased from Merck, Germany. All the used aldehydes and amines are purchased from Sigma-Aldrich.The X-ray diffraction (XRD) patterns of the samples are performed on Rigaku, Ultima IV X-ray diffractometer using Cu-Kα source (λ = 1.54 Å). Field-emission scanning electron microscopy (FE-SEM, SIGMA) is applied to investigate the size and morphology of the sample and EDS mapping was done in Oxford XMax 20 equipment. Specific surface area, pore volume, average pore diameter are measured with the Autosorb-1 (Quantachrome, USA). X-ray photoelectron spectroscopy is carried out on VG Microtech Multilab ESCA 3000 equipment with a non-monochromatized Mg Kα radiation (hν = 1253.6 eV). The size and morphology of the nanoparticles are observed by transmission electron microscopy (TEM) using JEOL (Model JEM-2011) equipment. The Pd content of the catalyst is determined through Atomic absorption spectroscopy (AAS) measurement using PerkinElmer (Model-AAnalyst-700) spectrometer. FT-IR spectra (4000–400 cm−1) are recorded on KBr discs in a Perkin-Elmer system 2000 FT-IR spectrophotometer. TGA analysis of the material has been carried out in a TA SDT-Q600 instrument under N2 atmosphere. 1H and 13C NMR spectra are recorded in a AV-500 Avance-III 500 MHz FT-NMR spectrometer using CDCl3 as a solvent. Mass spectra are recorded in a Mass spectrometer (Model: Trace DSQ GCMS Instument).
Catalyst preparation
The synthesis of amine functionalized MIL-101(Cr) is carried out according to the method described by Chen et al.77 Cr(NO3)3·9H2O (2 mmol), 2-aminoterephthalic acid (2 mmol) and sodium hydroxide (5 mmol) are dispersed in 15 ml of de-ionized water. After stirring for 5 minutes, the reaction solution is placed in a 100 ml Teflon-lined autoclave and heated at 150 °C for 12 h. After cooling to room temperature, the green precipitate is collected by centrifugation, washed it with DMF to remove the unreacted 2-aminoterephthalic acid and then further purified by solvothermal treatment in ethanol at 100 °C for 24 h. Finally, the product is dried at 80 °C in air.For preparing Pd/NH2-MIL-101(Cr), 0.5 g of activated NH2-MIL-101(Cr) is dispersed in 30 ml of ethylene glycol containing 0.0105 g of palladium(II) acetate in a 50 ml round bottom flask. The solution is then continuously stirred under reflux condition at 150 °C for 8 h. The colour of the resulting solid is changed from light green colour to dark green. The solid is then separated through centrifugation, repeatedly washed with ethanol and then dried under vacuum. The resulting solid thus prepared is designated as Pd/NH2-MIL-101(Cr). The Pd content (0.62 wt%) in the synthesized material has been calculated from AAS technique.
General procedure of the catalytic experiments
A mixture of aromatic aldehyde (1 mmol), aromatic amine (1 mmol), hydrogen peroxide (2 mmol) are refluxed at 60 °C in the presence of Pd/NH2-MIL-101(Cr) catalyst (0.094 mol%) for a desired reaction time. After the reaction is completed (monitored by TLC), the reaction mixture is allowed to cool to room temperature and then extracted it with ethyl acetate. The catalyst is recovered through centrifugation, washed it with acetone and dried. The organic layer is then dried over anhydrous Na2SO4 and the solvent is removed under vacuum through Rotavapor. The pure products are obtained by column chromatography on silica gel with hexane/ethyl acetate as eluent.Result and discussion
The chelating effect of the polyols controls the particles nucleation, growth and agglomeration of the nanoparticles by adhering on their surface and serve as colloidal stabilizers. In addition, high boiling point of the polyols makes the process much more advantageous as no high pressure or autoclave is required. Because of the water comparable polarity and solubility of polyols simple metal salts can be used as starting material rather than using volatile and reactive carbonyl metals or advanced organometalic compounds. Other advantages of this method include the formation of simple organic by products different from the residues of other reducing agents like borane derivatives,78 simple and reproducible procedure, small particle size and narrow particle size distribution showing high catalytic activity etc. Therefore, polyol can be considered as biocompatible, green, biodegradable and sustainable solvents which are also highly applicable for industrial synthesis of nanoparticles via polyol synthesis.The powder X-ray diffraction (PXRD) pattern of the NH2-MIL-101(Cr) (Fig. 1) clearly indicates the typical nanoscale MOF with broad Bragg reflections caused by the small nanometer range metal–organic frameworks.79 After the incorporation of palladium nanoparticles, the structure of the parent NH2-MIL-101(Cr) is mostly retained as demonstrated by the FE-SEM image (Fig. S1, ESI†) as well as the similar X-ray diffraction pattern of Pd/NH2-MIL-101(Cr) to that of the support which clearly indicates the preservation of the crystallinity of parent MOF. The incorporation of the well dispersed Pd Nps on NH2-MIL-101(Cr) is also illustrated by the TEM images (Fig. 2). The SAED pattern (Fig. 2(f)) clearly shows the crystallinity of Pd nanoparticles on NH2-MIL-101(Cr). The corresponding oxygen, carbon, nitrogen, chromium and palladium element mapping (Fig. 3(b)) and EDS data (Fig. 3(a)) further confirms the presence of different elements in Pd/NH2-MIL-101(Cr).
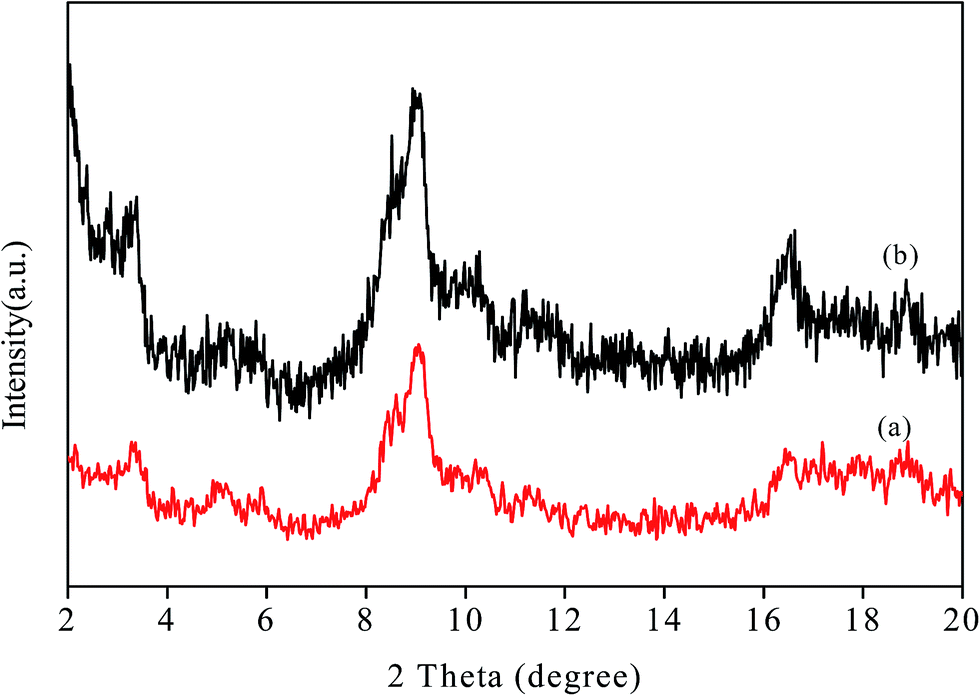 | ||
| Fig. 1 PXRD pattern of (a) MIL-101(Cr), (b) Pd/NH2-MIL-101(Cr). | ||
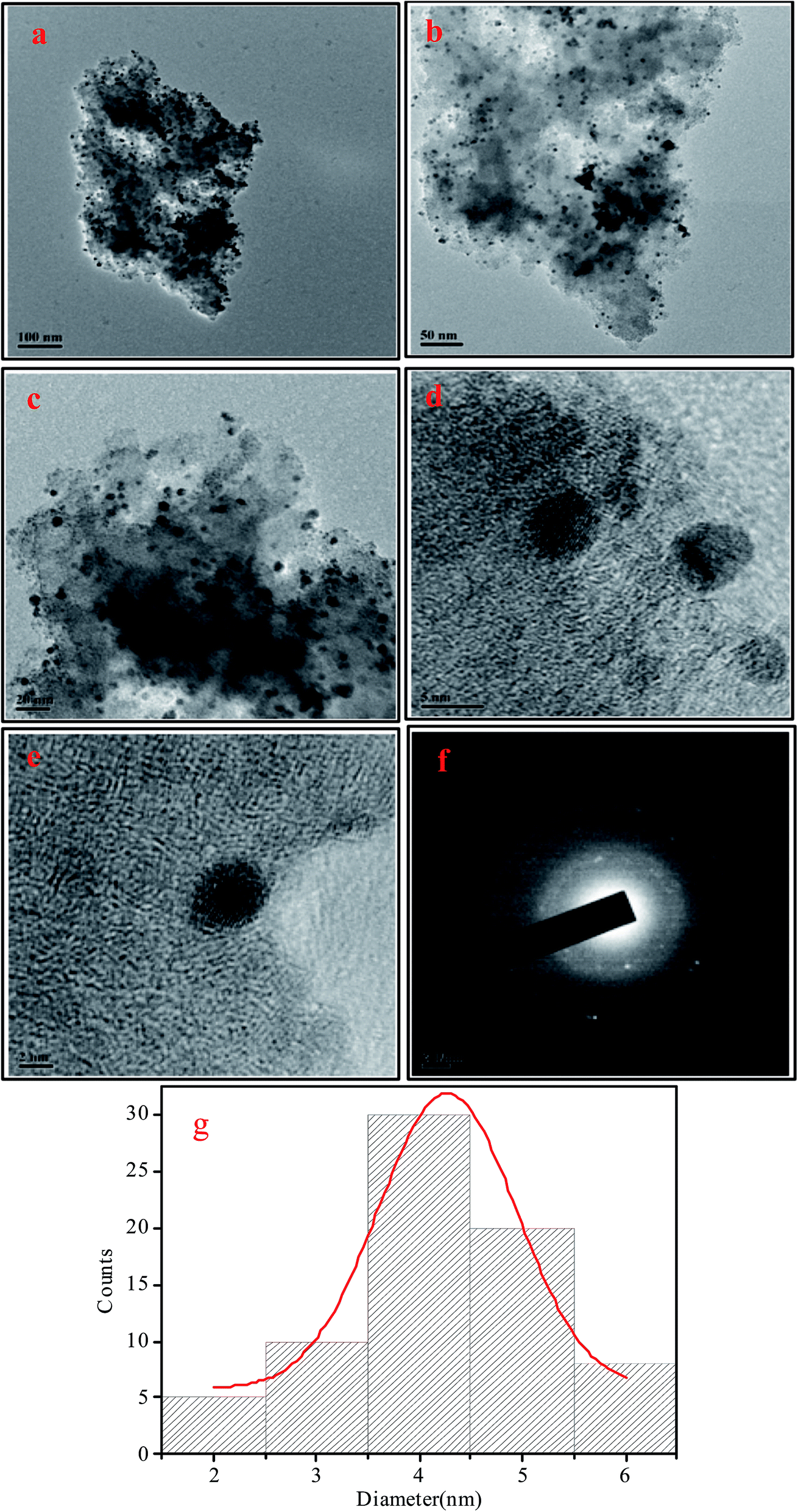 | ||
| Fig. 2 (a–e) TEM images of Pd/NH2-MIL-101(Cr), (f) SAED image of the NPs, (g) size distribution of Pd Nps. | ||
 | ||
| Fig. 3 (a) EDS analysis of Pd/NH2-MIL-101(Cr), (b) mapping of different elements of Pd/NH2-MIL-101(Cr). | ||
The specific surface area, pore size and pore volume of the composite material is measured by N2 physisorption analysis at 77 K (Fig. S2, ESI†). Both the Pd/NH2-MIL-101(Cr) and the parent material exhibit a typical completely reversible isotherm. The Brunauer–Emmett–Teller (BET) surface area (SBET) of NH2-MIL-101(Cr) and Pd/NH2-MIL-101(Cr) are found to be 1502 m2 g−1 and 1433 m2 g−1 with pore volumes (Vp) 1.674 cm−3 g−1 and 1.396 cm−3 g−1 respectively. After incorporating the palladium nanoparticles there is a decrease in the nitrogen adsorption amount and the surface area demonstrating that the cavities of NH2-MIL-101(Cr) are occupied either by the highly dispersed Pd nanoparticles or the nanoparticles may blocked the pores of the parent MOF. FT-IR spectra of the parent NH2-MIL-101(Cr) (Fig. S3(c), ESI†) exhibit double peaks at 3463 and 3397 cm−1, which can be ascribed to the asymmetrical and symmetrical stretching of the amine moieties.80,81 Two other characteristic bands of the amino group can be interpreted as the N–H bending (scissoring) vibration observed at 1624 cm−1 and the C–N stretching absorption typical of aromatic amines at 1338 cm−1.81 Like the parent one, Pd/NH2-MIL-101(Cr) nanocatalyst also shows vibrational bands in the N–H regions (Fig. S3(a), ESI†) that clearly indicate the presence of amino functionality in the resulting material after incorporating the palladium nanoparticles onto the matrix of NH2-MIL-101(Cr). Here, the amino group in the linkers of NH2-MIL-101(Cr) acts as stabilizers that provides a better dispersion for the nanoparticles on to the support as compared to that of the unfunctionalized MIL-101(Cr). The thermogravimetric analysis (TGA) of the synthesized Pd/NH2-MIL-101(Cr) (Fig. S4, ESI†) has been carried out to evaluate the thermal stability of the material. Two major weight loss step has been observed, the first step in the range 30–180 °C (calculated loss: 6%) is due the loss of water molecules and the second step in the range 180–500 °C (calculated loss: 74%) may be attributed to the framework decomposition which is well supported by the corresponding derivative curve.
X-ray photoelectron spectroscopy (XPS) (Fig. 4) indicates that Pd(0), Cr(III) and N coexist in the Pd/NH2-MIL-101(Cr) material. The Cr 2p3/2 and N 1s binding energies of 577.4 eV and 399.3 eV were designated for Cr–O and Ph-NH2 while the peaks appear at 336.7 eV and 341.9 eV were assigned to 3d5/2 and 3d3/2 peaks of Pd(0) with no obvious peak of Pd(II) that clearly shows the presence of palladium in the reduced form.37
 | ||
| Fig. 4 XPS spectrum of different elements of Pd/NH2-MIL-101(Cr). | ||
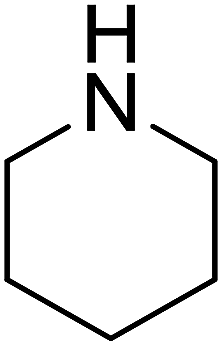
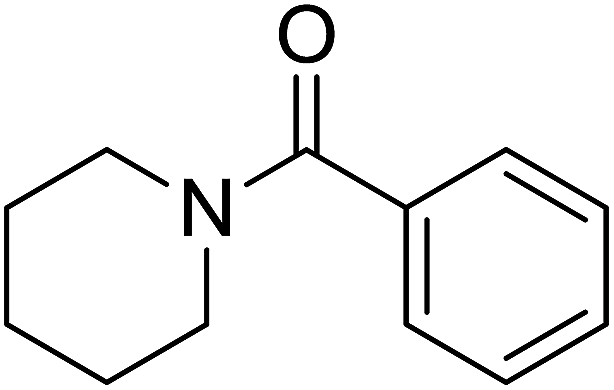
The catalytic activity of the prepared Pd/NH2-MIL-101(Cr) is explored in the direct transformation of aldehydes to amides. The effect of different mol% of the catalyst is studied by taking the benzaldehyde and piperidine as test substrate and hydrogen peroxide as the oxidant. The results are summarized in Table 1. The reaction proceed with a slower rate with neat NH2-MIL-101(Cr) (Table 1, entry 1) which clearly indicates the need of the active Pd centre to carry out the chemical transformation. On increasing the mol% of the catalyst the product yield increases, owing to the availability of more dispersed active catalytic sites on the catalyst surface. However, increase in the catalyst amount (Table 1, entry 5) beyond 0.094 mol% does not give any significant increase in the product yield. Therefore, 0.094 mol% Pd catalysts has been taken as an optimized catalyst amount which gives 85% yield of the desired product (Table 1, entry 4). The influence of the solvent on the catalytic activity is also investigated on the same substrate molecules. The catalytic activity of the catalyst in polar solvents (Table 2, entry 2–6) is higher than that in non polar solvent (Table 2, entry 1). In addition, we have carried out the reaction under solvent-free condition (Table 2, entry 7) and the result shows that the catalyst displays the best catalytic activity of 85% product yield without any solvent that can be assigned to the competitive adsorption of solvent molecules on the catalyst surface as compared to the substrate molecules. As the solvent-free condition is observed to be a quantitative and waste free process, so all the catalytic studies have been carried out under solvent-free condition.
| Entry | Solvent | Temperature (°C) | Time (h) | Yieldb% |
|---|---|---|---|---|
| a Reaction condition: benzaldehyde (1 mmol), piperidine (1.5 mmol), H2O2(2 mmol), solvent = 5 ml, Pd/NH2-MIL-101(Cr) (0.094 mol%).b Isolated yields are based on benzaldehydes. | ||||
| 1 | Toluene | 60 | 2 | 40 |
| 2 | Acetonitrile | 60 | 2 | 78 |
| 3 | Chloroform | 60 | 2 | 80 |
| 4 | THF | 60 | 2 | 75 |
| 5 | Ethanol | 60 | 2 | 80 |
| 6 | Ethyl acetate | 60 | 2 | 65 |
| 7 | Solvent free | 60 | 2 | 85 |
| 8 | Solvent free | RT | 2 | No product |
| 9 | Solvent free | 40 | 2 | 50 |
| 10 | Solvent free | 90 | 2 | 75 |
The effect of temperature on the oxidative amination of benzaldehydes is studied by performing the reaction at different temperatures (Table 2). No desired product is obtained at room temperature (Table 2, entry 8) confirming the requirement of high temperature to get the desired product. Hence, it is observed that the yield of the product increases with increase in the reaction temperature and a maximum yield of 85% of the desired product are obtained (Table 2, entry 7) when the reaction is carried out at 60 °C. At higher temperature (Table 2, entry 10) the yield of the desired product is found to be low as a result of self-decomposition of hydrogen-peroxide at higher temperature.
Under the optimized reaction conditions, the scope of the Pd/NH2-MIL-101(Cr) catalyzed oxidative amination of aldehydes is examined by employing a wide range of aldehydes. The results are summarized in Table 3. It has been observed that in all cases, the reaction progress in a smooth way with optimum yield of products. Nevertheless, effect of substituent on the aromatic ring of aldehydes is observed based on their electron withdrawing/donating capabilities. Aldehydes containing the electron withdrawing group (Table 3, entry 5, 12, 16) show a better product yield in comparison to that of electron donating group which can be attributed to the electronic and mesomeric effect offered by the electron withdrawing substituent that create a electron deficiency environment around the carbonyl carbon centre, thus providing an easy nucleophilic attack of the amines. Furthermore, the catalyst is also tolerable to the aldehydes containing the heterocycle (Table 3, entry 6) as well as fused ring system (Table 3, entry 7).
|
|||||
|---|---|---|---|---|---|
| Entrya | Aldehyde | Amine | Product | Time (h) | Yieldb (%) |
| a Reaction conditions: aldehyde (1 mmol), amine (1.5 mmol), H2O2 (2 mmol), Pd/NH2-MIL-101(Cr) (0.094 mol%), temperature = 60 °C.b Isolated yields are based on aldehydes. | |||||
| 1 |  |
 |
 |
2 | 85 |
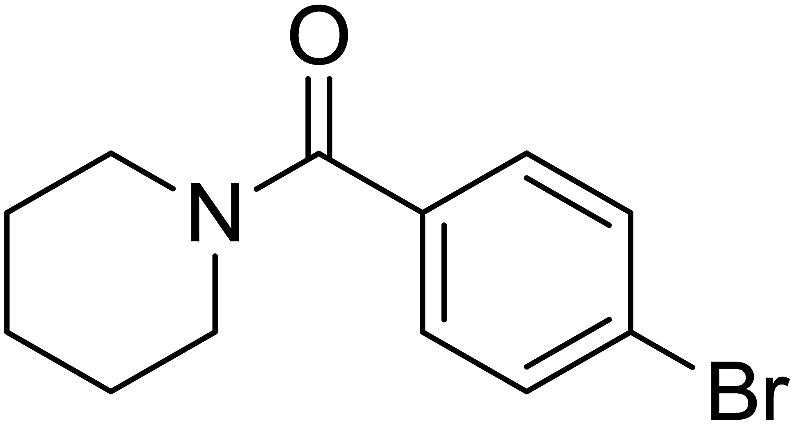
| 2 |  |
 |
 |
2 | 75 |
| 3 |  |
 |
 |
2.5 | 72 |
| 4 |  |
 |
 |
3 | 78 |
| 5 | 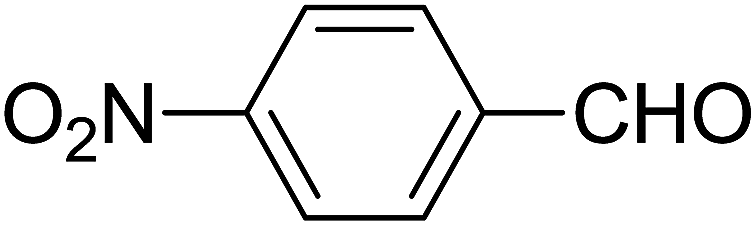 |
 |
 |
2 | 80 |
| 6 |  |
 |
 |
4 | 73 |
| 7 |  |
 |
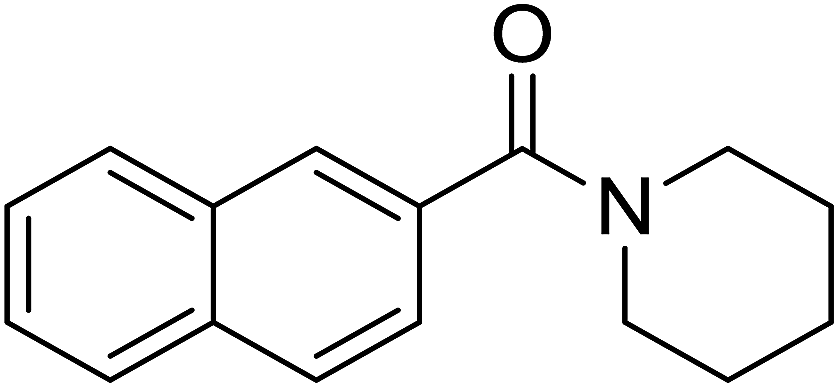 |
3.5 | 70 |
| 8 | 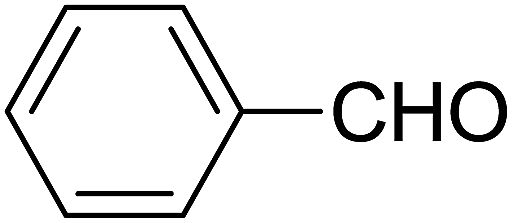 |
 |
 |
1.5 | 82 |
| 9 |  |
 |
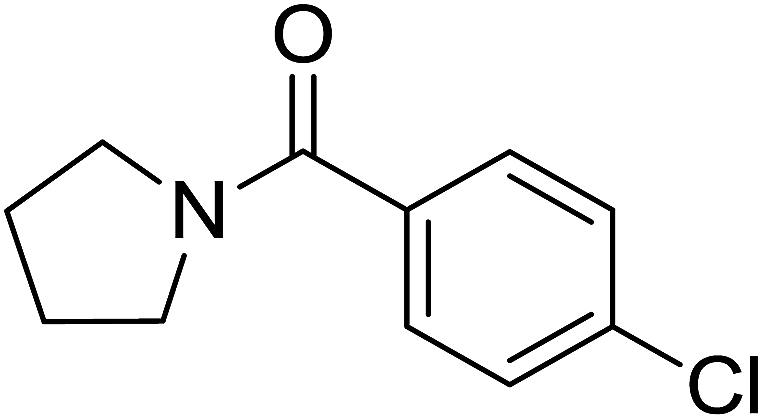 |
1.5 | 73 |
| 10 |  |
 |
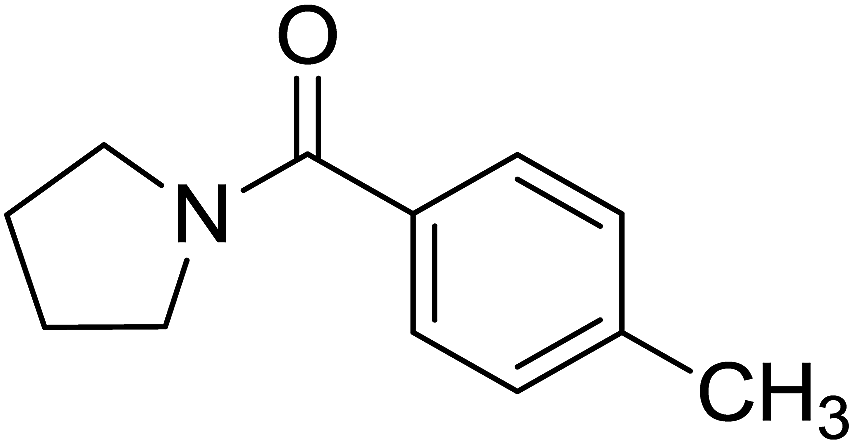 |
2 | 70 |
| 11 |  |
 |
 |
3.5 | 74 |
| 12 |  |
 |
 |
2 | 80 |
| 13 |  |
 |
 |
3 | 79 |
| 14 |  |
 |
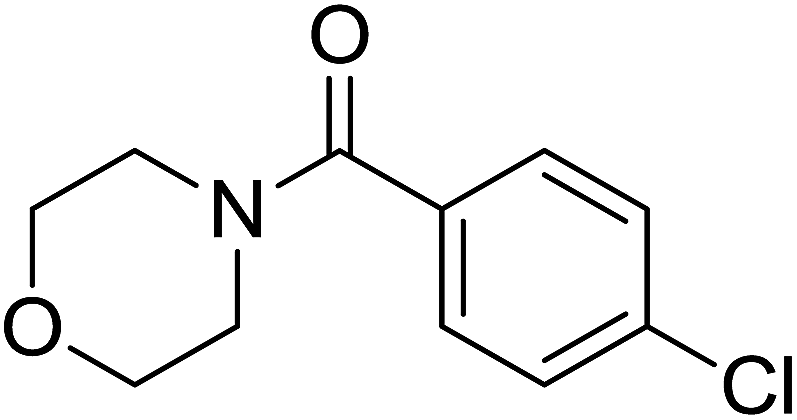 |
2.5 | 70 |
| 15 |  |
 |
 |
3 | 72 |
| 16 |  |
 |
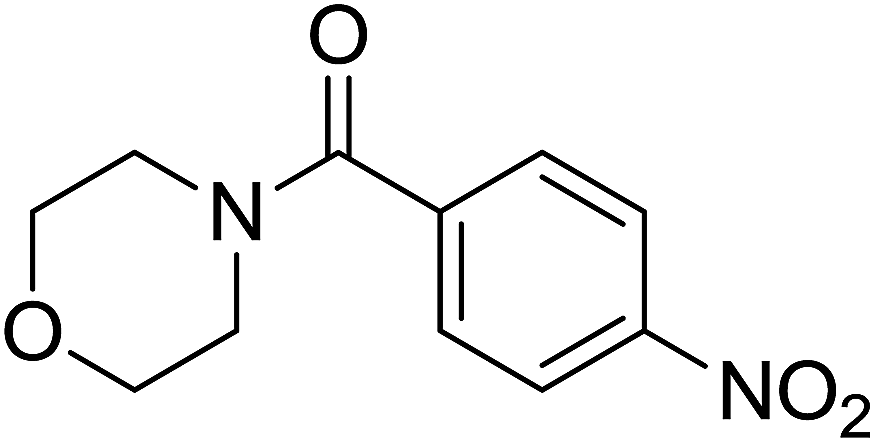 |
2.5 | 76 |
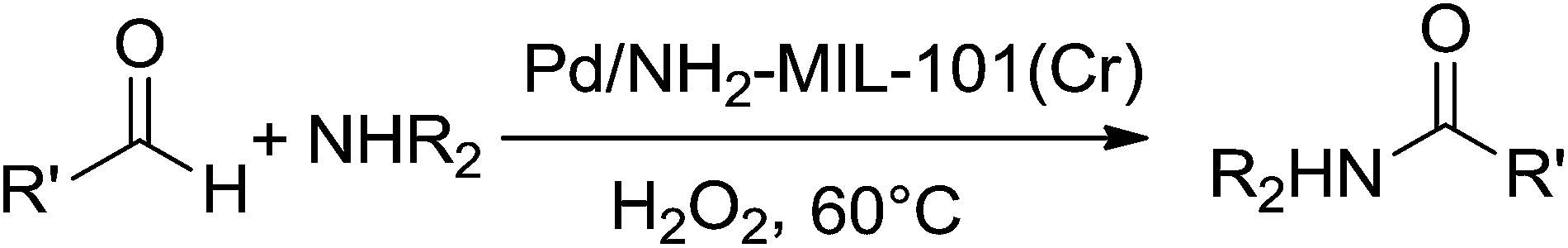
In order to evaluate the efficiency of the present catalytic system, a comparison of the catalytic activity of the present catalytic system is made with other reported catalyst system in terms of yield of the product, reaction temperature, time and dosage of catalyst (Table S1, ESI†). The data indicate that most of the catalytic systems available in the literature are homogeneous which have the basic drawback of catalyst recovery and reusability. The present protocol has the advantages of high product yield, short reaction time, low catalyst dosage, facile recovery and solvent-free condition. Further, the high surface area of the parent MOF provide a well dispersion of the Pd nanoparticles, thus enhancing the catalytic activity of the nanocatalyst.
The heterogeneous nature of catalyst is examined by taking the oxidative amination of benzaldehyde with piperidine as a model reaction. After 1 h of the reaction, the catalyst is removed from the reaction medium by filtration and allows the reaction to run for another 2 h. It is observed that no further conversion of benzaldehyde is detected after removal of the catalyst. The atomic absorption spectroscopic (AAS) analysis of the supernatant solution of the reaction mixture confirms the absence of palladium ion in the liquid phase. These results clearly demonstrate that Pd nanoparticles are not leached out from the MOF matrix during the reaction condition, indicating the true heterogeneous nature of the catalyst.
A plausible reaction mechanism is proposed (Scheme 1) based on the available literature77 and our experimental results. In fact, the reaction is preceded by forming a carbinolamine intermediate (Scheme 1, (III)) from aldehyde (Scheme 1, (I)) and amine (Scheme 1, (II)) which undergo oxidation to corresponding amide (Scheme 1, (IV)) in presence of Pd catalyst and H2O2. In this chemical transformation H2O2 gets decomposed upon contact with the active palladium sites to form water and oxygen and thus initiate the reaction. By virtue of its high surface area, the parent MOF act as stabilizers of the Pd nanoparticles that provide a better dispersion of the palladium active sites and facilitate the easy access of the reactive species.
 | ||
| Scheme 1 Plausible mechanism for the oxidative amination of aldehydes. | ||
From the industrial perspective, the recyclability of a catalyst is an important criterion for heterogeneous catalysis. The catalyst recycle experiment is performed on the same model reaction of oxidative amination of benzaldehyde with piperidine (Fig. 5). Upon completion of the reaction, the catalyst is separated from the reaction mixture by filtration, washed it properly with acetone and dried under vacuum desiccator. The recovered catalyst is then subsequently utilized for the same reaction up to four consecutive cycles which is well supported by the PXRD, FE-SEM-EDS, TEM and FT-IR analysis of the recovered catalyst. On comparing the PXRD patterns of the fresh and recovered catalyst (Fig. S5, ESI†), it is observed that the crystallinity of the support remain unaltered in the recovered catalyst upto forth catalytic run. FE-SEM image of the recovered catalyst (Fig. S1(c)†) demonstrate the conservation of the nanoscale morphology of the NH2-MIL-101(Cr). From the TEM analysis of the recovered catalyst (Fig. S6, ESI†) it has been observed that the dispersion of the nanoparticles on the support is well maintained throughout the chemical transformation. This observation is further clarified by the EDS spectrum of the recovered catalyst (Fig. S7, ESI†) which shows that the Pd nanoparticles remain intact with NH2-MIL-101(Cr) after the catalytic run. From FT-IR analysis of the recovered catalyst (Fig. S3(b), ESI†) it is found that the structural properties of the recovered catalyst remain same to that of the fresh one. Finally, all these results clearly display the heterogeneous and robustness of the Pd/NH2-MIL-101(Cr) nanocatalyst.
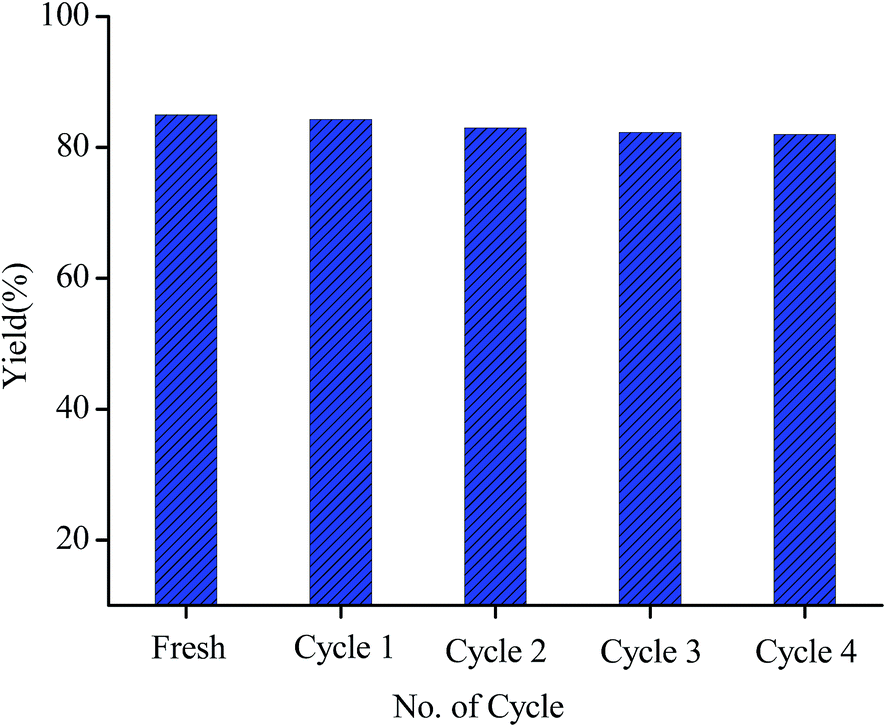 | ||
| Fig. 5 Catalyst recycling test for Pd/NH2-MIL-101(Cr). | ||
Conclusions
In conclusion, we have successfully immobilized the Pd nanoparticles on an amine-functionalized nanoscale metal–organic framework by a facile, greener polyol route. The catalytic activity of the resultant Pd nanocatalyst is explored through oxidative amination of aldehydes in presence of a green oxidant under solvent free condition. Short reaction time, solvent-free condition, low catalyst dosage and high product yields are the certain advantages that make our protocol somewhat superior to that of the reported catalytic methods available in the literature. The high catalytic activity of the Pd nanocatalyst may be attributed to the high surface of the support with the free amine groups of the ligands which provide a better dispersion of the catalytic active sites and make these active sites easily accessible towards the substrate and the oxidant species. Moreover, the catalyst can easily be recovered and recycled up to four catalytic cycles without any significant loss in catalytic activity.Acknowledgements
The authors are grateful to the Director of CSIR-North East Institute of Science and Technology, Jorhat, Assam, India for his kind permission to publish the work. We are thankful to Dr P. Sengupta, Head of Materials Science Division and Dr D. K. Dutta for support, and non network project MLP-6000(WP-1) for financial assistance. M. S. acknowledges DST New Delhi for an INSPIRE fellowship.References
- L. Schlapbach and A. Züttel, Nature, 2001, 414, 353–358 CrossRef CAS PubMed.
- C. Zlotea, F. Cuevas, V. P. Boncour, E. Leroy, P. Dibandjo, R. Gadiou, C. V. Guterl and M. Latroche, J. Am. Chem. Soc., 2010, 132, 7720–7729 CrossRef CAS PubMed.
- J. C. G. Martinez, R. Lezutekong and R. M. Crooks, J. Am. Chem. Soc., 2005, 127, 5097–5103 CrossRef PubMed.
- S. W. Kim, M. Kim, W. Y. Lee and T. Hyeon, J. Am. Chem. Soc., 2002, 124, 7642–7643 CrossRef CAS PubMed.
- F. Wang, C. Li, L. D. Sun, C. Xu, J. Wang, J. C. Yu and C. Yan, Angew. Chem., Int. Ed., 2012, 51, 4872–4876 CrossRef CAS PubMed.
- K. Nakao, O. Watanabe, T. Sasaki, S. Ito, K. Tomishige and K. Kunimori, Surf. Sci., 2007, 601, 3796–3800 CrossRef CAS.
- V. Mazumder and S. Sun, J. Am. Chem. Soc., 2009, 131, 4588–4589 CrossRef CAS PubMed.
- R. Long, K. Mao, X. Ye, W. Yan, Y. Huang, J. Wang, Y. Fu, X. Wang, X. Wu, Y. Xie and Y. Xiong, J. Am. Chem. Soc., 2013, 135, 3200–3207 CrossRef CAS PubMed.
- S. Bhattacharjee, D. M. Dotzauer and M. L. Bruening, J. Am. Chem. Soc., 2009, 131, 3601–3610 CrossRef CAS PubMed.
- O. M. Wilson, M. R. Knecht, J. C. G. Martinez and R. M. Crooks, J. Am. Chem. Soc., 2006, 128, 4510–4511 CrossRef CAS PubMed.
- A. M. Doyle, S. K. Shaikhutdinov and H. J. Freund, Angew. Chem., Int. Ed., 2005, 44, 629–631 CrossRef CAS PubMed.
- (a) F. Fievet, J. P. Lagier and M. Figlarz, MRS Bull., 1989, 29 CrossRef CAS; (b) F. Fievet, J. P. Lagier, B. Blin, B. Beaudoin and M. Figlarz, Solid State Ionics, 1989, 32/33 CrossRef.
- J. M. Humphery and A. R. Chamberlin, Chem. Rev., 1997, 97, 2243–2266 CrossRef.
- I. Johansson, Kirk-Othmer Encyclopedia of Chemical Technology: Amides, Fatty Acids, ed. J. I. Kroschwitz, Wiley, Hoboken, New Jersey, 5th edn, 2004, vol. 2, pp. 442–463 Search PubMed.
- E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606–631 RSC.
- (a) E. Saxo and C. Z. Bertozzi, Science, 2000, 287, 2007–2010 CrossRef; (b) F. Damkaci and P. de Shong, J. Am. Chem. Soc., 2003, 125, 4408–4409 CrossRef CAS PubMed; (c) Y. G. Gololobov and L. F. Kasukhin, Tetrahedron, 1992, 48, 1353–1406 CrossRef CAS.
- (a) T. Ribelin, C. E. Katz, D. G. English, S. Smith, A. K. Manukyan, V. W. Day, B. Neuenswander, J. L. Poutsma and J. Aube, Angew. Chem., Int. Ed., 2008, 47, 6233–6235 CrossRef CAS PubMed; (b) S. Lang and J. A. Murphy, Chem. Soc. Rev., 2006, 35, 146–156 RSC.
- (a) N. A. Owston, A. J. Parker and J. M. J. Williams, Org. Lett., 2007, 9, 3599–3601 CrossRef CAS PubMed; (b) M. Hashimoto, Y. Obora, S. Sakaguchi and Y. Ishii, J. Org. Chem., 2008, 73, 2894–2897 CrossRef CAS PubMed.
- (a) J. R. Martinelli, T. P. Clark, D. A. Watson, R. H. Munday and S. L. Buchwald, Angew. Chem., 2007, 119, 8612–8615 CrossRef; (b) P. Nanayakkara and H. Alper, Chem. Commun., 2003, 2384–2385 RSC.
- B. Shen, D. M. Makley and J. N. Johnston, Nature, 2010, 465, 1027–1032 CrossRef CAS PubMed.
- (a) C. Gunanathan, Y. Ben-David and D. Milstein, Science, 2007, 317, 790–792 CrossRef CAS PubMed; (b) L. U. Nordstom, H. Vogt and R. Madsen, J. Am. Chem. Soc., 2008, 130, 17672–17673 CrossRef PubMed; (c) T. Zweifel, J. V. Naubron and H. Grutzmacher, Angew. Chem., Int. Ed., 2009, 48, 559–563 CrossRef CAS PubMed; (d) S. C. Ghosh, S. Muthaiah, Y. Zhang, X. Xu and S. H. Homg, Adv. Synth. Catal., 2009, 351, 2643–2649 CrossRef CAS; (e) Y. Zhang, C. Chen, S. C. Ghosh, Y. Li and S. H. Hong, Organometallics, 2010, 29, 1374–1378 CrossRef CAS; (f) C. Cheng and S. H. Hong, Org. Biomol. Chem., 2011, 9, 20–26 RSC; (g) Y. Wang, D. Zhu, L. Tang, S. Wang and Z. Wang, Angew. Chem., Int. Ed., 2011, 50, 8917–8921 CrossRef CAS PubMed; (h) J.-F. Soule, H. Miyamura and S. Kobayashi, J. Am. Chem. Soc., 2011, 133, 18550–18553 CrossRef CAS PubMed; (i) S. Kegnaes, J. Mielby, U. V. Mentzel, T. Jensen, P. Fristrup and A. Riisager, Chem. Commun., 2012, 48, 2427–2429 RSC; (j) F. Xiao, Y. Liu, C. Tang and G.-J. Deng, Org. Lett., 2012, 14, 984–987 CrossRef CAS PubMed , and references herein.
- (a) S. Cho, E. Yoo, I. Bae and S. Chang, J. Am. Chem. Soc., 2005, 127, 16046–16047 CrossRef CAS PubMed; (b) Z.-W. Chen, H.-F. Jiang, X.-Y. Pan and Z.-J. He, Tetrahedron, 2011, 67, 5920–5927 CrossRef CAS.
- (a) R. V. Kolakowski, N. Shangguan, R. R. Sauers and L. J. Williams, J. Am. Chem. Soc., 2006, 128, 5695–5702 CrossRef CAS PubMed; (b) X. Zhang, F. Li, X. W. Lu and C. F. Liu, Bioconjugate Chem., 2009, 20, 197–200 CrossRef CAS PubMed.
- (a) T. A. Dineen, M. A. Zajac and A. G. Myers, J. Am. Chem. Soc., 2006, 128, 16046–16047 CrossRef PubMed; (b) C. L. Allen, B. N. Atkinson and J. M. J. Williams, Angew. Chem., Int. Ed., 2012, 51, 1383–1386 CrossRef CAS PubMed; (c) M. Tamura, T. Tonomura, K.-I. Shimizu and A. Satsuma, Green Chem., 2012, 14, 717–724 RSC; (d) M. Zhang, S. Imm, S. Bahn, L. Neubart, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2012, 51, 3905–3909 CrossRef CAS PubMed.
- A. Tillack, I. Rudloff and M. Beller, Eur. J. Org. Chem., 2001, 523–528 CrossRef CAS.
- R. Cadoni, A. Porcheddu, G. Giacomelli and L. de Luca, Org. Lett., 2012, 14, 5014–5017 CrossRef CAS PubMed.
- S. C. Ghosh, J. S. Y. Ngiam, A. M. Seayad, D. T. Tuan, C. L. L. Chai and A. Chen, J. Org. Chem., 2012, 77, 8007–8015 CrossRef CAS PubMed.
- S. C. Ghosh, J. S. Y. Ngiam, C. L. L. Chai, A. M. Seayad, T. T. Dang and A. Chena, Adv. Synth. Catal., 2012, 354, 1407–1412 CrossRef CAS.
- S. Muthaiah, S. C. Ghosh, J.-E. Jee, C. Chen, J. Zhang and S. H. Hong, J. Org. Chem., 2010, 75, 3002–3006 CrossRef CAS PubMed.
- W.-J. Yoo and C.-J. Li, J. Am. Chem. Soc., 2006, 128, 13064–13065 CrossRef CAS PubMed.
- C. L. Allen and J. M. J. Williams, Chem. Soc. Rev., 2011, 40, 3405–3415 RSC.
- Y. Suto, N. Yamagiwa and Y. Torisawa, Tetrahedron Lett., 2008, 49, 5732–5735 CrossRef CAS.
- Y. Tamaru, Y. Yamada and Z. Yoshida, Synthesis, 1983, 6, 474–476 CrossRef.
- K. Ekoue-Kovi and C. Wolf, Chem.–Eur. J., 2008, 14, 6302–6315 CrossRef CAS PubMed , and references therein.
- G. Ferey, C. M. Draznieks, C. Serre, F. Millange, J. Dutour, S. Surble and I. Margiolaki, Science, 2005, 309, 2040–2042 CrossRef CAS PubMed.
- L. Ma, C. Abney and W. Lin, Chem. Soc. Rev., 2009, 38, 1248–1256 RSC.
- A. Corma, H. Garcia and F. X. L. I. Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS PubMed.
- M. Meilikhov, K. Yusenko, D. Esken, S. Turner, G. V. Tendeloo and R. A. Fischer, Eur. J. Inorg. Chem., 2010, 24, 3701–3714 CrossRef.
- R. Ricco, L. Malfatti, M. Takahashi, A. J. Hill and P. Falcaro, J. Mater. Chem. A, 2013, 1, 13033–13045 CAS.
- Y. K. Hwang, D. Y. Hong, J. S. Chang, S. H. Jhung, Y. K. Seo, J. Kim, A. Vimont, M. Daturi, C. Serre and G. Férey, Angew. Chem., Int. Ed., 2008, 47, 4144–4148 CrossRef CAS PubMed.
- Y. Pan, B. Yuan, Y. Li and D. He, Chem. Commun., 2010, 46, 2280–2282 RSC.
- A. Henschel, K. Gedrich, R. Kraehnert and S. Kaskel, Chem. Commun., 2008, 4192–4194 RSC.
- A. Aijaz, T. Akita, N. Tsumori and Q. Xu, J. Am. Chem. Soc., 2013, 135, 16356–16359 CrossRef CAS PubMed.
- X. Zhao, Y. Jin, F. Zhang, Y. Zhong and W. Zhu, Chem. Eng. J., 2014, 239, 33 CrossRef CAS.
- B. Yuan, Y. Pan, Y. Li, B. Yin and H. Jiang, Angew. Chem., Int. Ed., 2010, 49, 4054–4058 CrossRef CAS PubMed.
- X. Gu, Z. H. Lu, H. L. Jiang, T. Akita and Q. Xu, J. Am. Chem. Soc., 2011, 133, 11822–11825 CrossRef CAS PubMed.
- F. G. Cirujano, F. X. Llabrés i Xamena and A. Corma, Dalton Trans., 2012, 41, 4249–4254 RSC.
- M. Yadav and Q. Xu, Chem. Commun., 2013, 49, 3327–3329 RSC.
- D. Zhang, Y. Guan, E. J. M. Hensen, L. Chen and Y. Wang, Catal. Commun., 2013, 41, 47–51 CrossRef CAS.
- H. Dai, J. Su, K. Hu, W. Luo and G. Cheng, Int. J. Hydrogen Energy, 2014, 39, 4947–4953 CrossRef CAS.
- J. Hermannsdörfer, M. Friedrich, N. Miyajima, R. Q. Albuquerque, S. Kummel and R. Kempe, Angew. Chem., Int. Ed., 2012, 51, 11473–11477 CrossRef PubMed.
- H. Li, Z. Zhu, F. Zhang, S. Xie, H. Li, P. Li and X. Zhou, ACS Catal., 2011, 1, 1604–1612 CrossRef CAS.
- J. J. Alcañiz, J. F. Soria, I. Luz, P. S. Crespo, E. Skupien, V. P. Santos, E. Pardo, F. X. L. I. Xamena, F. Kapteijn and J. Gascon, J. Catal., 2013, 307, 295–304 CrossRef.
- J. Hermannsdörfer and R. Kempe, Chem.–Eur. J., 2011, 17, 8071–8077 CrossRef PubMed.
- G. Chen, S. Wu, H. Liu, H. Jiang and Y. Li, Green Chem., 2013, 15, 230–235 RSC.
- J. J. Alcaniz, J. Gascon and F. Kapteijn, J. Mater. Chem., 2012, 22, 10102–10118 RSC.
- Q.-L. Zhu and Q. Xu, Chem. Soc. Rev., 2014, 43, 5468–5512 RSC.
- A. Aijaz, Q.-L. Zhu, N. Tsumori, T. Akita and Q. Xu, Chem. Commun., 2015, 51, 2577–2580 RSC.
- C. Rösler and R. A. Fischer, CrystEngComm, 2015, 17, 199–217 RSC.
- N. Shang, S. Gao, X. Zhou, C. Feng, Z. Wang and C. Wang, RSC Adv., 2014, 4, 54487–54493 RSC.
- S. Bhattacharjee, C. Chenab and W.-S. Ahn, RSC Adv., 2014, 4, 52500–52525 RSC.
- W. Qin, W. Cao, H. Liu, Z. Li and Y. Li, RSC Adv., 2014, 4, 2414–2420 RSC.
- Y. Huang, Z. Zheng, T. Liu, J. Lü, Z. Lin, H. Li and R. Cao, Catal. Commun., 2011, 14, 27–31 CrossRef CAS.
- Y. Huang, Z. Lin and R. Cao, Chem.–Eur. J., 2011, 17, 12706–12712 CrossRef CAS PubMed.
- Y. Huang, S. Liu, Z. Lin, W. Li, X. Li and R. Cao, J. Catal., 2012, 292, 111–117 CrossRef CAS.
- I. Luz, C. Rösler, K. Epp, F. X. L. I. Xamena and R. A. Fischer, Eur. J. Inorg. Chem., 2015, 23, 3904–3912 CrossRef.
- X. Li, Z. Guo, C. Xiao, T. W. Goh, D. Tesfagaber and W. Huang, ACS Catal., 2014, 4, 3490–3497 CrossRef CAS.
- R. Kardanpour, S. Tangestaninejad, V. Mirkhani, M. Moghadam, I. M- Baltork, A. R. Khosropour and F. Zadehahmadi, J. Organomet. Chem., 2014, 761, 127–133 CrossRef CAS.
- L. Shen, W. Wu, R. Liang, R. Lina and L. Wu, Nanoscale, 2013, 5, 9374–9382 RSC.
- Y. Huang, S. Gao, T. Liu, J. Lü, X. Lin, H. Li and R. Cao, ChemPlusChem, 2012, 77, 106–112 CrossRef CAS.
- N. Toshima and T. Yonezawa, New J. Chem., 1998, 11, 1179–1201 RSC.
- Y. Wang, S.-I. Choi, X. Zhao, S. Xie, H.-C. Peng, M. Chi, C. Z. Huang and Y. Xia, Adv. Funct. Mater., 2014, 24, 131–139 CrossRef CAS.
- X. Xia, S.-I. Choi, J. A. Herron, N. Lu, J. Scaranto, H.-C. Peng, J. Wang, M. Mavrikakis, M. J. Kim and Y. Xia, J. Am. Chem. Soc., 2013, 135, 15706–15709 CrossRef CAS PubMed.
- H. Huang, Y. Wang, A. Ruditskiy, H. C. Peng, X. Zhao, L. Zhang, J. Liu, Z. Ye and Y. Xia, ACS Nano, 2014, 8, 7041–7050 CrossRef CAS PubMed.
- L. Wang, J.-J. Zhai, K. Jiang, J.-Q. Wang and W.-B. Cai, Int. J. Hydrogen Energy, 2015, 40, 1726–1734 CrossRef CAS.
- R. K. Sharma and S. Sharma, Dalton Trans., 2014, 43, 1292–1304 RSC.
- Y. Lin, C. Kong and L. Chen, RSC Adv., 2012, 2, 6417–6419 RSC.
- B. Thiébaut, Platinum Met. Rev., 2004, 48, 62–63 Search PubMed.
- S. H. Jhung, J. H. Lee, J. W. Yoon, C. Serre, G. Ferey and J. S. Chang, Adv. Mater., 2007, 19, 121–124 CrossRef CAS.
- J. Gascon, U. Aktay, M. D. Hernandez-Alonso, G. P. M. van Klink and F. Kapteijn, J. Catal., 2009, 261, 75–87 CrossRef CAS.
- M. Kandiah, M. H. Nilsen, S. Usseglio, S. Jakobsen, U. Olsbye, M. Tilset, C. Larabi, E. A. Quadrelli, F. Bonino and K. P. Lillerud, Chem. Mater., 2010, 22, 6632–6640 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra27826k |
| This journal is © The Royal Society of Chemistry 2016 |