
Palladium nanoparticles in the catalysis of coupling reactions
Ansuman Beja,
Koena Ghoshab,
Amitabha Sarkar*a and
David. W. Knight*c
aDepartment of Organic Chemistry, Indian Association for the Cultivation of Science, Kolkata 700 032, India. E-mail: ocas@iacs.res.in; Fax: +91-33-2473-2805; Tel: +91-33-2473-4971
bPresidency University, Kolkata 700073, India
cSchool of Chemistry, Cardiff University, Cardiff CF10 3XQ, UK
First published on 18th January 2016
Abstract
Palladium catalyzed coupling reactions have emerged as a versatile, convenient, selective and mild protocol that can usually be adapted in any synthetic scheme for important target molecules with various degrees of structural complexity. While anchored catalysts offer recycling advantages, palladium nanoparticles display an impressive ability to catalyse coupling reactions. Along with their successful applications in organic synthesis, controversy has also arisen concerning the exact nature – heterogeneous or homogeneous – of the reaction catalyzed by palladium nanoparticles.
1. Introduction
Traditional heterogeneous catalysts are finely dispersed metal powders – supported on an oxide or similar non-participating matrix, or uncomplexed – that have the obvious advantages of separation from reactant-product mixtures and therefore potential recycling. The crystallinity of the metal determines the number of catalytically active atoms per unit area of the surface, which, in turn, governs its catalytic activity.1 Often these active centres are also sites of undesirable reactions (with acids or oxidants) detrimental for catalytic efficiency. Nanoparticles offer a miniaturized version of metal particles.2 Since the ratio of surface area to volume is large for nano-sized atomic clusters, these have been extensively investigated for improved catalytic function.3 Decades of research has shown that many such nanoparticles are indeed excellent catalysts for useful organic transformations. However, the actual nature of their involvement in catalysis has not been unambiguously established, although a large body of literature exists and several review articles have been published.4 The present article attempts to take a holistic view of the different thoughts and experiments reported so far, and to clarify conflicting notions. Attention is restricted to use of palladium nanoparticles (PdNps) in catalysis of Suzuki–Miyaura and Heck reactions, a representative area where a large body of data is available. Reactions in ionic liquids5 are deliberately omitted as this reaction medium is clearly different from organic solvents and a direct comparison of reactivity or mechanism can be misleading.2. Palladium nanoparticles
Palladium nanoparticles are easy to prepare and can be obtained in different sizes.6 The more common range of nanoparticle diameter used in organic synthesis is 6–12 (ref. 6b and c) nm while there are several reports of the use of particles with smaller sizes, e.g. 1–4 nm.6d–g Convenience, catalytic efficiency and recycling ability of palladium nanoparticles are well-established beneficial features, evident from the widely reported experimental data; yet, whether catalysis occurs on the nanoparticle surface and to what extent, remain an open question. Several groups believe that nano-sized palladium actually serves as a reservoir for smaller clusters or atomic palladium, which are catalytically active and which probably operate in the domain of a homogeneous catalytic cycle. A large number of palladium complexes with variously designed ligands, including cyclometalated complexes, give rise to palladium nanoparticles at elevated temperatures. Questions have been raised in many such instances regarding the relative importance of an anticipated homogeneous pathway vis-à-vis catalysis through a nanoparticle-mediated pathway. Evidence presented in the literature varies widely and much is circumstantial at best; hence the issue continues to be debated.3. Possible scenarios
The various possibilities that have been considered in trying to understand the process of palladium nanoparticle catalysis of coupling are summarized in the following sections:(i) Catalysis on a nanoparticle surface (heterogeneous catalysis)
Catalytic transformations of molecules occur on the nanoparticle surface, probably on defect sites.7 This thought, coupled with the expectation of high catalytic efficiency of nanoparticles with high surface/volume ratios, gained currency, particularly in the early literature. Two papers published in 1996 reported significant initial observations. Beller et al.8 obtained 97% conversion in a Heck reaction of 4-bromoacetophenone and butyl acrylate at 140 °C in the presence of 0.05 mol% of Pd-colloids in 5 min (Scheme 1). Addition of triphenylphosphine seriously retarded the rate of reaction (93% conversion after 5 h). | ||
| Scheme 1 | ||
Reetz reported9 a similar observation with preformed palladium nanoparticles (PdNps) stabilized by quaternary ammonium salt [pioneered by Jeffrey].10 Both research groups observed that chlorobenzene was not an ideal reaction partner. It was inferred that “catalysis is likely to occur at defect sites, steps and kinks on the surface of the colloidal metal particles, a process that is more closely related to heterogeneous catalysis.”9c In another paper of 2000, use of PdNps stabilized by N-vinyl-2-pyrrolidone (PVP) as catalyst (0.3 mol% of Pd) for Suzuki–Miyaura reactions in water was reported.11 The authors found “that the initial rate of Suzuki coupling reactions depends linearly on the concentration of the Pd catalyst, thus giving strong evidence that the catalysis occurs on the surface of the Pd nanoparticles.” This is echoed in a later work: “The product yield determined after 1 h decreased with decreasing catalyst concentration, from 96% for 1 mol% of palladium to 64% for 0.125 mol% of palladium”.12
Consistently high turnover for a large number of recycling experiments has been regarded as a reliable indication for catalysis via a heterogeneous pathway, that is, reactions occurring on the surface. This has also been reported for some supported catalysts.13 If atoms are steadily etched away by an initiation step in the catalytic cycle (oxidative addition, for example), it is conceivable that the surface morphology would change rapidly which could lead to a steady decline of catalytic efficiency. Such is the case with numerous in situ generated and moderately stabilized nanoparticles for which changes in size and contour are readily observed by TEM (Transmission Electron Microscopy). Catalysts performing with undiminished activity beyond fifteen or twenty recycles are rather scarce.13 Their robustness seems to underscore their similarity with truly heterogeneous catalysts. The smaller the size of the nanoparticles (1–3 nm), the greater is their catalytic (and recycling) efficiency.14 “In principle, the catalytic activity in nanocatalysis is determined by the size of nanoparticles. The smaller the nanoparticle is, the more effective the catalytic activity.” It is pertinent to recall that catalytically active PdNp of diameter 1.3–1.6 nm was reported by Reetz earlier.9c For nanoparticles of larger average diameter, leaching of smaller clusters or atoms might become important in promoting an alternative, homogeneous pathway.
When a bromoarene was part of a polystyrene backbone and the catalyst was PdNp (9–12 nm) supported on polyoxometalate, Suzuki–Miyaura products were not obtained.15 This ‘three-phase test’ rules out catalysis by leached palladium in this system. A similar three-phase test was reported independently for PdNp supported on extended pore MCM-41 (ref. 16) to conclude dominance of a heterogeneous pathway.
An overwhelming majority of reports would testify that unactivated chloroarenes are poor substrates for coupling reactions catalyzed by PdNps. This observation has been recorded also by groups that strongly favour catalysis by leached out palladium. Therefore, it is not unreasonable to assume that participation of chloroarenes in coupling reactions, a relatively rare event, would indicate an operative mechanism different from many. Significantly, the polyoxometalate supported PdNp catalyst described above15 is effective with chloroarenes having electron-donating substituents. It has been reported that PdNps supported on polyaniline, which acts both as host as well as reductant for palladium, catalyze Suzuki–Miyaura or Ullmann-type coupling reactions with chloroarenes including those with an ortho-substituent17 with high yield and efficiency. Following Buchwald's precedence, conversion of a chloro to an hydroxyl group was also accomplished using the same catalyst. The yield of Suzuki–Miyaura coupling after ten recycles was an impressive 89%. Another report where chloroarenes were successful substrates, catalytically active polyethylene glycol (PEG)400-stabilized PdNps were of smaller average size (1.3 nm) and were apparently activated (even stabilized) by oxygen.14 Stannoxane scaffolds featuring phosphines are reported to stabilize PdNps, which catalyze the coupling of iodo-, bromo- and chloroarenes; the last ones are probably made to participate by the smaller sized PdNps (3–4 nm).18 We shall return to this aspect in the context of supported nanoparticles as catalyst (vide infra).
In a much later paper by Fairlamb, Lee and others,19 strong evidence was put forward in supporting this notion of catalysis occurring on the surface of PdNps through defect sites based on an operando XAS (X-ray Absorption Spectroscopy) study, in tandem with time-dependent EXAFS (Extended X-ray Absorption Fine Structure) spectra. A drop in TOF (turnover frequency) corresponding to an increase in the size of the palladium nanoparticles is consistent with surface catalysis. The mercury poisoning test is also positive. Although the authors noted that the reaction rate did not pick up by spiking with Pd(OAc)2 (palladium(II) acetate) after mercury arrested the reaction, it has been argued3a that mercury could also poison molecular palladium species, especially when unprotected by ligands. There are several examples in the literature, however, attest to the fact that mercury does not generally inhibit homogeneously catalyzed reactions.20 Trapping nanoparticles in the core channels of Quadrasil MP (QuadraSil Mercaptopropyl) or flash silica retards the reaction considerably, supporting the idea of catalysis by nanoparticles rather than leached molecular palladium species. In a later in situ quick-scanning EXAFS study,21 palladium nanoclusters of diameter ∼2 nm were observed in the liquid phase during the entire “active phase” of catalysis, while homogeneous Pd(II) complexes were identified only towards the end of the reaction when the substrate molecules had been consumed. Monitoring the surface revealed an initial reduction step where all Pd(II) species were converted into Pd(0) prior to onset of the reaction. Palladium nanoparticles were detected in the liquid phase after the temperature had reached 150 °C. These experimental results are said to be consistent with a surface-catalyzed reaction when the nanoparticle size is rather small.
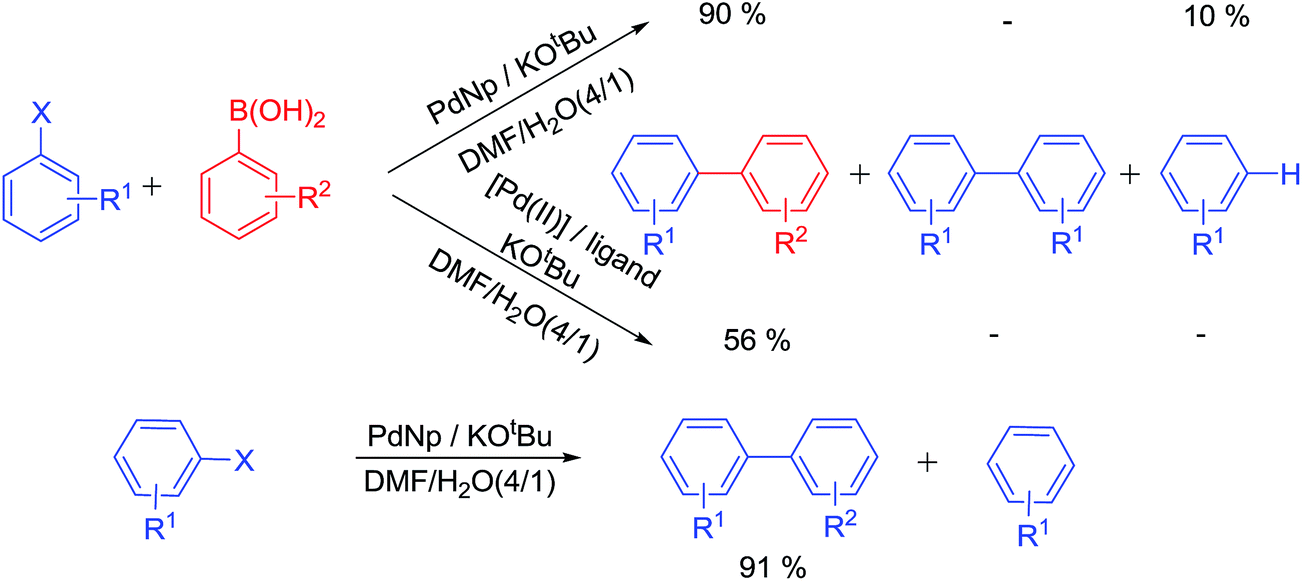
Ligands not only stabilize palladium atoms in a homogeneously catalyzed reaction, but they also modify the reactivity of metal centres. Hence, a particular ligand–metal complex displays well-defined reactivity profile and product selectivity. Palladium nanoparticles have their own reactivity–selectivity that is only slightly modified by additives. A recent article22 describes divergent product selectivity for homogeneous metal–ligand complex, nanoparticle and ligated nanoparticle (Scheme 2).
 | ||
| Scheme 2 | ||
The unique selectivity of a ligand–nanoparticle combination strongly supports surface catalysis where reactivity of the surface-bound palladium centre is altered by the ligand. An earlier report states that when optically pure diphosphines were used with PdNps at ambient or sub-ambient temperature, the enantiomeric excess of the product was found to be dependent on the modifying ligand used. With BINAP (2,2′-bis(diphenylphosphino)-1,1′-binaphthyl), the best yields and ees (enantiomeric excess) were obtained and these were even better than its homogeneous version.23 This also provides a strong evidence in favour of catalysis occurring on the ligand-modified surface (Scheme 3).24
 | ||
| Scheme 3 | ||
Such results should be contrasted with the observation of racemic products in a reaction where an optically pure palladacycle was used as the catalyst precursor;25 clearly, the optically active palladacycle generated PdNps that were no longer optically active.
(ii) Leached palladium from PdNp as active catalyst
An overwhelming data set suggests that palladium species leached out from nanoparticles into the reaction mixture are indeed the actual catalytic species rather than PdNps that are originally used and often recovered at the end of reaction and recycled. It is not always clearly stated whether palladium atoms or very small palladium clusters (<3 nm, at ppm level) are the catalyst. It is widely believed that oxidative addition on a suitable palladium atom on the PdNp is followed by detachment of that newly formed Pd(II) species from the cluster,26 which can trigger a homogeneous catalytic cycle that converts substrate molecules to products in solution. It is also presumed that Pd(0) could be leached out of a nanocluster (more likely from a silica or carbon support) prompted by base and/or solvent;27 oxidative addition would then occur in the solution phase as in homogeneous catalysis (see Fig. 1 below). It is not obvious why Pd(0) should leach out more easily than Pd(II) from PdNps as one report suggests.27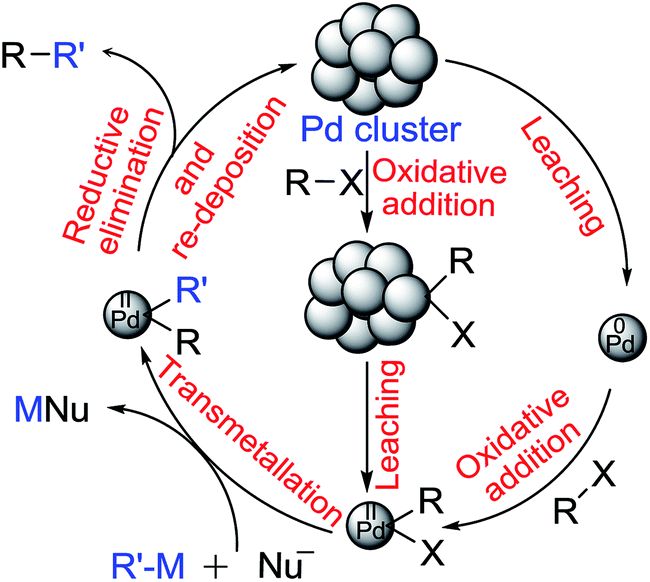 | ||
| Fig. 1 Proposed mechanism for the Pd catalyzed cross-coupling reaction involving homogeneous pathway. | ||
Collman's “three-phase test”28 is often conducted to settle such issues. While haloarenes tethered on a PEG backbone did not provide any Heck product with PdNps, addition of a small amount of free haloarene was found to reverse the situation – polymer-bound substrates were all converted to product, indicating catalysis by leached out soluble metal particles.29 The leached out species should be Pd(II) as the reaction did not proceed in the absence of added haloarene and the quantity of leached metal must be limited by the quantity of this additive. This would imply that a leaching mechanism must be available and operative so that catalysis by a homogeneous pathway becomes feasible.
Catalysis of Heck reaction by smaller palladium clusters in preference to larger nanoparticles was demonstrated by a unique experiment26c that has been cited in several articles.4a,5,6,30 A special reactor has two compartments separated by a membrane that allows particles smaller than 5 nm in diameter to pass from one compartment to the other (see schematic diagram below – Fig. 3). It was shown that the reaction took place gradually over a long period of time in a compartment that originally did not contain the PdNps; only after particles of a smaller size migrated into this compartment did the reaction commence. A solid base, sodium carbonate was used and added only to the compartment that did not contain the PdNps initially. Therefore, the reaction could not simultaneously take place in the other compartment containing large PdNps. Diffusion of smaller particles from one compartment to the other to catalyze the reaction required an unusually long reaction time (120 h). No experiment was performed with the orginal nanoparticles in a single pot for comparison of reaction kinetics. In a large number of reactions, even with homogeneous precursors, the nanoparticle size is 8–12 nm,6b,c,31 they are used in 1 mol% range and the reactions are over in 6–12 h. In contrast, the reaction described with the two-compartment reactor used an infinitesimally small amount of nanoparticles of smaller dimension which accumulated over a long period of time and the rate of catalytic reaction is significantly low. Despite the innovative design of the experiment, it only establishes that particles capable of diffusing through a 5 nm membrane catalyze the Heck coupling reaction. It clarifies neither the role of larger nanoparticles nor the involvement of atomic palladium in catalysis.
 | ||
| Fig. 2 Two-chamber reactor separated by a membrane. | ||
When the substrate is consumed, the ‘ligand-less’ palladium returns to the PdNp scaffold [Fig. 2: “release and capture”].27,29,32 Palladium nanoparticles supported on graphene afford coupling products from a polymer-bound iodoarene, indicating catalysis by leached palladium that returns to its support at the end of reaction.33 Returning to the scaffold is claimed to be faster at higher temperature27,32d and palladium is deposited on the Pd-cluster rather than an empty space on the support. In such a study, the soluble catalyst is allowed to perform in the presence of empty support and then the accumulation of palladium on the support is monitored at the end. Therefore, we are dealing with adsorption phenomena in tandem with catalysis and we do not know when or whether nanoparticles are involved (no TEM data given).
 | ||
| Fig. 3 Schematic representation of “release and capture” mechanism with leached palladium. | ||
(iii) “Homeopathic” concentration of catalyst more effective
Reetz reported34 that palladium at high dilution can catalyze Heck reactions in the presence of N,N-dimethylglycine (DMG); use of as little as 0.0009 mol% was effective. Later, de Vries along with Reetz highlighted the fact that high dilution of the catalyst solution (and popularized the term “homeopathic” to describe it) improves catalytic turnover and probably also suppresses atomic aggregation to form nanoparticles.35 This concept has been adapted for production of a drug intermediate on an industrial scale.36If “ligand-free” palladium atoms are indeed the catalyst in the reactions described by de Vries, the observation of an effective concentration range of palladium weighs in favour of molecular or homogeneous catalysis. A homeopathic concentration is expected to suppress aggregation, it is stated, but it does not reveal whether Pd(OAc)2 upon reduction forms nanoparticles promptly at that dilution and in the presence of a relatively large quantity of substrate. Homeopathic dilution also prevents smaller particles aggregating to form larger ones, so no precipitation of Pd black is observed. No stabilizer was used for the nanoparticles. It has always been assumed that nanoparticles exist in equilibrium with ‘molecular’ Pd (very high substrate/catalyst ratio leads to favourable kinetics), but no mention of the presence of PdNps in relation to the experiments of de Vries' group has been made36,37 It can also be argued that this particular example essentially represents a ‘homogeneous’ catalysis for the most part. The nanoparticles are formed only when the substrate has been nearly consumed. A significant feature is the existence of a well-defined range of catalyst concentrations that assures an acceptable level of product formation in a reasonable period of time. The same has been claimed to be true for a good number of instances of catalysis by “leached” palladium because the amount of palladium detected in solution is typically in the ppm range. A related study38 determined that the rate of reaction catalyzed by Pd(OAc)2 was independent of catalyst concentration (0.05 to 5 mol%), citing this as evidence of heterogeneous rather than homogeneous catalysis.
(iv) Simultaneous catalysis by Np surface and leached atom with rate difference
In principle, we could consider a situation where both the nanoparticle surface and leached palladium species are catalytically active. This is a possibility that is rarely considered,1c probably because the possibility is fraught with myriad complexities. Since there are several reports where leached out palladium is beyond the level of detection by ICP MS (Inductively Coupled Plasma Mass Spectrometry) or similar techniques but the product is formed, it may be assumed that the catalysis is occurring predominantly on the surface of the nanoparticles. When leached palladium is detected, variation of its concentration with time (progress of reaction) has been monitored, contours of the originally dispersed nanoparticles have been probed by analytical techniques, but only very limited studies exist where their catalytic role has been confirmed without ambiguity. Mere presence is not indicative of catalytic activity. If the surface catalysis rate is considerably higher than leached particle/atom catalysis, or vice versa, how accurately can we segregate the individual contributions? Literature reports seem to suggest that smaller nanoparticles (1–3 nm) may have very active surface atoms that can perform catalysis at a higher rate.(v) Palladium nanoparticles supported on solid matrix as catalyst
There are numerous reports of palladium nanoparticles being deposited, encapsulated or embedded on a solid matrix39 ranging from well-structured metal oxides,40 clay41 or zeolites42 to softer matrices such as dendrimers43 or polymers.44 Carbon nanotubes45 as well as graphene46 have also been used as supports. Additional stability, ease of separation and greater recyclability inspired these studies.47Often a palladium salt was reduced to Pd(0) in the presence of the solid support. In such cases, it is possible that a great deal of atomic palladium is physically adsorbed on the matrix. This could explain the ready leaching from such a support and re-adsorption. For oxides, clays or zeolites, a calcinations step often follows adsorption of the nascent metal. Nanoparticles are believed to form during this calcination step and their dimensions can be measured by TEM. Sometimes, the nanoparticles are first produced and then allowed to nest in porous supports. Irrespective of the mode of preparation or nature of support, and independent of the individual (atomic) or collective (nano-cluster) existence of palladium, catalytic reactions with these supported palladium species are popular though they frequently invoke the issue of leaching and leached material being considered the ‘actual’ catalyst.29,48
It is only in the case of a few supported palladium catalysts that coupling reactions of chloroarenes, even with electron-donating substituents, have been reported.14,17,30 In such examples, anionic oxygen-donor ligands appear to render the catalytically active Pd centre substantially more electron-rich so that chloroarenes undergo oxidative addition at reasonable rates. This should be true, then, largely for palladium atoms, but not necessarily for clusters.49 An example is provided by ‘layered double hydroxide supported nanopalladium catalyst’ for coupling reactions.50 TEM studies determined the size of the Pd clusters as 4–6 nm (diameter), which showed no change over several catalytic cycles; further, minimal leaching of Pd was identified during the course of reaction (Fig. 4).
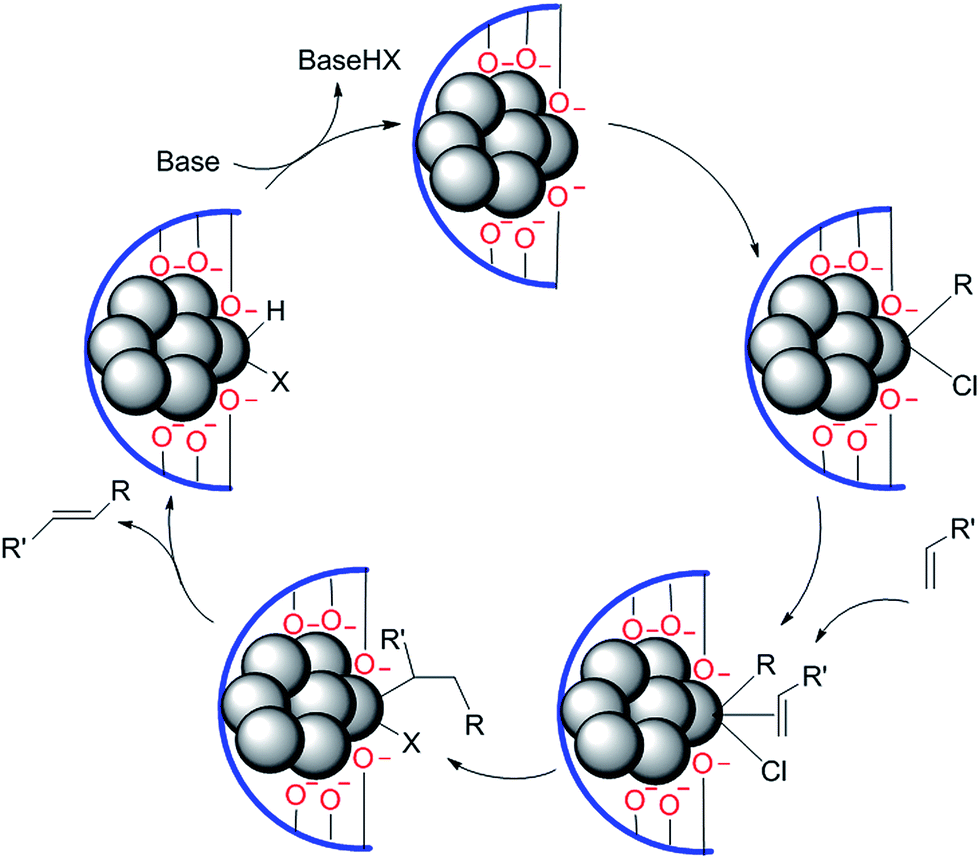 | ||
| Fig. 4 Plausible mechanism for the layered double hydroxide (LDH-Pd0) catalyzed Heck olefination of chloroarene. | ||
Triggering of several coupling reactions by this catalyst points to a common reaction mechanism. This may not be considered as catalysis by clusters; it probably follows a mechanism as proposed by Amatore and Jutand51 involving palladium as anionic dimers. The palladium centre is activated through participation of neighbouring oxide anions. PEG has no such ionic oxides despite high oxygen donor concentration on its surface, and it alone cannot impart such reactivity to palladium.52 On the other hand, anchored ligands with donor groups can activate a specific palladium centre in a cluster to enable involvement of choroarenes in coupling reaction.53 These should more appropriately be considered as ‘immobilized’ homogeneous catalysts. Leaching is not a significant issue for such catalysts as leached PdNps do not interact with chloroarenes at any useful rate.
When Pd(0) is allowed to be deposited on a surface, leaching is a common phenomenon. Ligand-less palladium is dispersed in a solution phase after release from the support and can commences a catalytic cycle. When the substrate is consumed, the catalytically active palladium reassembles onto the matrix. Even for dendrimeric scaffolds, leaching has been shown to be the most viable pathway for catalysis, which can operate at a very high dilution (‘homeopathic’ concentration, ppm level).54 However, when PdNps were encapsulated in a PEG matrix grafted on silica, the reaction continued unabated even in the presence of thiolated silica gel, suggesting insignificant contribution to catalysis by leached entities.55
Catalysis of coupling reactions using Pd/C as catalyst has been explored extensively because of its ready availability and generally good activity for a range of substrates.56 It is easy to recover and because charcoal is an excellent adsorbent, the availability of free palladium species in solution at the end of reaction is minimized. Since PdNps are seldom mentioned in the context of catalysis by Pd/C, we prefer to keep it outside the purview of the present discussion.
Examples of polymer-encapsulated PdNps described in the review by Molnár47 reveals that smaller-sized nanoparticles (1–3 nm) efficiently catalyze coupling reactions with minimal leaching and practically no change in nanoparticle shape or size. They can be recycled several times (7–10 times are most common) without loss of yield. These observations are consistent with heterogeneous catalysis on a nanoparticle surface.
4. Conclusion
The large body of literature offers a few general agreements: (a) smaller nanoparticles (<5 nm) are better as catalysts and with such smaller nanoparticles, catalysis probably occurs on the nanoparticle surface; (b) palladium nanoparticles generally do not catalyse coupling reactions of chloroarenes; (c) high dilution or ‘homeopathic’ concentration of ligand-less palladium display useful levels of catalysis, especially at higher temperatures; and, (d) palladium nanoparticles immobilized on a solid matrix are prone to leaching unless stabilized by co-ordinating functional groups.Catalysis by leached palladium only, even when a heterogeneous platform is used, is still a contentious issue. Some groups failed to detect significant levels of palladium during a reaction. Proponents of ‘catalysis by leached palladium’ theory would explain such observation as a ‘rapid release and capture of leached palladium’ by the matrix. Some groups, on the other hand, profiled the growth of palladium concentration with progress of reaction and claimed it as evidence for catalysis solely by leached palladium. It is indeed possible that there is no one universal mechanism. Variations of nanoparticle size and their varied origins, the nature of the diverse supports and scaffolds that house them, reaction parameters and substrate functionality – all contribute towards determining the course of catalysis. What stands out beyond dispute is the efficiency and advantage of such nanoparticle catalysis that led to their wide acceptability in organic synthesis.
Acknowledgements
Research fellowship from CSIR (A. B.) and IFCPAR (K. G.), New Delhi, and IACS, Kolkata (K. G.), are gratefully acknowledged. A. S. wishes to thank the Leverhulme Trust, UK, for a visiting fellowship to the laboratory of D. W. K.References
- (a) L. Yin and J. Liebscher, Chem. Rev., 2007, 107, 133 CrossRef CAS PubMed; (b) S. K. Beaumont, J. Chem. Technol. Biotechnol., 2012, 87, 595 CrossRef CAS; (c) R. M. Crooks, M. Zhao, L. Sun, V. Chechik and L. K. Yeung, Acc. Chem. Res., 2001, 34, 181 CrossRef CAS PubMed; (d) A. Roucoux, J. Schulz and H. Patin, Chem. Rev., 2002, 102, 3757 CrossRef CAS PubMed.
- M. Moreno-Manas and R. Pleixats, Acc. Chem. Res., 2003, 36, 638 CrossRef CAS PubMed.
- (a) N. T. S. Phan, M. V. Sluys and C. W. Jones, Adv. Synth. Catal., 2006, 348, 609 CrossRef CAS; (b) J. Durand, E. Teuma and M. Gmez, Eur. J. Inorg. Chem., 2008, 3577 CrossRef CAS.
- (a) A. Balanta, C. Godard and C. Claver, Chem. Soc. Rev., 2011, 40, 4973 RSC; (b) A. Fihri, M. Bouhrara, B. Nekoueisharki, J. M. Basset and V. Polshettiwar, Chem. Soc. Rev., 2011, 40, 5181 RSC; (c) D. Astruc, F. Lu and J. R. Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852 CrossRef CAS PubMed; (d) M. Pérez-Lorenzo, J. Phys. Chem. Lett., 2012, 3, 167 CrossRef.
- J. D. Scholten, B. C. Leal and J. Dupont, ACS Catal., 2012, 2, 184 CrossRef CAS.
- (a) L. D. Pachón and G. Rothenberg, Appl. Organomet. Chem., 2008, 22, 288 CrossRef; (b) S. Sawoo, D. Srimani, P. Dutta, R. Lahiri and A. Sarkar, Tetrahedron, 2009, 65, 4367 CrossRef CAS; (c) A. Bej, D. Srimani and A. Sarkar, Green Chem., 2012, 14, 661 RSC; (d) M. T. Reetz, M. Winter, R. Breinbauer, T. Thurn-Albrecht and W. Vogel, Chem.–Eur. J., 2001, 7, 1084 CrossRef CAS; (e) N. Erathodiyil, S. Ooi, A. M. Seayad, Y. Han, S. S. Lee and J. Y. Ying, Chem.–Eur. J., 2008, 14, 3118 CrossRef CAS PubMed; (f) M. Hyotanishi, Y. Isomura, H. Yamamoto, H. Kawasaki and Y. Obora, Chem. Commun., 2011, 47, 5750 RSC; (g) K. Wada, K. Yano, T. Kondo and T. Mitsudo, Catal. Lett., 2006, 112, 63 CrossRef CAS.
- (a) A. F. Lee, P. J. Ellis, I. J. S. Fairlamb and K. Wilson, Dalton Trans., 2010, 39, 10473 RSC; (b) B. Veisz, Z. Kiràly, L. Tóth and B. Pécz, Chem. Mater., 2002, 14, 2882 CrossRef CAS.
- M. Beller, H. Fischer, K. Kiihlein, C.-P. Reisinger and W. A. Herrmarm, J. Organomet. Chem., 1996, 520, 257 CrossRef CAS.
- (a) M. T. Reetz and G. Lohmer, Chem. Commun., 1996, 1921 RSC; (b) M. T. Reetz, R. Breinbauer and K. Wannlnger, Tetrahedron Lett., 1996, 37, 4499 CrossRef CAS; (c) M. T. Reetz and E. Westermann, Angew. Chem., Int. Ed., 2000, 39, 165 CrossRef CAS.
- (a) T. Jeffery, Tetrahedron, 1996, 52, 10113 CrossRef CAS; (b) T. Jeffery and M. David, Tetrahedron Lett., 1998, 39, 5751 CrossRef CAS.
- Y. Li, X. M. Hong, D. M. Collard and M. A. El-Sayed, Org. Lett., 2000, 2, 2385 CrossRef CAS PubMed.
- A. Gniewek, J. J. Ziółkowski, A. M. Trzeciak, M. Zawadzki, H. Grabowsk and J. Wrzyszcz, J. Catal., 2008, 254, 121 CrossRef CAS.
- (a) R. Bernini, S. Cacchi, G. Fabrizi, G. Forte, F. Petrucci, A. Prastaro, S. Niembro, A. Shafird and A. Vallribera, Green Chem., 2010, 12, 150 RSC; (b) H. Liu, L. Wang and P. Li, Synthesis, 2008, 2405 CAS; (c) P.-H. Li and L. Wang, Adv. Synth. Catal., 2006, 348, 681 CrossRef CAS; (d) P. G. Steel and C. W. T. Teasdale, Tetrahedron Lett., 2004, 45, 8977 CrossRef CAS; (e) J.-N. Young, T.-C. Chang, S.-C. Tsai, L. Yang and S. J. Yu, Journal of Catalysis, 2010, 272, 253 CrossRef CAS; (f) L. Zhang, L. Wang, H. Li and P. Li, Synth. Commun., 2008, 38, 1498 CrossRef CAS; (g) F. Durapa, M. Rakapb, M. Aydemira and S. Özkar, Appl. Catal., A, 2010, 382, 339 CrossRef; (h) D. Rosario-Amorin, X. Wang, M. Gaboyard, R. Clérac, S. Nlate and K. Heuzé, Chem.–Eur. J., 2009, 15, 12636 CrossRef CAS PubMed; (i) J. C. Xiao and J. M. Shreeve, J. Org. Chem., 2005, 70, 3072 CrossRef CAS PubMed.
- W. Han, C. Liu and Z.-L. Jin, Org. Lett., 2007, 9, 4005 CrossRef CAS PubMed.
- V. Kogan, Z. Aizenshtat, R. Popovitz-Biro and R. Neumann, Org. Lett., 2002, 4, 3529 CrossRef CAS PubMed.
- D. D. Das and A. Sayari, J. Catal., 2007, 246, 60 CrossRef CAS.
- B. J. Gallon, R. W. Kojima, R. B. Kaner and P. L. Diaconescu, Angew. Chem., Int. Ed., 2007, 46, 7251 CrossRef CAS PubMed.
- V. Chandrasekhar, R. S. Narayanan and P. Thilagar, Organometallics, 2009, 28, 5883 CrossRef CAS.
- P. J. Ellis, I. J. S. Fairlamb, S. F. J. Hackett, K. Wilson and A. F. Lee, Angew. Chem., Int. Ed., 2010, 49, 1820 CrossRef CAS PubMed.
- (a) G. M. Whitesides, M. Hackett, R. L. Brainard, J.-P. P. M. Lavalleye, A. F. Sowinski, A. N. Izumi, S. S. Moore, D. W. Brown and E. M. Staudt, Organometallics, 1985, 4, 1819 CrossRef CAS; (b) A. R. Hajipour and F. Rafiee, Synth. Commun., 2013, 43, 1314 CrossRef CAS; (c) K. Q. Yu, W. Sommer, J. M. Richardson, M. Weck and C. W. Jones, Adv. Synth. Catal., 2005, 347, 161 CrossRef CAS; (d) K. Q. Yu, W. Sommer, M. Weck and C. W. Jones, J. Catal., 2004, 226, 101 CrossRef CAS; (e) M. R. Eberhard, Org. Lett., 2004, 6, 2125 CrossRef CAS PubMed; (f) W. J. Sommer, K. Q. Yu, J. S. Sears, Y. Y. Ji, X. L. Zheng, R. J. Davis, C. D. Sherrill, C. W. Jones and M. Weck, Organometallics, 2005, 24, 4351 CrossRef CAS; (g) D. Olsson, P. Nilsson, M. El Masnaouy and O. F. Wendt, Dalton Trans., 2005, 1924 RSC.
- S. Reimann, J. Stötzel, R. Frahm, W. Kleist, J.-D. Grunwaldt and A. Baiker, J. Am. Chem. Soc., 2011, 133, 3921 CrossRef CAS PubMed.
- D. Peral, F. G. Villarraga, X. Sala, J. Pons, J. C. Bayón, J. Ros, M. Guerrero, L. Vendier, P. Lecante, J. G. Antón and K. Philippot, Catal. Sci. Technol., 2013, 3, 475 CAS.
- A. N. Cammidge and K. V. L. Crépy, Chem. Commun., 2000, 1723 RSC.
- K. Sawai, R. Tatumi, T. Nakahodo and H. Fujihara, Angew. Chem., Int. Ed., 2008, 47, 6917 CrossRef CAS.
- J. Dupont, A. S. Gruber, G. S. Fonseca, A. L. Monteiro and G. Ebeling, Organometallics, 2001, 20, 171 CrossRef CAS.
- (a) J. Le Bars, U. Specht, J. S. Bradley and D. G. Blackmond, Langmuir, 1999, 15, 7621 CrossRef CAS; (b) A. Gniewek, A. M. Trzeciak, J. J. Ziólkowsky, L. Kepinski, J. Wrzyszcz and W. Tylus, J. Catal., 2005, 229, 332 CrossRef CAS; (c) M. B. Thathagar, J. E. ten Elshof and G. Rothenberg, Angew. Chem., Int. Ed., 2006, 45, 2886 CrossRef CAS PubMed.
- F. Zhao, B. M. Bhanage, M. Shirai and M. Arai, Chem.–Eur. J., 2000, 6, 843 CrossRef CAS.
- J. P. Collman, K. M. Kosydar, M. Bressan, W. Lamanna and T. Garrett, J. Am. Chem. Soc., 1984, 106, 2569 CrossRef CAS.
- I. W. Davies, L. Matty, D. L. Hughes and P. J. Reider, J. Am. Chem. Soc., 2001, 123, 10139 CrossRef CAS.
- (a) A. Kumar, G. K. Rao, S. Kumar and A. K. Singh, Dalton Trans., 2013, 42, 5200 RSC; (b) A. F. Schmidt and A. A. Kurokhtina, Kinet. Catal., 2012, 53, 714 CrossRef CAS; (c) M. H. G. Prechtl, J. D. Scholten and J. Dupont, Molecules, 2010, 15, 3441 CrossRef CAS PubMed; (d) R. Chinchilla and C. Nájera, Chem. Rev., 2007, 107, 874 CrossRef CAS PubMed.
- (a) D. Srimani, S. Sawoo and A. Sarkar, Org. Lett., 2007, 9, 3639 CrossRef CAS PubMed; (b) D. Srimani, A. Bej and A. Sarkar, J. Org. Chem., 2010, 75, 4296 CrossRef CAS PubMed; (c) A. Bej, A. Chakraborty and A. Sarkar, RSC Adv., 2013, 3, 15812 RSC.
- (a) F. Zhao, M. Shirai and M. Arai, J. Mol. Catal. A: Chem., 2000, 154, 39 CrossRef CAS; (b) F. Zhao, K. Murakami, M. Shirai and M. Arai, J. Catal., 2000, 194, 479 CrossRef CAS; (c) L. Djakovitch and K. Koehler, J. Mol. Catal. A: Chem., 1999, 142, 275 CrossRef CAS; (d) M. Wagner, K. Köhler, L. Djakovitch, S. Weinkauf, V. Hagen and M. Muhler, Top. Catal., 2000, 13, 319 CrossRef CAS; (e) F. Zhao, M. Shirai, Y. Ikushima and M. Arai, J. Mol. Catal. A: Chem., 2002, 180, 211 CrossRef CAS; (f) A. Biffis, J. Mol. Catal. A: Chem., 2001, 165, 303 CrossRef CAS; (g) A. Biffis, M. Zecca and M. Basato, Eur. J. Inorg. Chem., 2001, 1131 CrossRef CAS.
- G. M. Scheuermann, L. Rumi, P. Steurer, W. Bannwarth and R. Mülhaupt, J. Am. Chem. Soc., 2009, 131, 8262 CrossRef CAS PubMed.
- M. T. Reetz, E. Westermann, R. Lohmer and G. Lohmer, Tetrahedron Lett., 1998, 39, 8449 CrossRef CAS.
- (a) M. T. Reetz and J. G. de Vries, Chem. Commun., 2004, 1559 RSC; (b) J. G. de Vries, Dalton Trans., 2006, 421 RSC.
- A. H. M. de Vries, J. M. C. A. Mulders, J. H. M. Mommers, H. J. W. Henderickx and J. G. de Vries, Org. Lett., 2003, 5, 3285 CrossRef CAS PubMed.
- (a) A. Alimardanov, L. S. de Vondervoort, A. H. M. de Vries and J. G. de Vriesa, Adv. Synth. Catal., 2004, 346, 1812 CrossRef CAS; (b) A. H. M. de Vries, F. J. Parlevliet, L. S. de Vondervoort, J. H. M. Mommers, H. J. W. Henderickx, M. A. M. Walet and J. G. de Vries, Adv. Synth. Catal., 2002, 344, 996 CrossRef CAS; (c) J. G. de Vries and A. H. M. de Vries, Eur. J. Org. Chem., 2003, 799 CrossRef CAS.
- J. Liu, Y. Deng, H. Wang, H. Zhang, G. Yu, B. Wu, H. Zhang, Q. Li, T. B. Marder, Z. Yang and A. Lei, Org. Lett., 2008, 10, 2661 CrossRef CAS PubMed.
- J. M. Campelo, D. Luna, R. Luque, J. M. Marinas and A. A. Romero, ChemSusChem, 2009, 2, 18 CrossRef CAS PubMed.
- (a) M. L. Kantam, S. Roy, M. Roy, B. Sreedhar and B. M. Choudary, Adv. Synth. Catal., 2005, 347, 2002 CrossRef CAS; (b) M. L. Kantam, S. Roy, M. Roy, M. S. Subhas, P. R. Likhar, B. Sreedhar and B. M. Choudary, Synlett, 2006, 2747 CrossRef CAS; (c) A. Gniewek, J. J. Ziółkowski, A. M. Trzeciak, M. Zawadzki, H. Grabowska and J. Wrzyszcz, J. Catal., 2008, 254, 121 CrossRef CAS; (d) A. Monopoli, A. Nacci, V. Calò, F. Ciminale, P. Cotugno, A. Mangone, L. C. Giannossa, P. Azzone and N. Cioffi, Molecules, 2010, 15, 4511 CrossRef CAS PubMed; (e) G. Budroni, A. Corma, H. García and A. Primo, J. Catal., 2007, 251, 345 CrossRef CAS.
- (a) A. V. Martínez, J. A. Mayoral and J. I. García, Appl. Catal., A, 2014, 472, 21 CrossRef; (b) G. B. B. Varadwaj, S. Rana and K. Parida, J. Phys. Chem. C, 2014, 118, 1640 CrossRef CAS; (c) B. J. Borah, S. J. Borah, K. Saikia and D. K. Dutta, Appl. Catal., A, 2014, 479, 350 CrossRef.
- (a) N. Ren, Y.-H. Yang, Y.-H. Zhang, Q.-R. Wang and Y. Tang, J. Catal., 2007, 246, 215 CrossRef CAS; (b) M. Choi, D.-H. Lee, K. Na, B.-W. Yu and R. Ryoo, Angew. Chem., Int. Ed., 2009, 48, 3673 CrossRef CAS PubMed; (c) K. Okumura, H. Matsui, T. Tomiyama, T. Sanada, T. Honma, S. Hirayama and M. Niwa, ChemPhysChem, 2009, 10, 3265 CrossRef CAS PubMed; (d) F. Durapa, M. Rakap, M. Aydemir and S. Ozkar, Appl. Catal., A, 2010, 382, 339 CrossRef; (e) K. Okumura, T. Tomiyama, S. Okuda, H. Yoshida and M. Niwa, J. Catal., 2010, 273, 156 CrossRef CAS.
- (a) J. C. Garcia-Martinez, R. Lezutekong and R. M. Crooks, J. Am. Chem. Soc., 2005, 127, 5097 CrossRef CAS PubMed; (b) C. Ornelas, J. Ruiz, L. Salmon and D. Astruc, Adv. Synth. Catal., 2008, 350, 837 CrossRef CAS; (c) E. Murugan, J. N. Jebaranjitham and A. Usha, Appl. Nanosci., 2012, 2, 211 CrossRef CAS; (d) C. Deraedt, L. Salmon and D. Astruc, Adv. Synth. Catal., 2014, 356, 2525 CrossRef CAS; (e) J.-H. Noh and R. Meijboom, J. Colloid Interface Sci., 2014, 415, 57 CrossRef CAS PubMed; (f) M. Pittelkow, K. M. Poulsen, U. Boas and J. B. Christensen, Langmuir, 2003, 19, 7682 CrossRef CAS.
- (a) S. Ogasawara and S. Kato, J. Am. Chem. Soc., 2010, 132, 4608 CrossRef CAS PubMed; (b) B. J. Gallon, R. W. Kojima, R. B. Kaner and P. L. Diaconescu, Angew. Chem., Int. Ed., 2007, 46, 7251 CrossRef CAS PubMed; (c) A. Ohtaka, T. Teratani, R. Fujii, K. Ikeshita, O. Shimomura and R. Nomura, Chem. Commun., 2009, 7188 RSC; (d) C. Evangelisti, N. Panziera, P. Perticib, G. Vitulli, P. Salvadori, C. Battocchio and G. Polzonetti, Journal of Catalysis, 2009, 262, 287 CrossRef CAS; (e) C.-A. Lin and F.-T. Luo, Tetrahedron Lett., 2003, 44, 7565 CrossRef CAS; (f) X. Jiang, G. Wei, X. Zhang, W. Zhang, P. Zheng, F. Wen and L. Shi, J. Mol. Catal. A: Chem., 2007, 277, 102 CrossRef CAS; (g) S. Schweizer, J.-M. Becht and C. L. Drian, Tetrahedron, 2010, 66, 765 CrossRef CAS; (h) J.-H. Kim, J.-W. Kim, M. Shokouhimehr and Y.-S. Lee, J. Org. Chem., 2005, 70, 6714 CrossRef CAS PubMed.
- (a) X. R. Ye, Y. Linb and C. M. Wai, Chem. Commun., 2003, 642 RSC; (b) X. Chen, Y. Hou, H. Wang, Y. Cao and J. He, J. Phys. Chem. C, 2008, 112, 8172 CrossRef CAS; (c) H.-B. Pan, C. H. Yen, B. Yoon, M. Sato and C. M. Wai, Synth. Commun., 2006, 36, 3473 CrossRef CAS.
- (a) A. R. Siamaki, A. E. R. S. Khder, V. Abdelsayed, M. S. El-Shall and B. F. Gupton, Journal of Catalysis, 2011, 279, 1 CrossRef CAS; (b) Y. Li, X. Fan, J. Qi, J. Ji, S. Wang, G. Zhang and F. Zhang, Nano Res., 2010, 3, 429 CrossRef CAS; (c) S. Moussa, A. R. Siamaki, B. F. Gupton and M. S. El-Shall, ACS Catal., 2012, 2, 145 CrossRef CAS; (d) G. M. Scheuermann, L. Rumi, P. Steurer, W. Bannwarth and R. Mülhaupt, J. Am. Chem. Soc., 2009, 131, 8262 CrossRef CAS PubMed.
- Á. Molnár, Chem. Rev., 2011, 111, 2251 CrossRef PubMed.
- (a) J. M. Richardson and C. W. Jones, J. Catal., 2007, 251, 80 CrossRef CAS; (b) A. Biffis, M. Zecca and M. Basato, Eur. J. Inorg. Chem., 2001, 1131 CrossRef CAS; (c) S. S. Pröckl, W. Kleist and K. Köhler, Tetrahedron, 2005, 61, 9855 CrossRef; (d) S. S. Prcckl, W. Kleist, M. A. Gruber and K. Köhler, Angew. Chem., Int. Ed., 2004, 43, 1881 CrossRef PubMed; (e) K. Köhler, W. Kleist and S. S. Pröckl, Inorg. Chem., 2007, 46, 1876 CrossRef; (f) M. Lysén and K. Köhler, Synthesis, 2006, 692 CrossRef; (g) W. Kleist, S. S. Pröckl1 and K. Köhler, Catal. Lett., 2008, 125, 197 CrossRef CAS.
- S. R. Sanjaykumar, B. D. Mukri, S. Patil, G. Madras and M. S. Hegde, J. Chem. Sci., 2011, 123, 47 CrossRef CAS.
- B. M. Choudary, S. Madhi, N. S. Chowdari, M. L. Kantam and B. Sreedhar, J. Am. Chem. Soc., 2002, 124, 14127 CrossRef CAS PubMed.
- C. Amatore and A. Jutand, Acc. Chem. Res., 2000, 33, 314 CrossRef CAS PubMed.
- C. Luo, Y. Zhang and Y. Wang, J. Mol. Catal. A: Chem., 2005, 229, 7 CrossRef CAS.
- (a) M.-J. Jin and D.-H. Lee, Angew. Chem., Int. Ed., 2010, 49, 1119 CrossRef CAS PubMed; (b) D. A. Alonso, C. Nàjera and M. C. Pacheco, Org. Lett., 2000, 2, 1823 CrossRef CAS; (c) R. B. Bedford, S. L. Hazelwood, P. N. Horton and M. B. Hursthouse, Dalton Trans., 2003, 4164 RSC.
- (a) A. K. Diallo, C. Ornelas, L. Salmon, J. R. Aranzaes and D. Astruc, Angew. Chem., Int. Ed., 2007, 46, 8644 CrossRef CAS PubMed; (b) D. Astruc, Inorg. Chem., 2007, 46, 1884 CrossRef CAS PubMed; (c) C. Deraedt and D. Astruc, Acc. Chem. Res., 2014, 47, 494 CrossRef CAS.
- P. Dutta and A. Sarkar, Adv. Synth. Catal., 2011, 353, 2814 CrossRef CAS.
- (a) H. Sakurai, T. Tsukuda and T. Hirao, J. Org. Chem., 2002, 67, 2721 CrossRef CAS PubMed; (b) T. Maegawa, Y. Kitamura, S. Sako, T. Udzu, A. Sakurai, A. Tanaka, Y. Kobayashi, K. Endo, U. Bora, T. Kurita, A. Kozaki, Y. Monguchi and H. Sajiki, Chem.–Eur. J., 2007, 13, 5937 CrossRef CAS PubMed; (c) M. Lamblin, H. Bares, J. Dessolin, C. Marty, N. Bourgougnon and F.-X. Felpin, Eur. J. Org. Chem., 2012, 5525 CrossRef CAS; (d) T. Yanase, Y. Monguchi and H. Sajiki, RSC Adv., 2012, 2, 590 RSC.
| This journal is © The Royal Society of Chemistry 2016 |