
Development of a new version of homogenous liquid–liquid extraction based on an acid–base reaction: application for extraction and preconcentration of aryloxyphenoxy-propionate pesticides from fruit juice and vegetable samples
Abstract
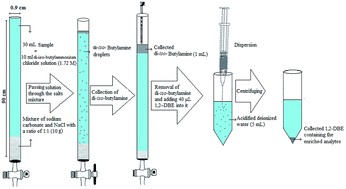
In this paper, a new version of homogeneous liquid–liquid extraction based on an acid–base reaction combined with dispersive liquid–liquid microextraction followed by gas chromatography-flame ionization detection has been developed for the extraction and determination of aryloxyphenoxy-propionate pesticides in fruit juice and vegetable samples. In this method, initially a basic solvent (amine) is mixed with acidified deionized water spiked with the analytes, which leads to the formation of a homogenous solution. Afterward, the homogenous solution is transferred into a narrow bore tube containing the mixture of sodium chloride and sodium carbonate. As the solution is passed through the filled portion of the tube, fine droplets of the organic solvent (amine) are released and move through the aqueous phase to the top of the tube and form a separated phase in which the target analytes are extracted. In the second step, the collected organic phase is removed and mixed with a preconcentration solvent and injected into acidified deionized water for more enrichment of the analytes. Effective parameters controlling the performance of the proposed method, such as type and volume of extraction solvent and flow rate in homogenous liquid–liquid extraction step and type and volume of preconcentration solvent, ionic strength and centrifugation rate and time in the dispersive liquid–liquid extraction step were optimized. Under the optimum extraction conditions, the method showed low limits of detection and quantification within the ranges of 0.12–0.40 and 0.40–1.3 μg L−1, respectively. Relative standard deviations were in the range of 3–5% (n = 6, C = 50 or 100 μg L−1). Comparison of the proposed method with the existing reports in the literature demonstrates that it is simple, rapid, selective, and sensitive, and has high enrichment factors and low detection limits for determining aryloxyphenoxy-propionate pesticides in the studied samples.
 Please wait while we load your content...
Please wait while we load your content...

